
TRPM8-Rap1A Interaction Sites as Critical Determinants for Adhesion and Migration of Prostate and Other Epithelial Cancer Cells


 , ,
, ,  ,
,  , , , , , ,
, , , , , ,  and
and 
Abstract
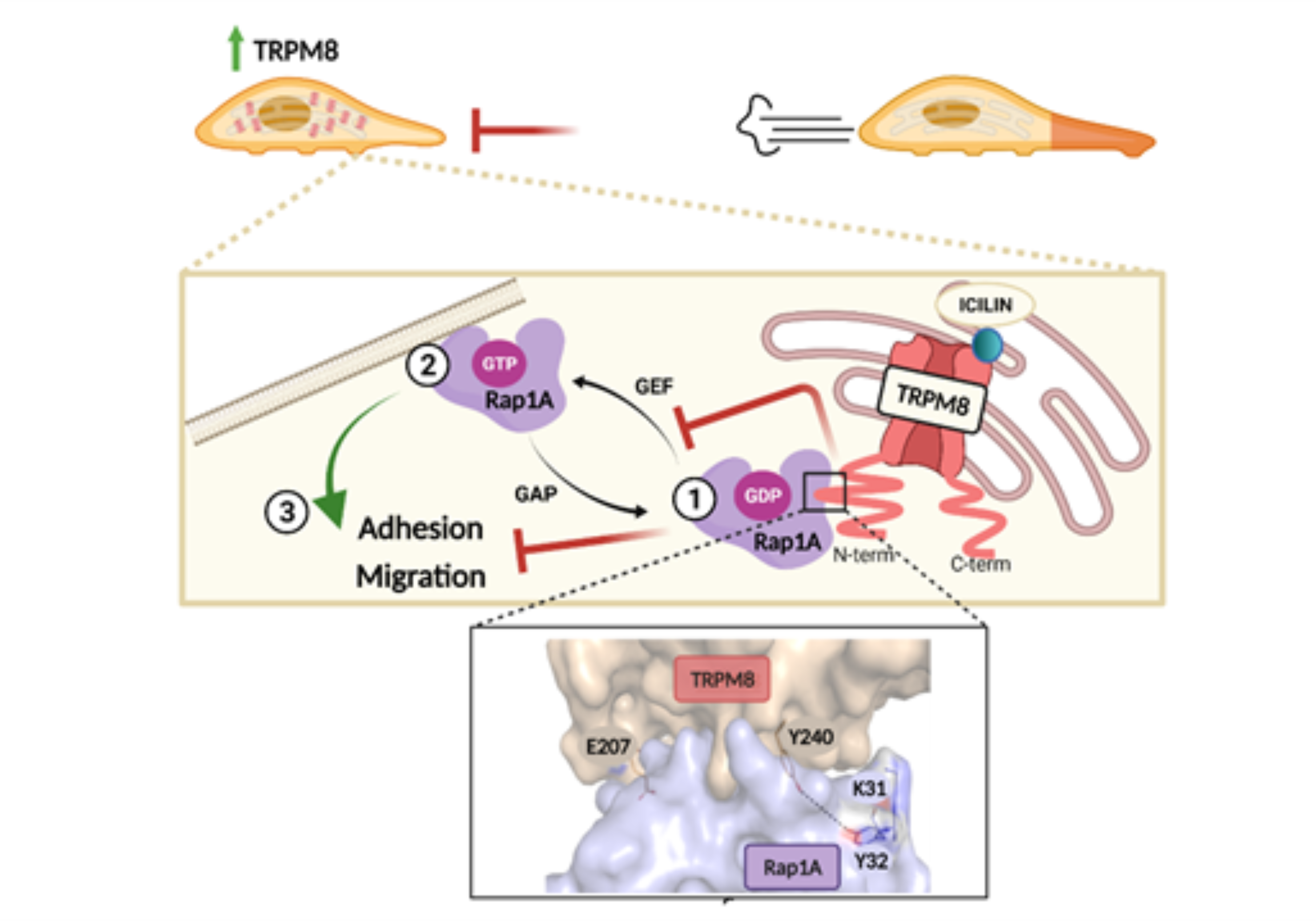
:Simple Summary
Abstract
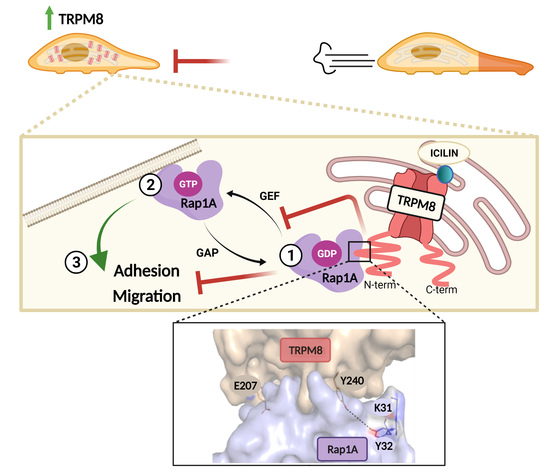
1. Introduction
2. Materials and Methods
2.1. Chemicals and Drugs
2.2. Molecular Modeling
2.3. Cell Cultures
2.4. Molecular Biology
2.5. Western Blot Analysis
2.6. GST-Fusion Protein and Pull-Down Assay
2.7. Co-Immunoprecipitation Assay
2.8. Proximity Ligation Assay (PLA)
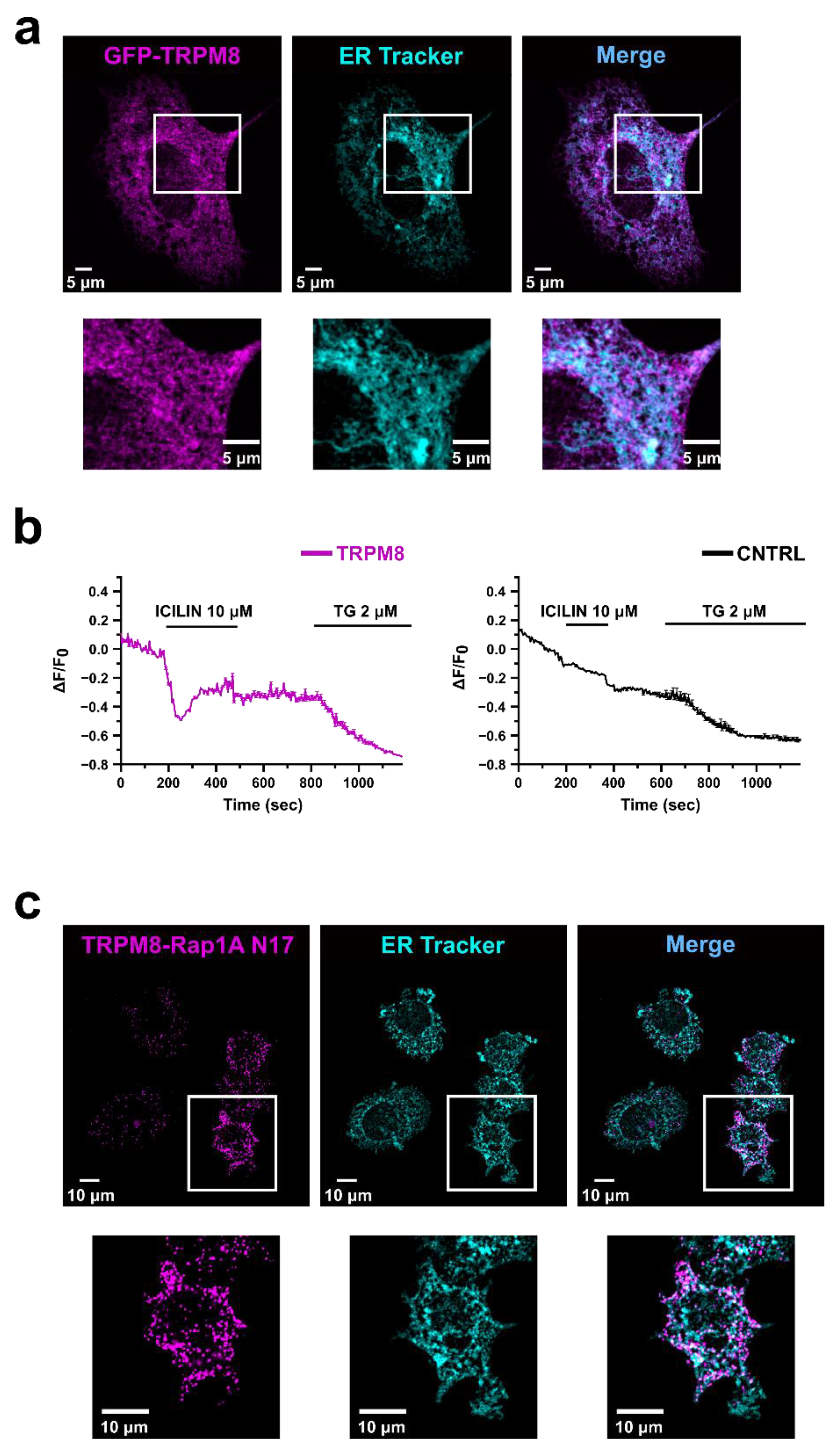
2.9. Live-Cell Imaging Experiments
2.10. Active Rap1 Pull-Down and Detection Assay
2.11. Cytosolic and ER Ca2+ Imaging
2.12. Random Migration Assay
2.13. Transwell Migration Assay
2.14. Adhesion Assay
2.15. Cell Viability Assay
2.16. Confocal Analysis
2.17. Immunohistochemistry
2.18. Bioinformatic Analysis
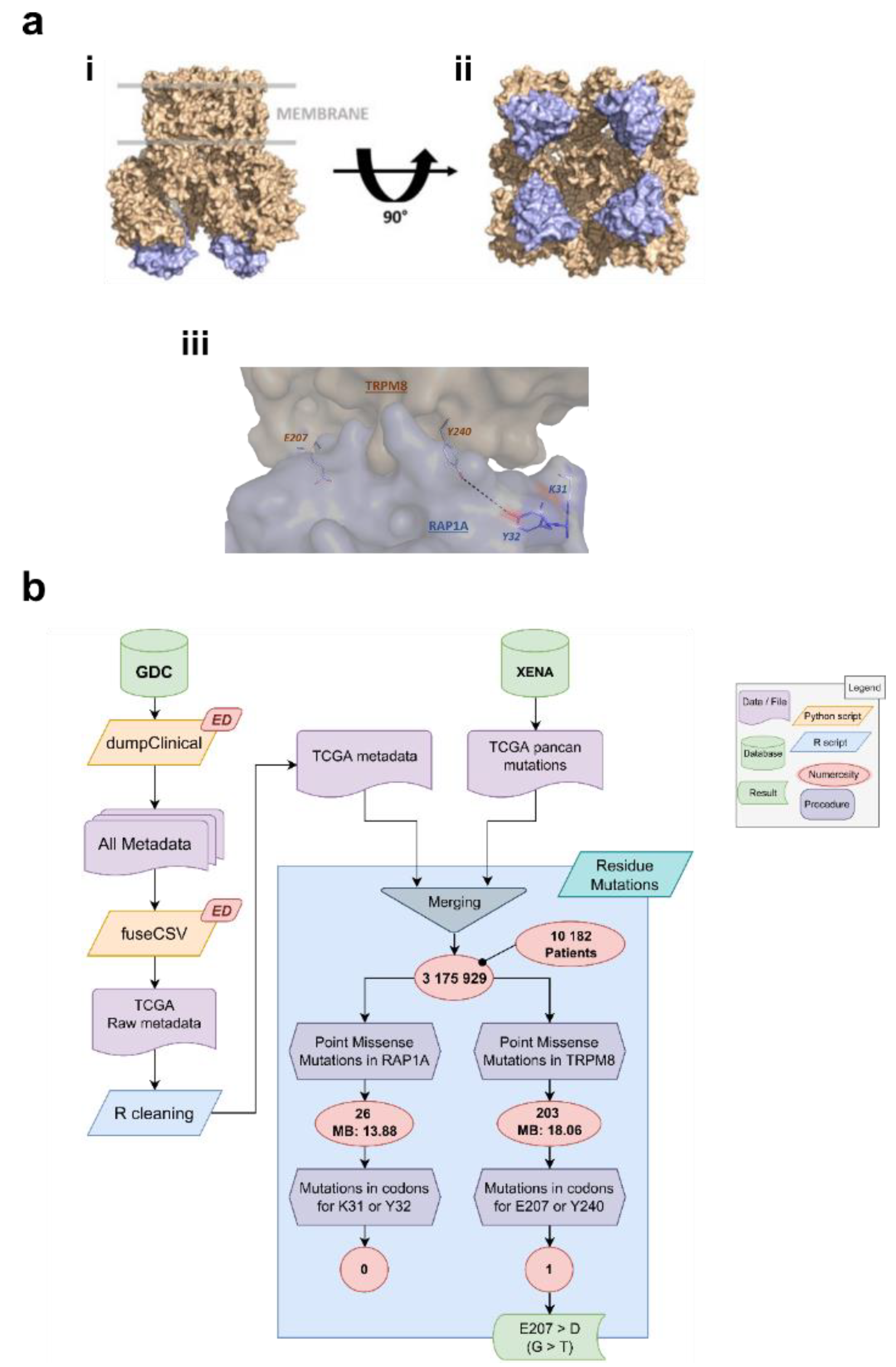
2.18.1. Detection of Mutations
2.18.2. Differential Expression Analysis
- GTEx samples labeled as “Breast” and TCGA samples labeled as “TCGA-BRCA” composed the “Breast” dataset;
- GTEx samples labeled as “Prostate” and TCGA samples labeled as “TCGA-PRAD” composed the “Prostate” dataset;
- GTEx samples labeled as “Uterus” or “Cervix Uteri” and TCGA samples labeled as “TCGA-UCEC” composed the “Uterus” dataset.
- The samples were labeled as “healthy”, “metastatic”, or “not metastatic” based on these criteria (“Metastatic” labeling):
