
Optimizing Rhabdomyosarcoma Treatment in Adolescents and Young Adults


Simple Summary
Abstract
1. Introduction
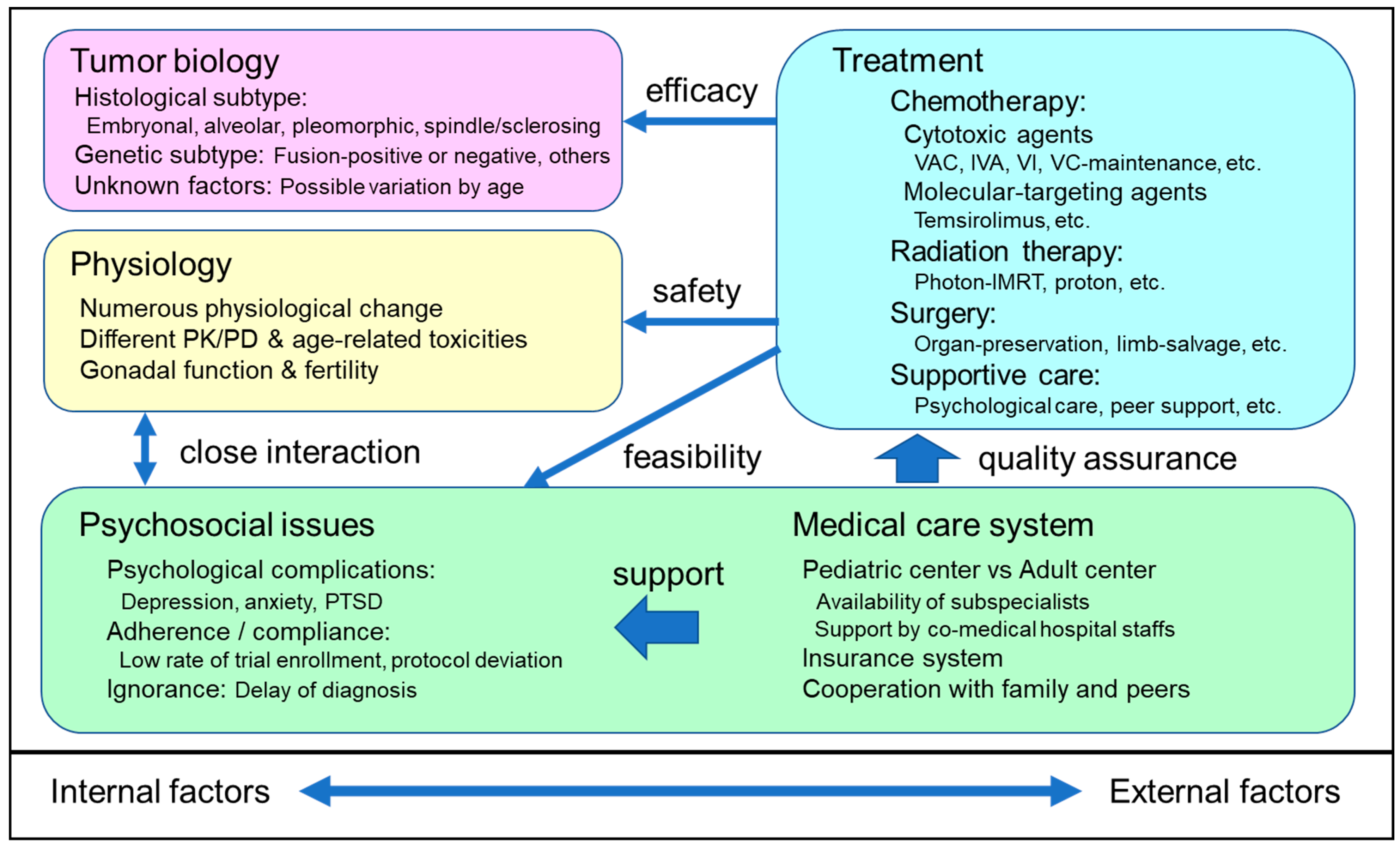
2. Tumor Biology of RMS
3. Physiology of AYA
4. Development of Optimal Multimodal Treatment for AYA RMS
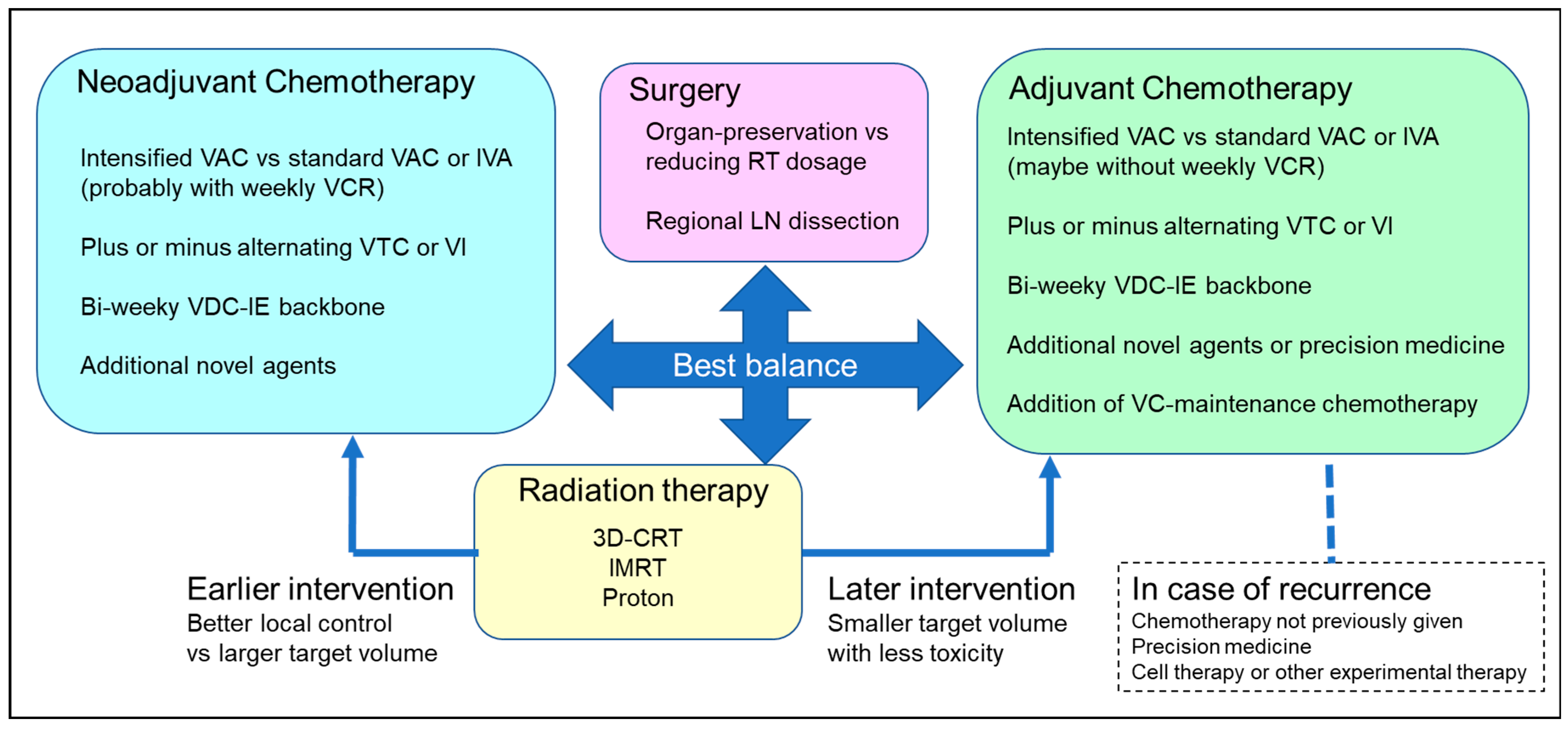
4.1. Differentiating Neoadjuvant and Adjuvant Chemotherapy
4.2. Optimal Dosing of Alkylating Agents
4.3. Optimal Chemotherapy Duration
4.4. VAC, Alternating VAC, and Other Regimens
4.5. Possible Re-Evaluation of Cytotoxic Chemotherapy Agents
4.6. Incorporating Molecularly Targeted Agents, Immunotherapy, and Cell Therapy
4.7. Special Considerations Regarding Local Treatment
4.8. Psychosocial Issues in AYA RMS and the Perspective of Clinical Researchers
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Linardie, C.M.; Wexler, L.H. Rhabdomyosarcoma. In Pizzo and Poplack’s Pediatric Oncology, 8th ed.; Blaney, S.M., Adamson, P.C., Helman, L.J., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2020; pp. 693–720. [Google Scholar]
- Surveillance, Epidemiology and End Results (SEER) Program. Cancer Epidemiology in Older Adolescents and Young Adults 15 to 29 Years of Age. Available online: https://seer.cancer.gov/archive/publications/aya/aya_mono_complete.pdf (accessed on 25 March 2022). (In Japanese)
- Joshi, D.; Anderson, J.R.; Paidas, C.; Breneman, J.; Parham, D.M.; Crist, W. Age is an independent prognostic factor in rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Pediatr. Blood Cancer 2004, 42, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: An analysis of 2,600 patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Trama, A.; Botta, L.; Foschi, R.; Ferrari, A.; Stiller, C.; Desandes, E.; Maule, M.M.; Merletti, F.; Gatta, G.; EUROCARE-5 Working Group. Survival of European adolescents and young adults diagnosed with cancer in 2000-07: Population-based data from EUROCARE-5. Lancet Oncol. 2016, 17, 896–906. [Google Scholar] [CrossRef]
- Ferrari, A.; Bleyer, A.; Patel, S.; Chiaravalli, S.; Gasparini, P.; Casanova, M. The challenge of the management of adolescents and young adults with soft tissue sarcomas. Pediatr. Blood Cancer 2018, 65, e27013. [Google Scholar] [CrossRef] [PubMed]
- Guersant, M.P. Polypes du vagin chez une petite fille de treize mois. Monit. Hop. 1854, 2, 187–189. [Google Scholar]