- If the sample was from the TCGA cohort and had an entry in the AJCC “m” category (both clinical or pathologic) reflecting the metastatic nature of the sample, it was labeled as “metastatic”;
- If the sample was labeled as having AJCC stage “IV” or equivalent, it was labeled as “metastatic”;
- If the sample was labeled with a FIGO stage of “III” or “IV”, it was labeled as “metastatic”;
- If the sample was from the GTEx cohort, it was labeled as “healthy”;
- All other samples were labeled as “not metastatic”.
- Produced a non-significant p-value under the Kolmogorov-Smirnov two-sided test with the null hypothesis of being drawn from a normal distribution;
- Had sample numerosity greater than 30.
2.19. Statistical Analysis
3. Results
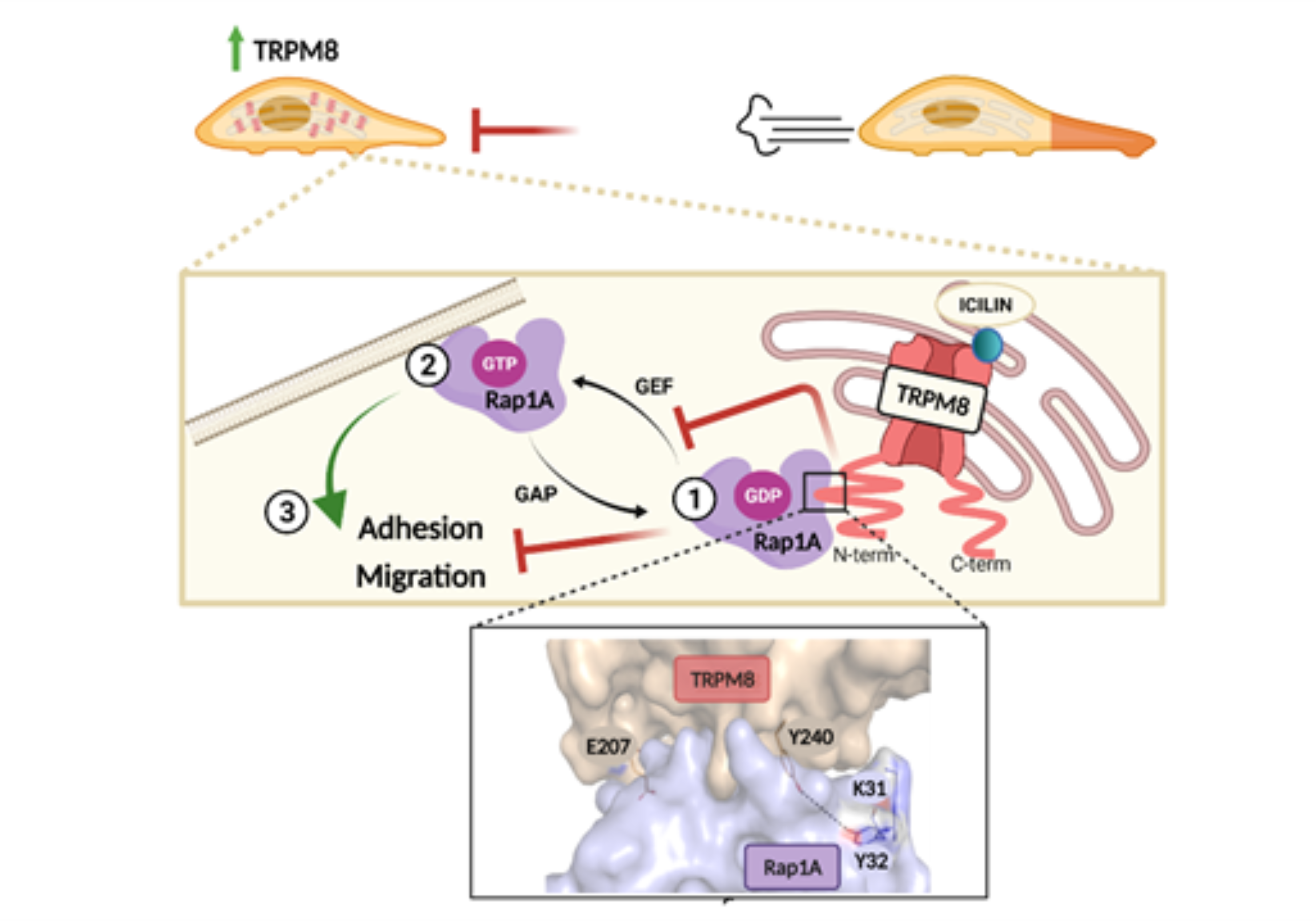
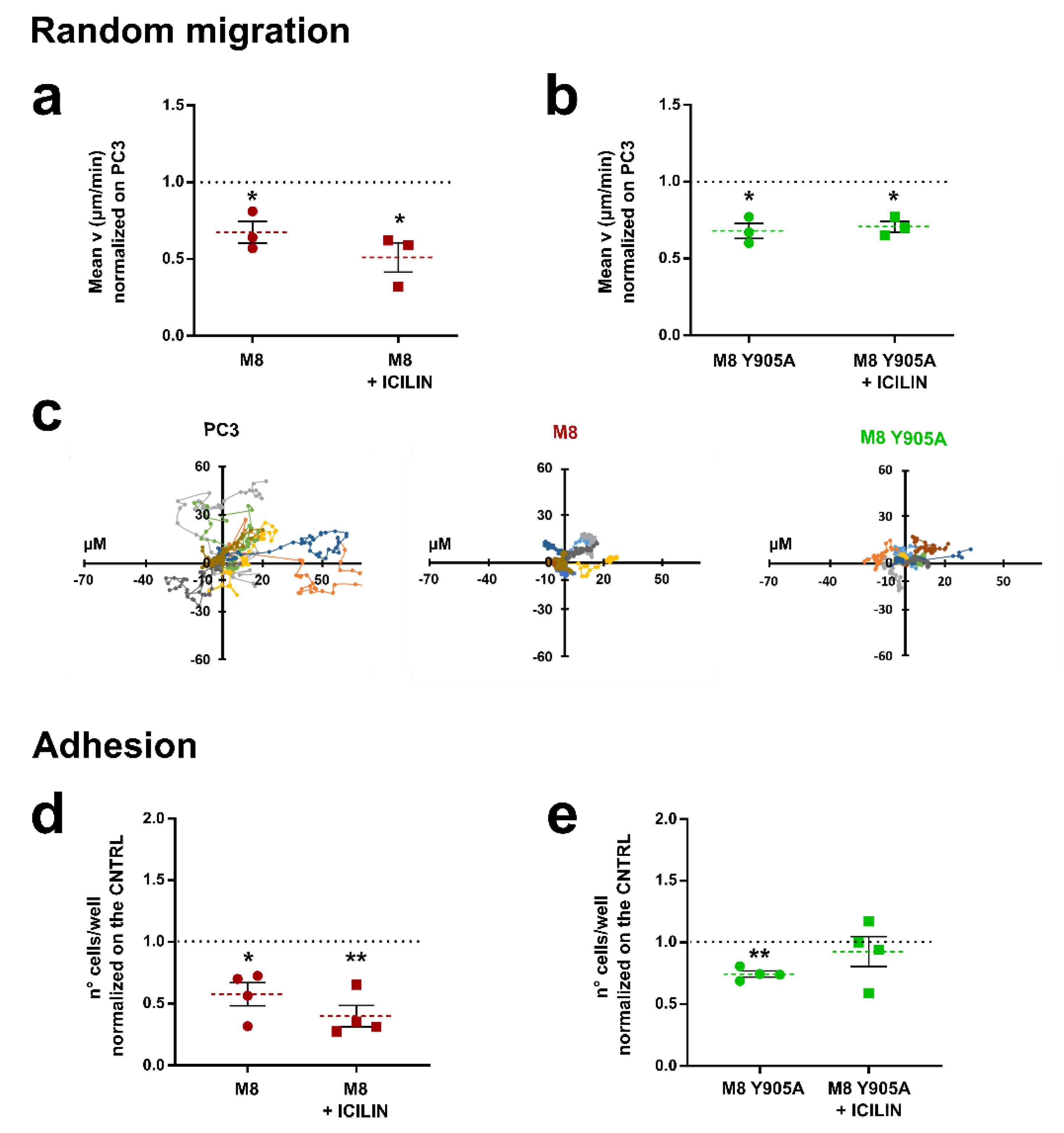
3.1. TRPM8 Inhibits PC3 Migration and Adhesion Regardless of Channel Activity
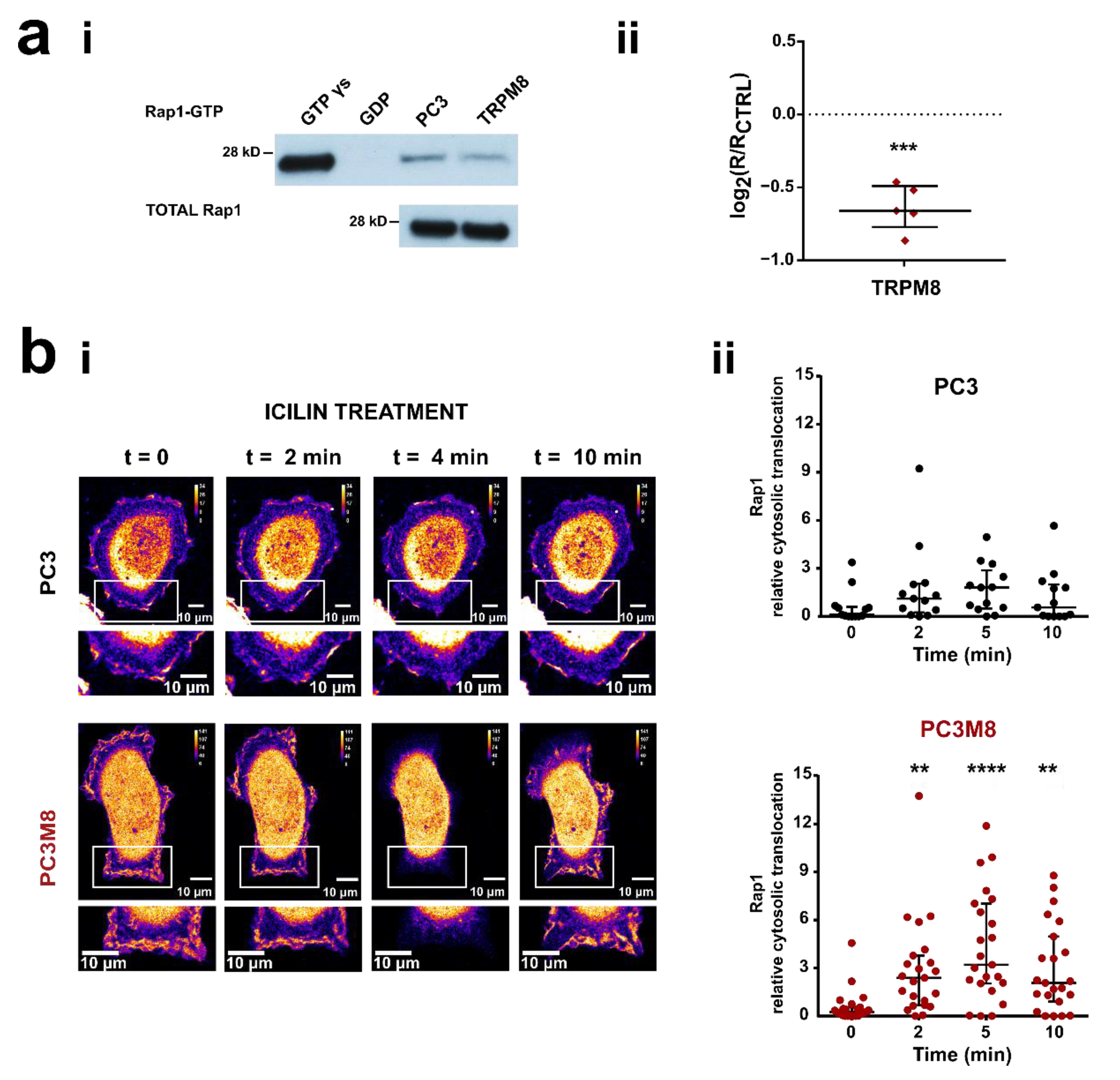
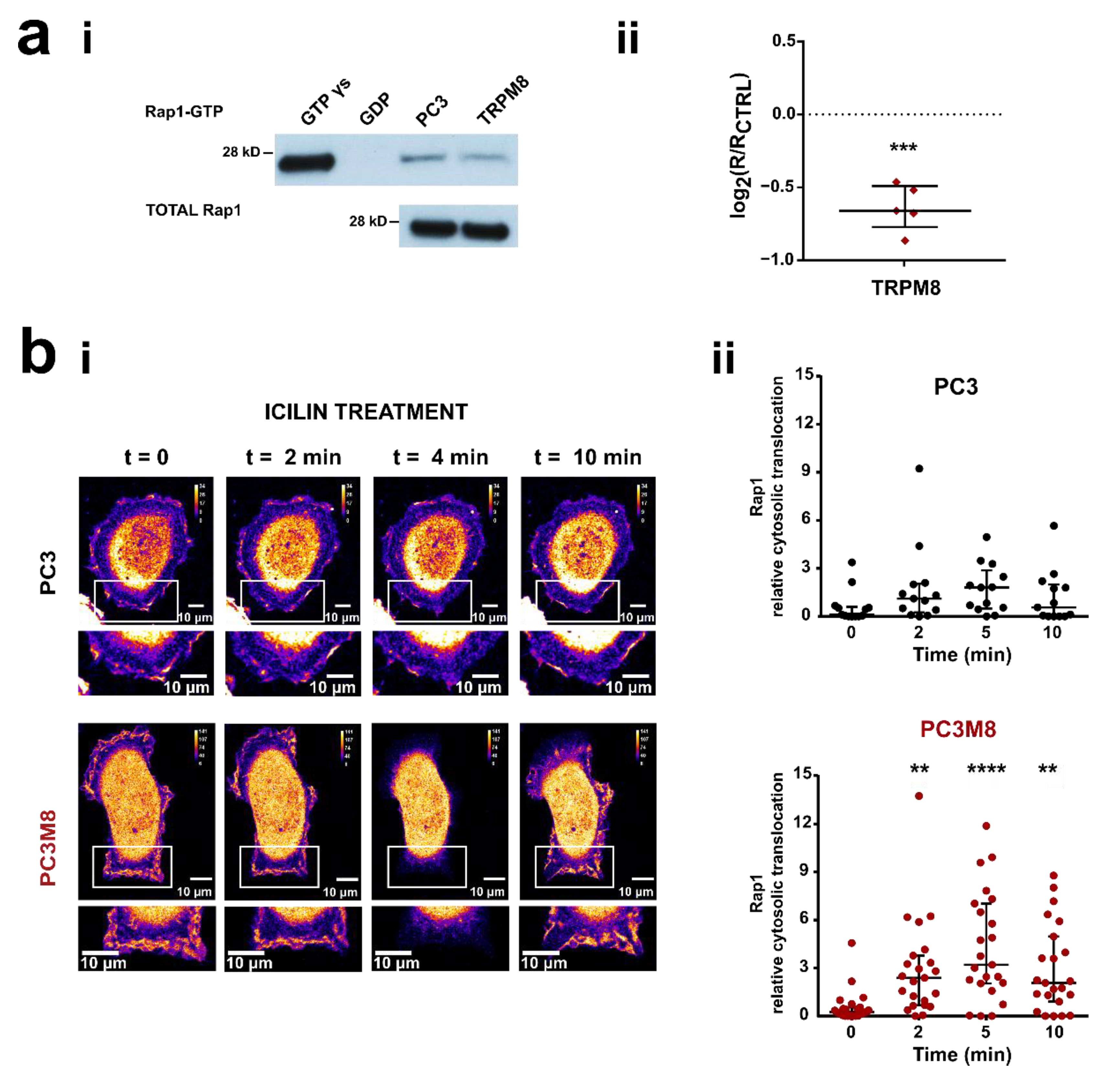
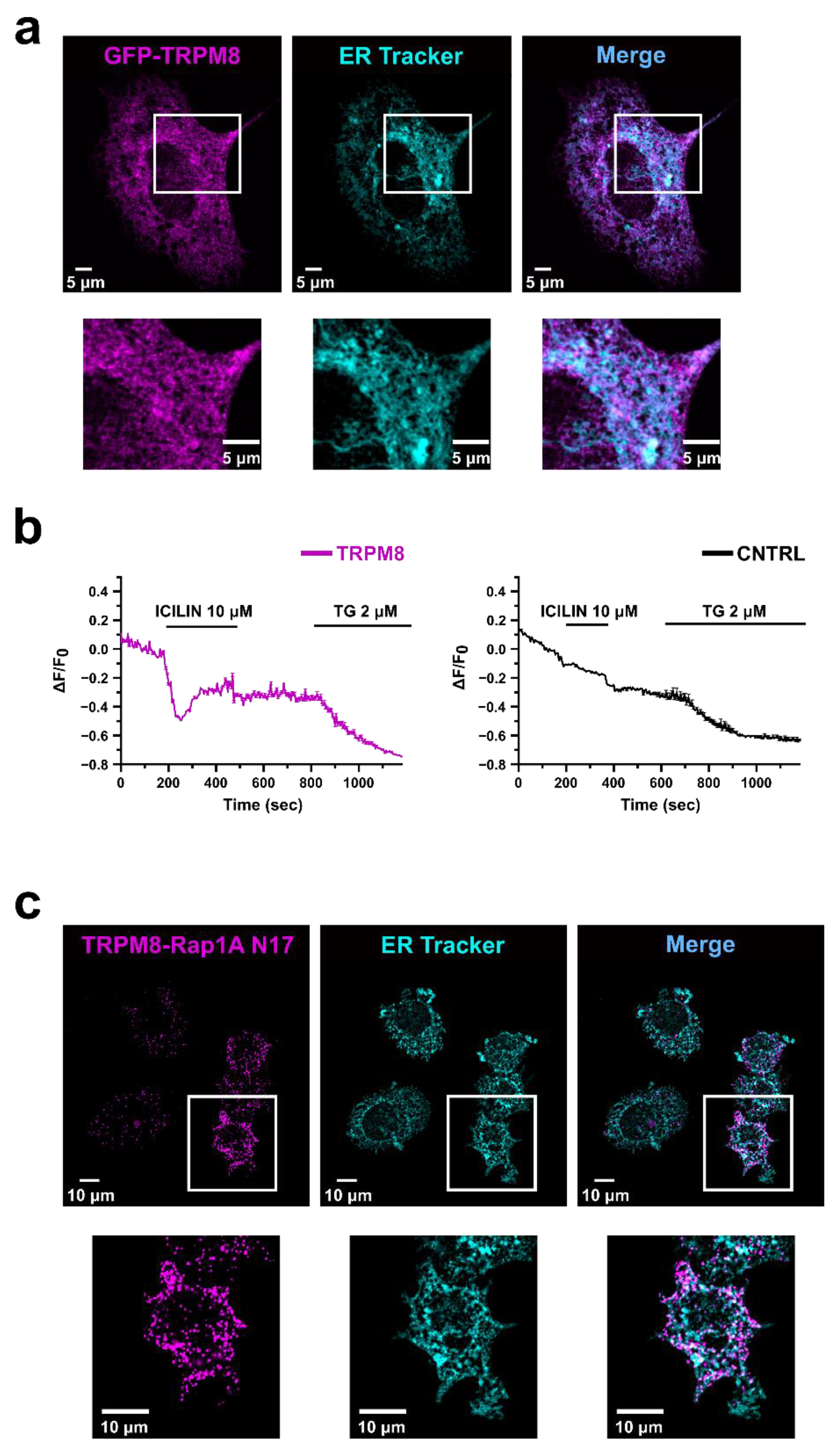
3.2. TRPM8 Inhibits Rap1′s Activity by Intracellularly Sequestering GDP-Bound Rap1
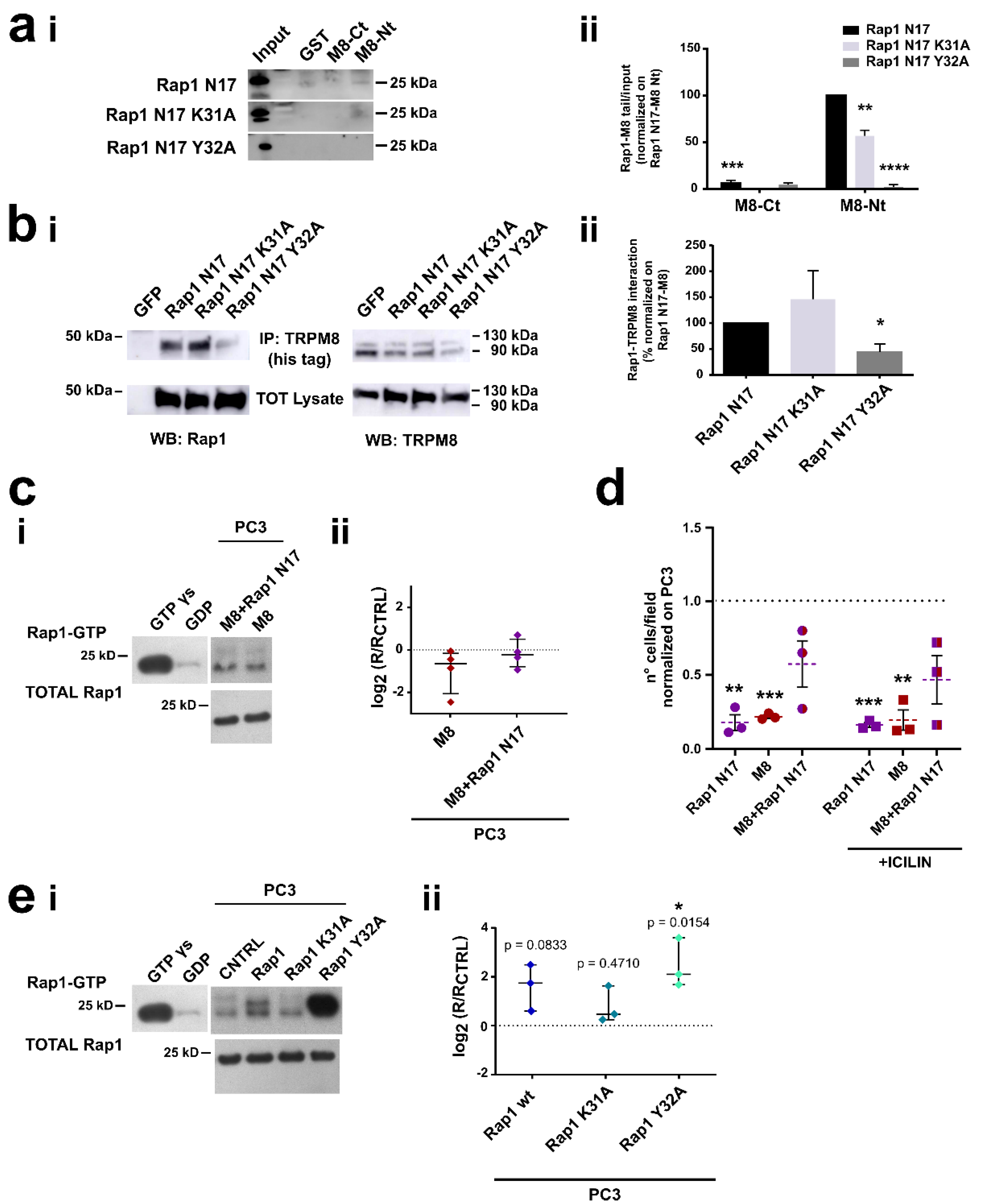
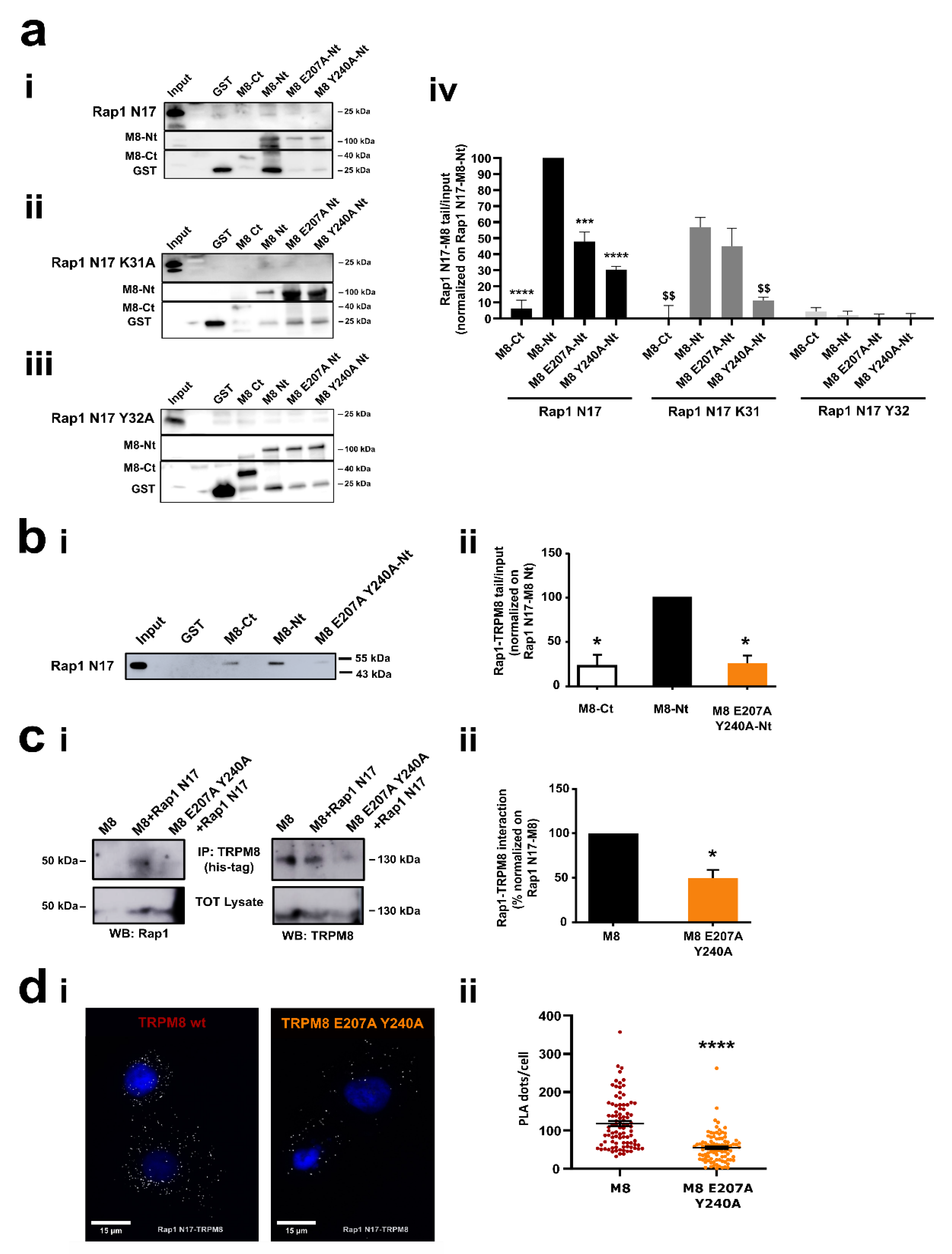
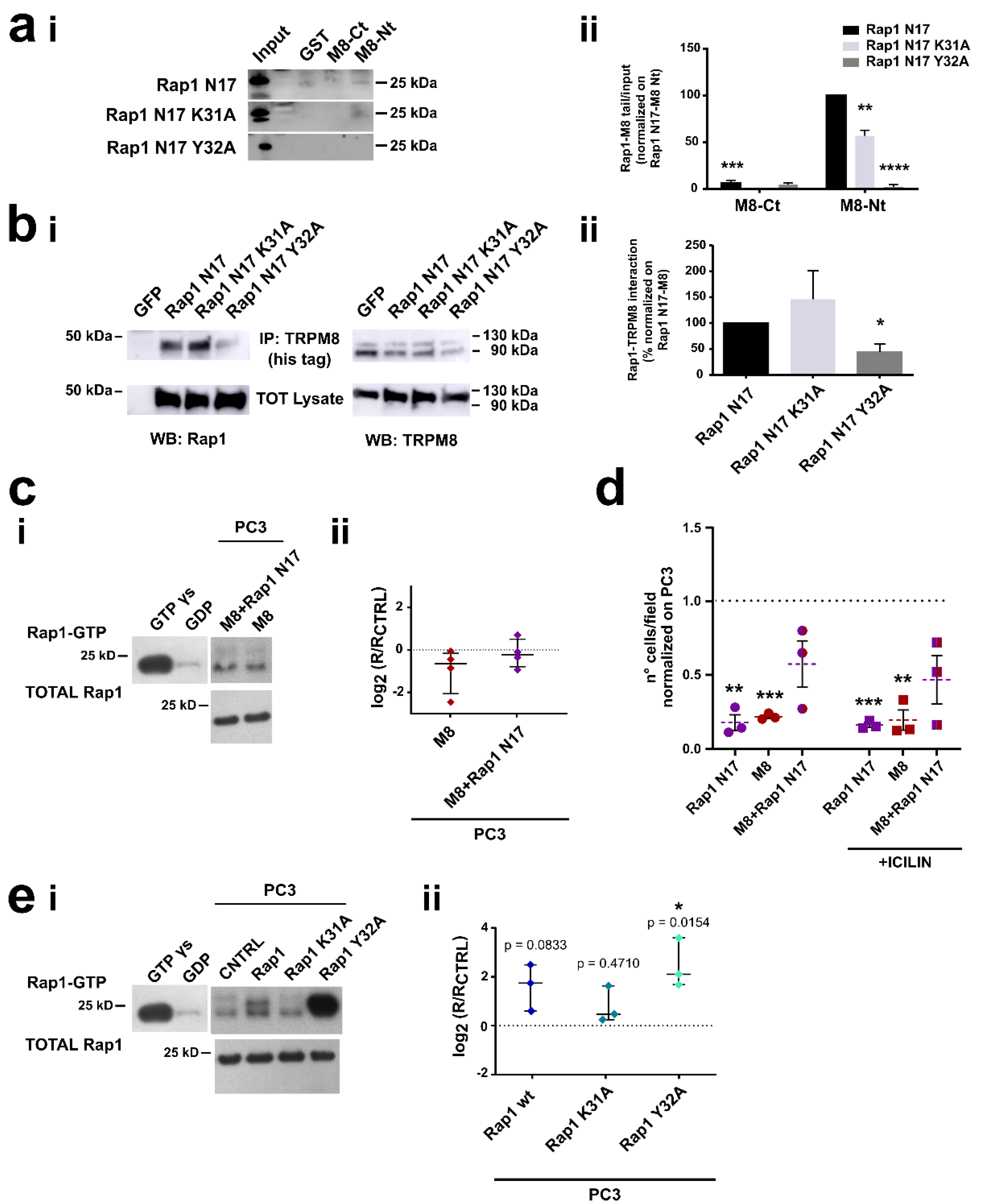
3.3. Identification of the Residues Involved in TRPM8-Rap1 Interaction
3.4. Characterization of the Residues Involved in TRPM8-Rap1 Interaction
3.4.1. Role of the Residue Y32 in the Interaction of GDP-Bound Rap1 with TRPM8 Cytosolic N-Terminal Tail
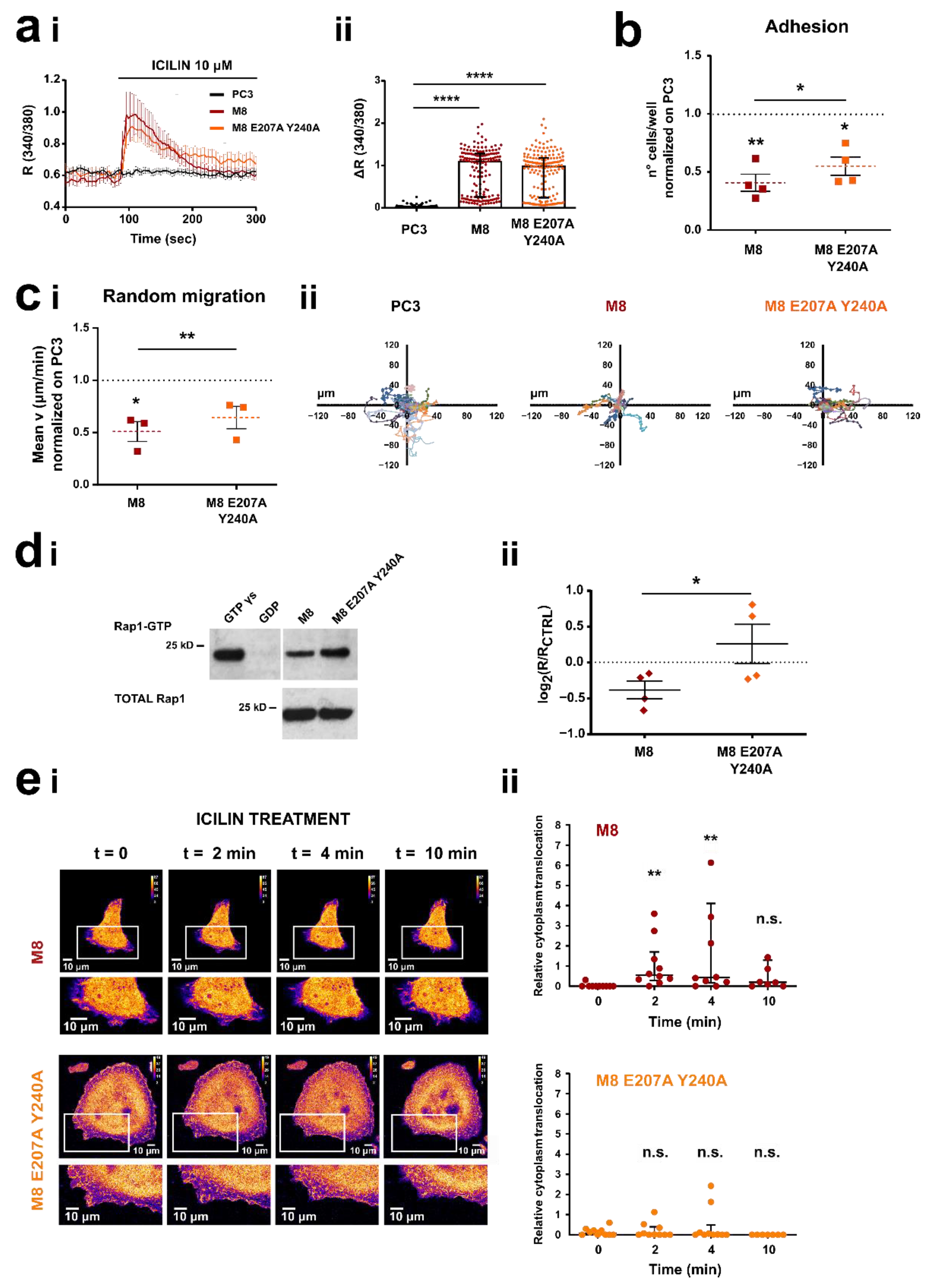
3.4.2. Role of Residues E207 and Y240 in the Interaction of TRPM8 Cytosolic N-Terminal Tail with GDP-Bound Rap1
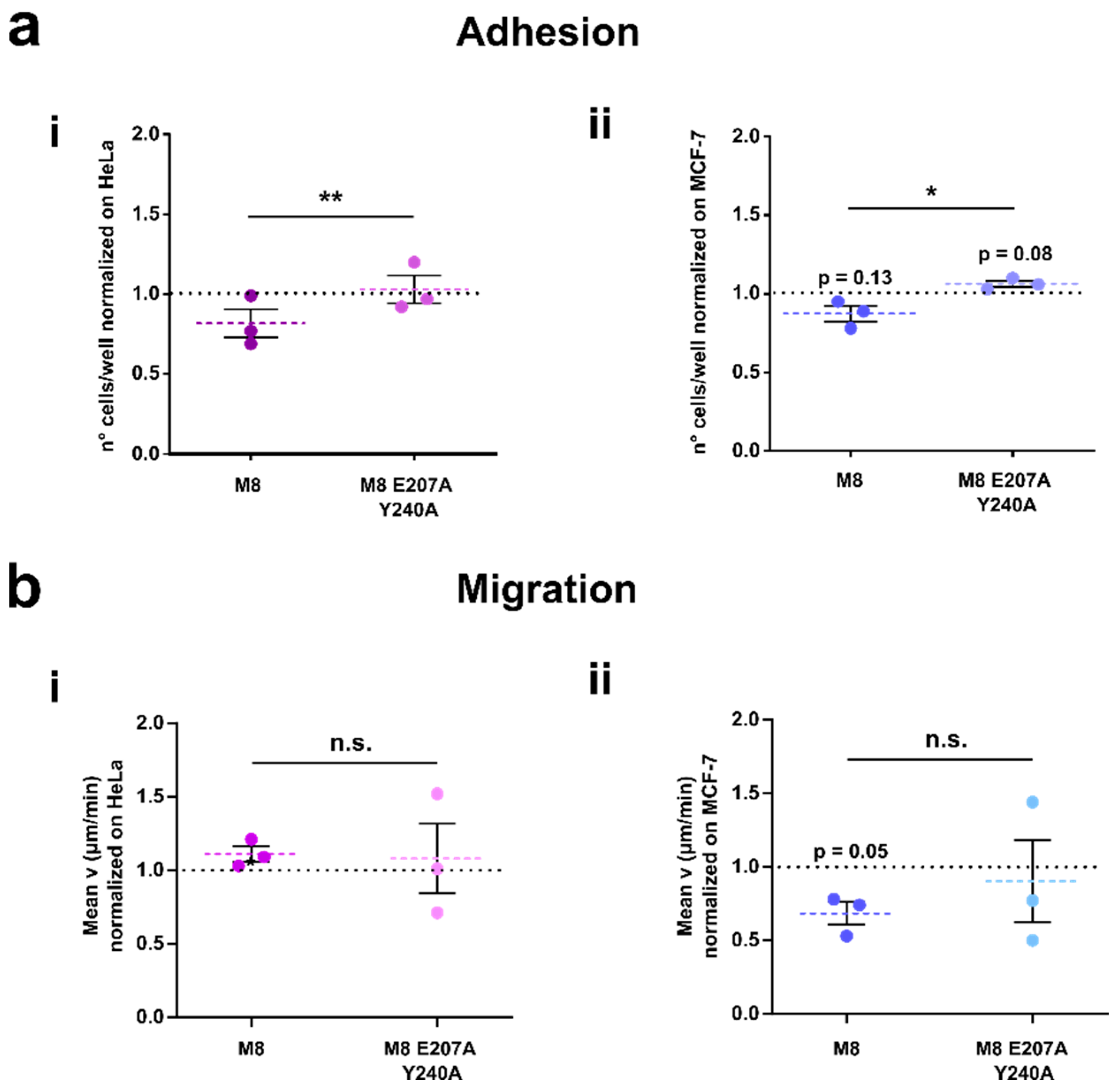
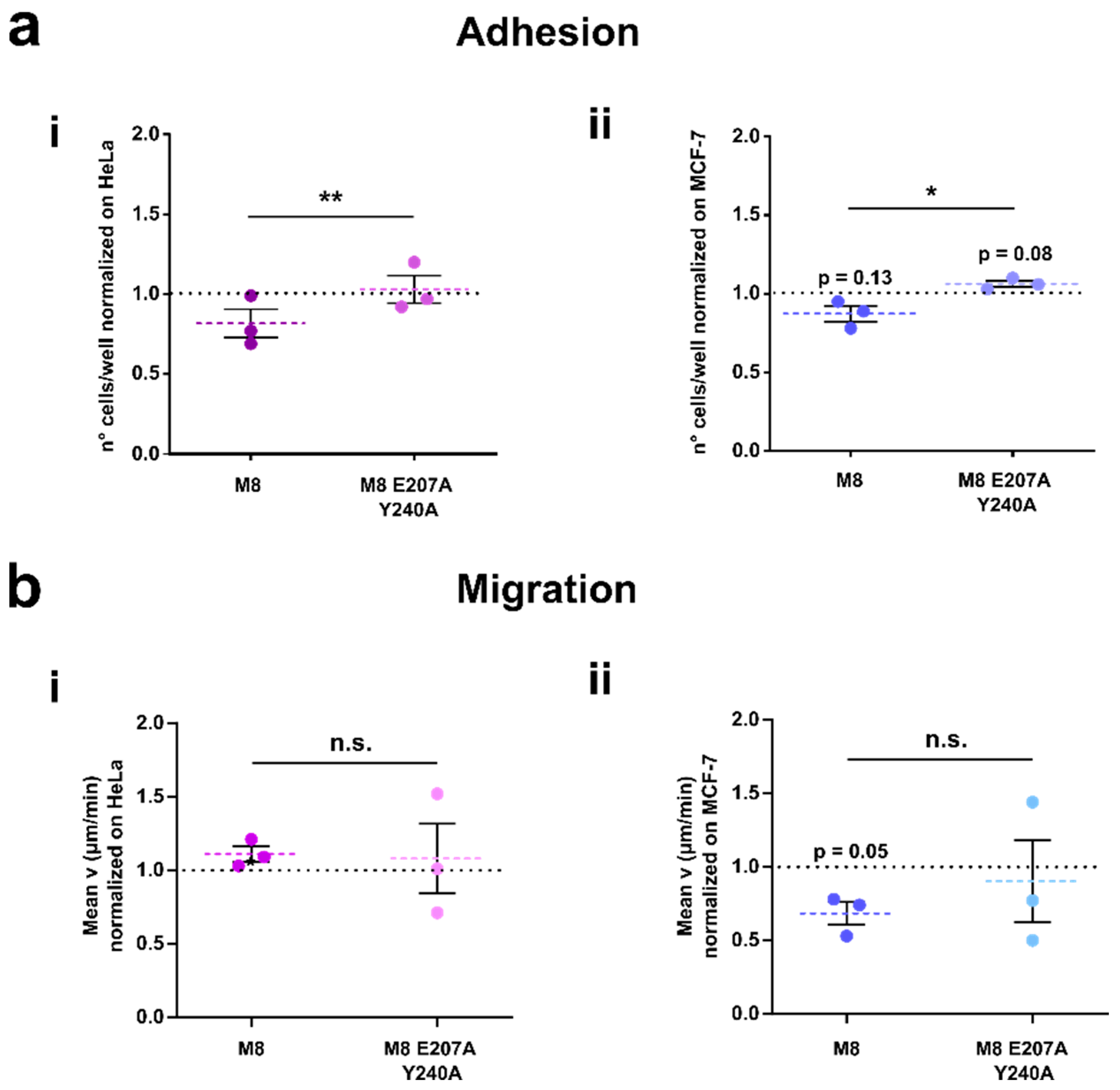
3.5. TRPM8 E207 Y240 Residues Revert Rap1-Mediated Inhibition of Cancer Cell Adhesion and Migration
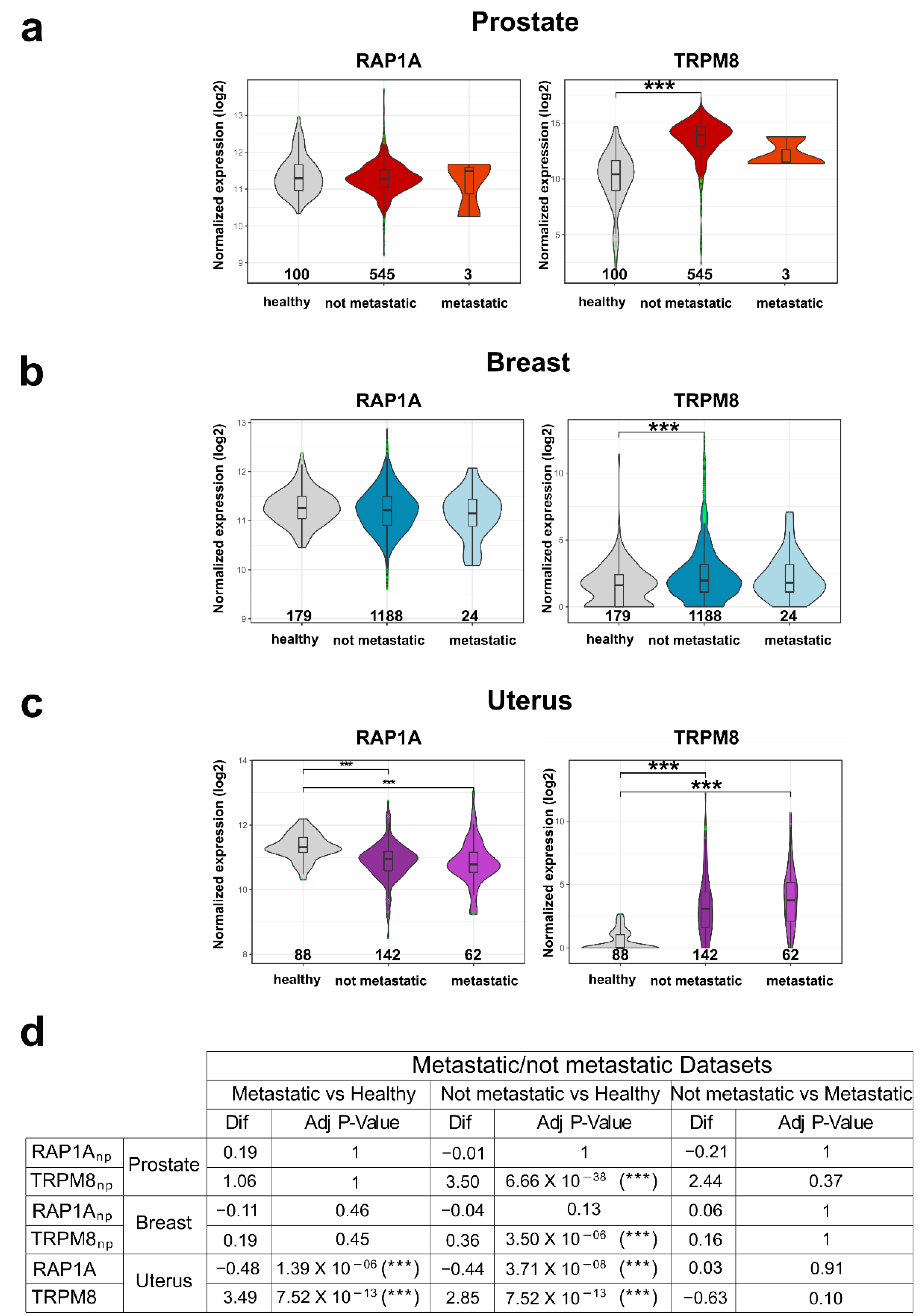
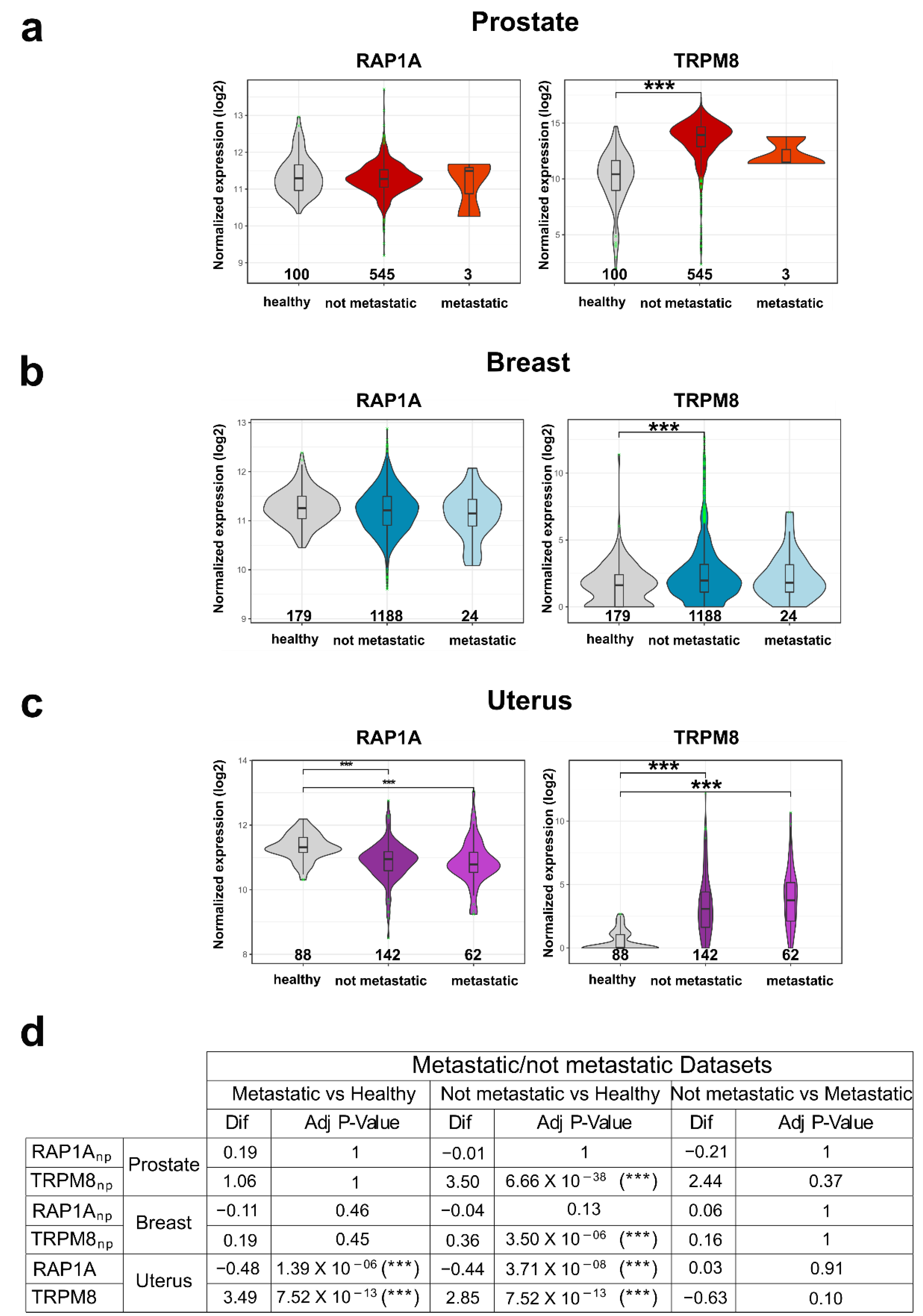
3.6. TRPM8-Rap1 Interaction in Breast and Cervical Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Gardel, M.L.; Schneider, I.C.; Aratyn-Schaus, Y.; Waterman, C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010, 26, 315–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Gkika, D.; Prevarskaya, N. TRP channels in prostate cancer: The good, the bad and the ugly? Asian J. Androl. 2011, 13, 673–676. [Google Scholar] [CrossRef] [Green Version]
- Fiorio Pla, A.; Gkika, D. Emerging role of TRP channels in cell migration: From tumor vascularization to metastasis. Front. Physiol. 2013, 4, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardini, M.; Fiorio Pla, A.; Prevarskaya, N.; Gkika, D. Human transient receptor potential (TRP) channels expression profiling in carcinogenesis. Int. J. Dev. Biol. 2015, 59, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta—Biomembr. 2015, 1848, 2685–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chinigò, G.; Castel, H.; Chever, O.; Gkika, D. TRP Channels in Brain Tumors. Front. Cell Dev. Biol. 2021, 9, 617801. [Google Scholar] [CrossRef]