- Raney, R.B.; Bergeron, C.; Parham, D. English translation of M. Berard: Tumeur Embryonnaire Du Muscle Strie. [Embryonal tumor of striated muscle]. Lyon Med. 1894;77:52. Fetal Pediatr. Pathol. 2019, 38, 182–184. [Google Scholar] [CrossRef]
- Raney, R.B.; Oberlin, O.; Parham, D.M. An English translation of Joseph Luc Riopelle, MD, (Hotel-Dieu of Montreal), and Jean Paul Theriault (Hopital General of Verdun, Quebec, Canada): Sur une forme meconnue de sarcome des partiesmolles: Le rhabdomyosarcome alveolaire (concerning an unrecognized form of sarcoma of the soft tissues: Alveolar rhabdomyosarcoma). annales d’anatomie pathologique. 1956;1:88-111. Pediatr. Dev. Pathol. 2012, 15, 407–416. [Google Scholar]
- WHO Classification of Tumours Editorial Board. World Health Organization Classification of Tumours: Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020; pp. 201–213. [Google Scholar]
- Maurer, H.M.; Beltangady, M.; Gehan, E.A.; Crist, W.; Hammond, D.; Hays, D.M.; Heyn, R.; Lawrence, W.; Newton, W.; Ortega, J.; et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 1988, 61, 209–220. [Google Scholar] [CrossRef]
- Maurer, H.M.; Gehan, E.A.; Beltangady, M.; Crist, W.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer 1993, 71, 1904–1922. [Google Scholar] [CrossRef]
- Crist, W.; Gehan, E.A.; Ragab, A.H.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; Heyn, R.; et al. The Third Intergroup Rhabdomyosarcoma Study. J. Clin. Oncol. 1995, 13, 610–630. [Google Scholar] [CrossRef]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; Breneman, J.; Qualman, S.J.; Wiener, E.; Wharam, M.; et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J. Clin. Oncol. 2001, 19, 3091–3102. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.A.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; Parham, D.M.; Teot, L.A.; Wharam, M.D.; Breneman, J.C.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s oncology group study D9803. J. Clin. Oncol. 2009, 27, 5182–5188. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, D.S.; Chi, Y.Y.; Anderson, J.R.; Tian, J.; Arndt, C.A.S.; Bomgaars, L.; Donaldson, S.S.; Hayes-Jordan, A.; Mascarenhas, L.; McCarville, M.B.; et al. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2018, 36, 2770–2777. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, G.; Jenney, M.; Bergeron, C.; Gallego Melcón, S.; Ferrari, A.; Oberlin, O.; Carli, M.; Stevens, M.; Kelsey, A.; De Paoli, A.; et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): A multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1061–1071. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Gallego Melcón, S.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomized, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wie, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef]