- Tsavaler, L.; Shapero, M.H.; Morkowski, S.; Laus, R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 2001, 61, 3760–3769. [Google Scholar]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Iamshanova, O.; Fiorio Pla, A.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrenken, K.S.; Jalink, K.; van Leeuwen, F.N.; Middelbeek, J. Beyond ion-conduction: Channel-dependent and -independent roles of TRP channels during development and tissue homeostasis. Biochim. Biophys. Acta—Mol. Cell Res. 2015, 1863, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Bödding, M. TRP proteins and cancer. Cell. Signal. 2007, 19, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Van De Graaf, S.F.J.; Hoenderop, J.G.J.; Bindels, R.J.M. Regulation of TRPV5 and TRPV6 by associated proteins. Am. J. Physiol. Renal Physiol. 2006, 290, F1295–F1302. [Google Scholar] [CrossRef] [PubMed]
- Gkika, D.; Mahieu, F.; Niliur, B.; Hoenderop, J.G.J.; Bindels, R.J.M. 80K-H as a new Ca2+ sensor regulating the activity of the epithelial Ca2+ channel transient receptor potential cation channel V5 (TRPV5). J. Biol. Chem. 2004, 279, 26351–26357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkika, D.; Topala, C.N.; Hoenderop, J.G.J.; Bindels, R.J.M. The immunophilin FKBP52 inhibits the activity of the epithelial Ca2+ channel TRPV5. Am. J. Physiol. Renal Physiol. 2006, 290, F1253–F1259. [Google Scholar] [CrossRef]
- Gkika, D.; Topala, C.N.; Chang, Q.; Picard, N.; Thébault, S.; Houillier, P.; Hoenderop, J.G.J.; Bindels, R.J.M. Tissue kallikrein stimulates Ca2+ reabsorption via PKC-dependent plasma membrane accumulation of TRPV5. EMBO J. 2006, 25, 4707–4716. [Google Scholar] [CrossRef] [Green Version]
- Sinkins, W.G.; Goel, M.; Estacion, M.; Schilling, W.P. Association of immunophilins with mammalian TRPC channels. J. Biol. Chem. 2004, 279, 34521–34529. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Hoefs, S.; Van Der Kemp, A.W.; Topala, C.N.; Bindels, R.J.; Hoenderop, J.G. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 2005, 310, 490–493. [Google Scholar] [CrossRef]
- Chinigò, G.; Fiorio Pla, A.; Gkika, D. TRP Channels and Small GTPases Interplay in the Main Hallmarks of Metastatic Cancer. Front. Pharmacol. 2020, 11, 1657. [Google Scholar] [CrossRef] [PubMed]
- Guéguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantôme, A.; Haelters, J.P.; Jaffrès, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzerides, V.J.; Ramsey, I.S.; Kotecha, S.; Greka, A.; Clapham, D.E. Rapid vesicular translocation and insertion of TRP channels. Nat. Cell Biol. 2004, 6, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Knezevic, N.; Ahmmed, G.U.; Kini, V.; Malik, A.B.; Mehta, D. Gαq-TRPC6-mediated Ca 2+ Entry Induces RhoA Activation and Resultant Endothelial Cell Shape Change in Response to Thrombin. J. Biol. Chem. 2007, 282, 7833–7843. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Jacobo, S.M.P.; Billing, D.; Rozkalne, A.; Gage, S.D.; Anagnostou, T.; Pavenstädt, H.; Hsu, H.; Schlondorff, J.; Ramos, A.; et al. Antagonistic regulation od actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci. Signal. 2010, 3, ra77. [Google Scholar] [CrossRef] [Green Version]