- Shern, J.F.; Selfe, J.; Izquierdo, E.; Patidar, R.; Chou, H.C.; Song, Y.K.; Yohe, M.E.; Sindiri, S.; Wei, J.; Wen, X.; et al. Genomic classification and clinical outcome in rhabdomyosarcoma: A report from an international consortium. J. Clin. Oncol. 2021, 39, 2859–2871. [Google Scholar] [CrossRef]
- Casey, D.L.; Wexler, L.H.; Pitter, K.L.; Samstein, R.M.; Slotkin, E.K.; Wolden, S.L. Genomic determinants of clinical outcomes in rhabdomyosarcoma. Clin. Cancer Res. 2020, 26, 1135–1140. [Google Scholar] [CrossRef]
- Agaram, N.P.; LaQuaglia, M.P.; Alaggio, R.; Zhang, L.; Fujisawa, Y.; Ladanyi, M.; Wexler, L.H.; Antonescu, C.R. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod. Pathol. 2019, 32, 27–36. [Google Scholar] [CrossRef]
- Furlong, M.A.; Mentzel, T.; Fanburg-Smith, J.C. Pleomorphic rhabdomyosarcoma in adults: A clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod. Pathol. 2001, 14, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, G.; Compostella, A.; Ferrari, A.; Pastore, G.; Cecchetto, G.; Garaventa, A.; Indolfi, P.; De Sio, L.; Carli, M. Rhabdomyosarcoma in adolescents: A report from the AIEOP Soft Tissue Sarcoma Committee. Cancer 2012, 118, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Veal, G.J.; Hartford, C.M.; Stewart, C.F. Clinical pharmacology in the adolescent oncology patient. J. Clin. Oncol. 2010, 28, 4790–4799. [Google Scholar] [CrossRef] [PubMed]
- Crom, W.R.; de Graaf, S.S.; Synold, T.; Uges, D.R.; Bloemhof, H.; Rivera, G.; Christensen, M.L.; Mahmoud, H.; Evans, W.E. Pharmacokinetics of vincristine in children and adolescents with acute lymphocytic leukemia. J. Pediatr. 1994, 125, 642–649. [Google Scholar] [CrossRef]
- Frost, B.M.; Lönnerholm, G.; Koopmans, P.; Abrahamsson, J.; Behrendtz, M.; Castor, A.; Forestier, E.; Uges, D.R.; de Graaf, S.S. Vincristine in childhood leukaemia: No pharmacokinetic rationale for dose reduction in adolescents. Acta Pædiatrica 2003, 92, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Veal, G.J.; Cole, M.; Errington, J.; Parry, A.; Hale, J.; Pearson, A.D.; Howe, K.; Chisholm, J.C.; Beane, C.; Brennan, B.; et al. Pharmacokinetics of dactinomycin in a pediatric patient population: A United Kingdom Children’s Cancer Study Group Study. Clin. Cancer Res. 2005, 11, 5893–5899. [Google Scholar] [CrossRef] [PubMed]