- Su, L.T.; Liu, W.; Chen, H.C.; González-Pagán, O.; Habas, R.; Runnels, L.W. TRPM7 regulates polarized cell movements. Biochem. J. 2011, 434, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Vaughan, M.B.; Kropp, B.P.; Gabbiani, G.; Martin, M.D.; Haaksma, C.J.; Hinz, B. Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair. Regen. 2006, 14, 313–320. [Google Scholar] [CrossRef]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. Cellular Cardiology TRPV4 channels mediate cardiac fi broblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Sri, V.; Krishna, R.; Singh, H.; et al. TRPV4 regulates breast cancer cell extravasation, stiffness and actin cortex. Sci. Rep. 2016, 6, 27903. [Google Scholar] [CrossRef] [Green Version]
- Ou-yang, Q.; Li, B.; Xu, M.; Liang, H. TRPV4 promotes the migration and invasion of glioma cells via AKT/Rac1 signaling. Biochem. Biophys. Res. Commun. 2018, 503, 876–881. [Google Scholar] [CrossRef]
- Gao, G.; Wang, W.; Tadagavadi, R.K.; Briley, N.E.; Love, M.I.; Miller, B.A.; Reeves, W.B. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Investig. 2014, 124, 4989–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laragione, T.; Harris, C.; Gulko, P.S. TRPV2 suppresses Rac1 and RhoA activation and invasion in rheumatoid arthritis fibroblast-like synoviocytes. Int. Immunopharmacol. 2019, 70, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Ahmmed, G.U.; Paria, B.C.; Holinstat, M.; Voyno-Yasenetskaya, T.; Tiruppathi, C.; Minshall, R.D.; Malik, A.B. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry: Role in signaling increased endothelial permeability. J. Biol. Chem. 2003, 278, 33492–33500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.K.; Rathor, N.; Wang, S.R.; Wang, J.Y.; Rao, J.N. RhoA enhances store-operated Ca2+ entry and intestinal epithelial restitution by interacting with TRPC1 after wounding. Am. J. Physiol.—Gastrointest. Liver Physiol. 2015, 309, G759–G767. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, M.; Kojima, I. Translocation of TRPV2 channel induced by focal administration of mechanical stress. Physiol. Rep. 2015, 3, e12296. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Yang, T.; Wang, P.; Ma, S.; Zhu, Z.; Pu, Y.; Li, L.; Zhao, Y.; Xiong, S.; Liu, D.; et al. Activation of cold-sensing transient receptor potential melastatin subtype 8 antagonizes vasoconstriction and hypertension through attenuating RhoA/Rho kinase pathway. Hypertension 2014, 63, 1354–1363. [Google Scholar] [CrossRef]
- Thoppil, R.J.; Cappelli, H.C.; Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Thodeti, C.K. TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 2016, 7, 25849–25861. [Google Scholar] [CrossRef] [Green Version]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef]
- Henshall, S.M.; Afar, D.E.H.; Hiller, J.; Horvath, L.G.; Quinn, D.I.; Rasiah, K.K.; Gish, K.; Willhite, D.; Kench, J.G.; Gardiner-Garden, M.; et al. Survival analysis of genome-wide gene expression profiles of prostate cancers identifies new prognostic targets of disease relapse. Cancer Res. 2003, 63, 4196–4203. [Google Scholar]