- Tasso, M.J.; Boddy, A.V.; Price, L.; Wyllie, R.A.; Pearson, A.D.; Idle, J.R. Pharmacokinetics and metabolism of cyclophosphamide in paediatric patients. Cancer Chemother. Pharmacol. 1992, 30, 207–211. [Google Scholar] [CrossRef]
- Moore, M.J. Clinical pharmacokinetics of cyclophosphamide. Clin. Pharmacokinet. 1991, 20, 194–208. [Google Scholar] [CrossRef]
- Gupta, A.A.; Anderson, J.R.; Pappo, A.S.; Spunt, S.L.; Dasgupta, R.; Indelicato, D.J.; Hawkins, D.S. Patterns of chemotherapy-induced toxicities in younger children and adolescents with rhabdomyosarcoma: A report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. Cancer 2012, 118, 1130–1137. [Google Scholar] [CrossRef]
- Altaf, S.; Enders, F.; Lyden, E.; Donaldson, S.S.; Rodeberg, D.; Arndt, C. Age-related toxicity in patients with rhabdomyosarcoma: A report from the children’s oncology group. J. Pediatr. Hematol. Oncol 2014, 36, 599–604. [Google Scholar] [CrossRef]
- Gupta, A.A.; Chi, Y.Y.; Anderson, J.R.; Lyden, E.; Weigel, B.; Arndt, C.; Meyer, W.H.; Rosenberg, A.; Hawkins, D.S. Patterns of chemotherapy-induced toxicities and outcome in children and adolescents with metastatic rhabdomyosarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2017, 64, e26479. [Google Scholar] [CrossRef] [PubMed]
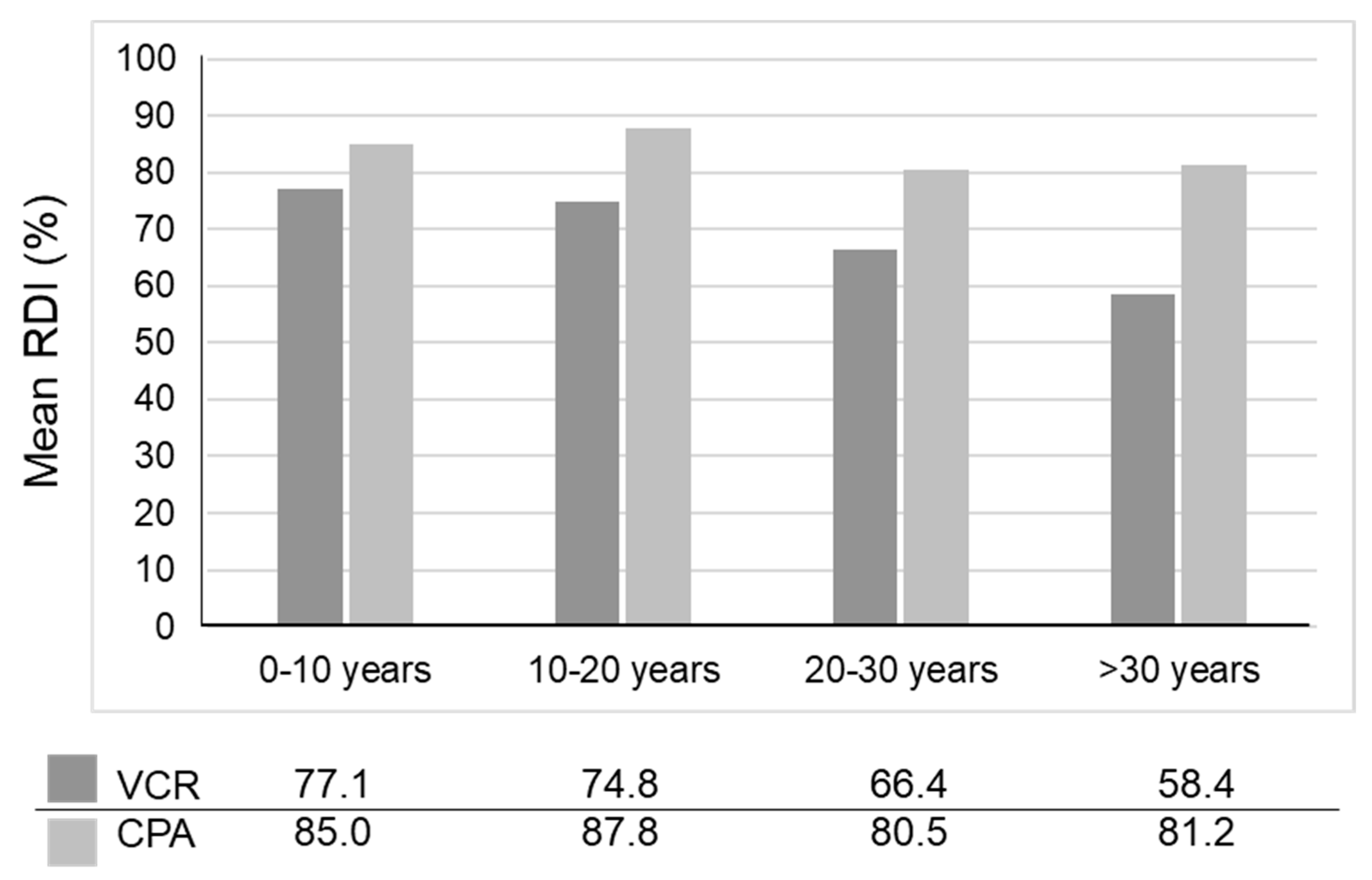
- Kojima, Y.; Hashimoto, K.; Ando, M.; Yonemori, K.; Yamamoto, H.; Kodaira, M.; Yunokawa, M.; Shimizu, C.; Tamura, K.; Hosono, A.; et al. Comparison of dose intensity of vincristine, d-actinomycin, and cyclophosphamide chemotherapy for child and adult rhabdomyosarcoma: A retrospective analysis. Cancer Chemother. Pharmacol. 2012, 70, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.; Anderson, J.R.; Kao, S.C.; Rodeberg, D.; Qualman, S.J.; Wolden, S.L.; Meyer, W.H.; Breitfeld, P.P.; Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Assessment of response to induction therapy and its influence on 5-year failure-free survival in group III rhabdomyosarcoma: The Intergroup Rhabdomyosarcoma Study-IV experience—A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 4909–4913. [Google Scholar]
- Pappo, A.S.; Lyden, E.; Breitfeld, P.; Donaldson, S.S.; Wiener, E.; Parham, D.; Crews, K.R.; Houghton, P.; Meyer, W.H.; Children’s Oncology Group. Children’s Oncology Group. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: The Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.J.; Lyden, E.; Anderson, J.R.; Meyer, W.H.; Parham, D.M.; Rodeberg, D.A.; Michalski, J.M.; Hawkins, D.S.; Arndt, C.A. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2016, 34, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.S.; Anderson, J.R.; Link, M.P.; Grier, H.E.; Qualman, S.J.; Maurer, H.M.; Breneman, J.C.; Wiener, E.S.; Crist, W.M. Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: Results from the Intergroup Rhabdomyosarcoma Study IV. J. Clin. Oncol. 2000, 18, 2427–2434. [Google Scholar] [CrossRef]
- Norton, L. A Gompertzian model of human breast cancer growth. Cancer Res. 1988, 48, 7067–7071. [Google Scholar]
- Goldie, J.H.; Coldman, A.J. A mathematic model for relating the drug sensitivity of tumors to their spontaneous mutation rate. Cancer Treat. Rep. 1979, 63, 1727–1733. [Google Scholar]
- Dantonello, T.M.; Stark, M.; Timmermann, B.; Fuchs, J.; Selle, B.; Linderkamp, C.; Handgretinger, R.; Hagen, R.; Feuchtgruber, S.; Kube, S.; et al. Tumour volume reduction after neoadjuvant chemotherapy impacts outcome in localised embryonal rhabdomyosarcoma. Pediatr. Blood Cancer 2015, 62, 16–23. [Google Scholar] [CrossRef]
- Dantonello, T.M.; Int-Veen, C.; Harms, D.; Leuschner, I.; Schmidt, B.F.; Herbst, M.; Juergens, H.; Scheel-Walter, H.G.; Bielack, S.S.; Klingebiel, T.; et al. Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J. Clin. Oncol. 2009, 27, 1446–1455. [Google Scholar] [CrossRef]
- Koscielniak, E.; Blank, B.; Vokuhl, C.; Kazanowska, B.; Ladenstein, R.; Niggli, F.; Ljungman, G.; Handgretinger, R.; Seitz, G.; Fuchs, J.; et al. Long-Term Clinical Outcome and Prognostic Factors of Children and Adolescents with Localized Rhabdomyosarcoma Treated on the CWS-2002P Protocol. Cancers 2022, 14, 899. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.L.; Chi, Y.Y.; Donaldson, S.S.; Hawkins, D.S.; Tian, J.; Arndt, C.A.; Rodeberg, D.A.; Routh, J.C.; Lautz, T.B.; Gupta, A.A.; et al. Increased loca l failure for patients with intermediate-risk rhabdomyosarcoma on ARST0531: A report from the Children’s Oncology Group. Cancer 2019, 125, 3242–3248. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, G.; Hawkins, D.S. An unsolved issue in rhabdomyosarcoma treatment: The duration of chemotherapy. Pediatr. Blood Cancer 2020, 67, e28174. [Google Scholar] [CrossRef] [PubMed]
- Haduong, J.H.; Heske, C.M.; Allen-Rhoades, W.; Xue, W.; Teot, L.A.; Rodeberg, D.A.; Donaldson, S.S.; Weiss, A.; Hawkins, D.S.; Venkatramani, R. An update on rhabdomyosarcoma risk stratification and the rationale for current and future Children’s Oncology Group clinical trials. Pediatr. Blood Cancer 2022, 69, e29511. [Google Scholar] [CrossRef]
- Chisholm, J.C.; Marandet, J.; Rey, A.; Scopinaro, M.; de Toledo, J.S.; Merks, J.H.; O’Meara, A.; Stevens, M.C.; Oberlin, O. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: A nomogram to better define patients who can be salvaged with further therapy. J. Clin. Oncol. 2011, 29, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Judson, I.; Radford, J.A.; Harris, M.; Blay, J.Y.; van Hoesel, Q.; le Cesne, A.; van Oosterom, A.T.; Clemons, M.J.; Kamby, C.; Hermans, C.; et al. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: A study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer 2001, 37, 870–877. [Google Scholar] [CrossRef]
- Makimoto, A.; Fang, J.; Maeda, H. Development of a Selective Tumor-Targeted Drug Delivery System: Hydroxypropyl-Acrylamide Polymer-Conjugated Pirarubicin (P-THP) for Pediatric Solid Tumors. Cancers 2021, 13, 3698. [Google Scholar] [CrossRef]
- Baum, E.S.; Gaynon, P.; Greenberg, L.; Krivit, W.; Hammond, D. Phase II trial cisplatin in refractory childhood cancer: Children’s Cancer Study Group Report. Cancer Treat. Rep. 1981, 65, 815–822. [Google Scholar] [PubMed]
- Matsui, M.; Saito, Y.; Yamaoka, S.; Yokokawa, Y.; Morikawa, Y.; Makimoto, A.; Yuza, Y. Kidney-protective effect of magnesium supplementation in cisplatin-containing chemotherapy for pediatric cancer: A retrospective study. J. Pediatr. Hematol. Oncol. 2018, 40, 379–381. [Google Scholar] [CrossRef]
- Makimoto, A.; Matsui, M.; Chin, M.; Koh, K.; Tomotsune, M.; Kaneko, T.; Morikawa, Y.; Yuza, Y. Magnesium supplementation therapy to prevent cisplatin-induced acute nephrotoxicity in pediatric cancer: A protocol for a randomized phase 2 trial. Contemp. Clin. Trials Commun. 2019, 16, 100440. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Chi, Y.Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized phase II trial of bevacizumab or temsirolimus in combination with chemotherapy for first relapse rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- ARST1431. Children’s Oncology Group. Available online: https://childrensoncologygroup.org/arst1431 (accessed on 25 March 2022).
- Makimoto, A.; Kami, M.; Mineishi, S.; Tanosaki, R.; Kanda, Y.; Kim, S.W.; Hori, A.; Heike, Y.; Takaue, Y.; Kakizoe, T. Reduced-intensity allogeneic stem cell transplantation (RIST) for patients including children with refractory sarcomas. In Proceedings of the 39th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 31 May–3 June 2003; Abstract Number 3355. p. 835. [Google Scholar]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; DeRenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C.; et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 3549. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Tlemsani, C.; Leroy, K.; Gimenez-Roqueplo, A.P.; Mansuet-Lupo, A.; Pasmant, E.; Larousserie, F.; Boudou-Rouquette, P.; Vidaud, M.; Cadranel, J.; Blons, H.; et al. Chemoresistant pleomorphic rhabdomyosarcoma: Whole exome sequencing reveals underlying cancer predisposition and therapeutic options. J. Med. Genet. 2020, 57, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.H.; Thomson, A.B.; Kelsey, T.W. The radiosensitivity of the human oocyte. Hum. Reprod. 2003, 18, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Lautz, T.B.; Martelli, H.; Fuchs, J.; Chargari, C.; Smeulders, N.; Granberg, C.F.; Wolden, S.L.; Sparber-Sauer, M.; Hawkins, D.S.; Bisogno, G.; et al. Local treatment of rhabdomyosarcoma of the female genital tract: Expert consensus from the Children’s Oncology Group.; the European Soft-Tissue Sarcoma Group.; and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2020, 67, e28601. [Google Scholar] [CrossRef]
- Morris, C.D.; Tunn, P.U.; Rodeberg, D.A.; Terwisscha van Scheltinga, S.; Binitie, O.; Godzinski, J.; Dall’Igna, P.; Million, L.; Hawkins, D.S.; Koscielniak, E.; et al. Surgical management of extremity rhabdomyosarcoma: A consensus opinion from the Children’s Oncology Group, the European Pediatric Soft-Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2020, e28608. [Google Scholar] [CrossRef]
- Rogers, T.N.; Seitz, G.; Fuchs, J.; Martelli, H.; Dasgupta, R.; Routh, J.C.; Hawkins, D.S.; Koscielniak, E.; Bisogno, G.; Rodeberg, D.A. Surgical management of paratesticular rhabdomyosarcoma: A consensus opinion from the Children’s Oncology Group, European paediatric Soft tissue sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2021, 68, e28938. [Google Scholar] [CrossRef]
- Raney, R.B.; Anderson, J.R.; Andrassy, R.J.; Crist, W.M.; Donaldson, S.S.; Maurer, H.M. Soft-tissue sarcomas of the diaphragm: A report from the Intergroup rhabdomyosarcoma study group from 1972 to 1997. J. Pediatr. Hematol. Oncol. 2000, 22, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, P.; Cremers, M.; van der Meer, S.; Bot, F.; Bras, J. Cytologic diagnosis of rhabdomyosarcoma in a child with a pleural effusion. A case report. Acta Cytol. 2004, 48, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Shimojima, N.; Makimoto, A.; Yokokawa, Y.; Miyaguni, K.; Tsukizaki, A.; Hashimoto, M.; Ishikawa, M.; Ishihama, H.; Tomita, H.; et al. Primary alveolar rhabdomyosarcoma of the diaphragm requiring proximal gastrectomy. J. Pediatr. Surg. Case Re.p 2022, 78, 102206. [Google Scholar]
- McMulkin, H.M.; Yanchar, N.L.; Fernandez, C.V.; Giacomantonio, C. Sentinel lymph node mapping and biopsy: A potentially valuable tool in the management of childhood extremity rhabdomyosarcoma. Pediatr. Surg. Int. 2003, 19, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Turpin, B.; Pressey, J.G.; Nagarajan, R.; Weiss, B.D.; Trout, A.T.; Gelfand, M.J.; Pater, L.; Vatner, R.E.; Breneman, J.C.; Dasgupta, R. Sentinel lymph node biopsy in head and neck rhabdomyosarcoma. Pediatr. Blood Cancer 2019, 66, e27532. [Google Scholar] [CrossRef]
- Lin, C.; Donaldson, S.S.; Meza, J.L.; Anderson, J.R.; Lyden, E.R.; Brown, C.K.; Morano, K.; Laurie, F.; Arndt, C.A.; Enke, C.A.; et al. Effect of radiotherapy techniques (IMRT vs. 3D-CRT) on outcome in patients with intermediate-risk rhabdomyosarcoma enrolled in COG D9803—A report from the Children’s Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 1764–1770. [Google Scholar] [CrossRef]
- Ladra, M.M.; Szymonifka, J.D.; Mahajan, A.; Friedmann, A.M.; Yong Yeap, B.; Goebel, C.P.; MacDonald, S.M.; Grosshans, D.R.; Rodriguez-Galindo, C.; Marcus, K.J.; et al. Preliminary results of a phase II trial of proton radiotherapy for pediatric rhabdomyosarcoma. J. Clin. Oncol. 2014, 32, 3762–3770. [Google Scholar] [CrossRef]
- Ferrari, A.; Miceli, R.; Casanova, M.; Meazza, C.; Favini, F.; Luksch, R.; Catania, S.; Fiore, M.; Morosi, C.; Mariani, L. The symptom interval in children and adolescents with soft tissue sarcomas. Cancer 2010, 116, 177–183. [Google Scholar] [CrossRef]
- Davis, L.E.; Janeway, K.A.; Weiss, A.R.; Chen, Y.E.; Scharschmidt, T.J.; Krailo, M.; Glade Bender, J.L.; Kopp, L.M.; Patel, S.R.; Schwartz, G.K.; et al. Clinical trial enrollment of adolescents and young adults with sarcoma. Cancer 2017, 123, 3434–3440. [Google Scholar] [CrossRef]
- De, R.; Sutradhar, R.; Kurdyak, P.; Aktar, S.; Pole, J.D.; Baxter, N.; Nathan, P.C.; Gupta, S. Incidence and predictors of mental health outcomes among survivors of adolescent and young adult cancer: A population-based study using the impact cohort. J. Clin. Oncol. 2021, 39, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- AYA Oncology Alliance. Available online: https://aya-ken.jp/line (accessed on 25 March 2022). (In Japanese).
- WMA Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 25 March 2022).




{kind=link}
{kind=link}
{kind=link}
| Drug | Treatment Phase | Adult (%) | Child (%) | p-Value |
|---|---|---|---|---|
| Vincristine | Induction phase | 77.7 | 86.1 | 0.109 |
| Maintenance phase | 71.9 | 100.1 | 0.042 | |
| Total phase | 76.8 | 90.2 | 0.040 | |
| Cyclophosphamide | Induction phase | 87.1 | 88.2 | 0.820 |
| Maintenance phase | 69.7 | 86.4 | 0.011 | |
| Total phase | 80.0 | 86.9 | 0.156 |
| Regimen | Trial | Dosage (mg/m2) and Schedule | Ref. |
|---|---|---|---|
| VAC | IRS-IV | VCR 1.5 on days 1, 8, 15; ACD 0.015/kg on days 1–5; CPA 2200 on day1; every 3 weeks | [14] |
| VAC | D9802/ D9803 | VCR 1.5 on days 1, 8, 15; ACD 1.5 on day 1; CPA 2200 on day1; every 3 weeks | [15,37] |
| VAC | ARST0531 | VCR 1.5 on days 1, 8, 15; ACD 1.5 on day 1; CPA 1200 on day 1; every 3 weeks | [16] |
| VIE | IRS-IV | VCR 1.5 on days 1, 8, 15; IFM 1800 on days 1–5; ETP 100 on days 1–5; every 3 weeks | [14] |
| VAI | IRS-IV | VCR 1.5 on days 1, 8, 15; ACD 1.5 on day 1; IFM 1800 on days 1–5; every 3 weeks | [14] |
| VTC | D9803 | VCR 1.5 on days 1, 8, 15; Topo 250 on days 1–5; CPA 250 on days 1–5; every 3 weeks | [15] |
| VI | ARST0431/ ARST0531 | VCR 1.5 on days 1, 8, 15; IRI 50 on days 1–5; every 3 weeks | [16,38] |
| VDC | ARST0431 | VCR 1.5 on days 1, 8, 15; DXR 37.5 on days 1, 2; CPA 1200 on day 1; every 2 weeks alternating with IE | [39] |
| IE | ARST0431 | IFM 1800 on days 1–5; ETP 100 on days 1–5; every 2 weeks alternating with VDC | [39] |
| IVA | RMS2005 | IFM 3000 on days 1–2; VCR 1.5 on days 1, 8, 15; ACD 1.5 on day 1; every 3 weeks | [17] |
| VC maintenance | RMS2005 | VNR 25 on days 1, 8, 15; CPA (po) 25 daily; for 4 weeks cycles × 6 cycles | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makimoto, A. Optimizing Rhabdomyosarcoma Treatment in Adolescents and Young Adults. Cancers 2022, 14, 2270. https://doi.org/10.3390/cancers14092270
Makimoto A. Optimizing Rhabdomyosarcoma Treatment in Adolescents and Young Adults. Cancers. 2022; 14(9):2270. https://doi.org/10.3390/cancers14092270
Chicago/Turabian StyleMakimoto, Atsushi. 2022. "Optimizing Rhabdomyosarcoma Treatment in Adolescents and Young Adults" Cancers 14, no. 9: 2270. https://doi.org/10.3390/cancers14092270
APA StyleMakimoto, A. (2022). Optimizing Rhabdomyosarcoma Treatment in Adolescents and Young Adults. Cancers, 14(9), 2270. https://doi.org/10.3390/cancers14092270