- Grolez, G.P.; Gordiendko, D.V.; Clarisse, M.; Hammadi, M.; Desruelles, E.; Fromont, G.; Prevarskaya, N.; Slomianny, C.; Gkika, D. TRPM8-androgen receptor association within lipid rafts promotes prostate cancer cell migration. Cell Death Dis. 2019, 10, 652. [Google Scholar] [CrossRef] [Green Version]
- Bidaux, G.; Roudbaraki, M.; Merle, C.; Crépin, A.; Delcourt, P.; Slomianny, C.; Thebault, S.; Bonnal, J.L.; Benahmed, M.; Cabon, F.; et al. Evidence for specific TRPM8 expression in human prostate secretory epithelial cells: Functional androgen receptor requirement. Endocr. Relat. Cancer 2005, 12, 367–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Barritt, G.J. Evidence that TRPM8 is an androgen-dependent Ca2+ channel required for the survival of prostate cancer cells. Cancer Res. 2004, 64, 8365–8373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkika, D.; Flourakis, M.; Lemonnier, L.; Prevarskaya, N. PSA reduces prostate cancer cell motility by stimulating TRPM8 activity and plasma membrane expression. Oncogene 2010, 29, 4611–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkika, D.; Lemonnier, L.; Shapovalov, G.; Gordienko, D.; Poux, C.; Bernardini, M.; Bokhobza, A.; Bidaux, G.; Degerny, C.; Verreman, K.; et al. TRP channel-associated factors are a novel protein family that regulates TRPM8 trafficking and activity. J. Cell Biol. 2015, 208, 89–107. [Google Scholar] [CrossRef] [Green Version]
- Grolez, G.P.; Hammadi, M.; Barras, A.; Gordienko, D.; Slomianny, C.; Völkel, P.; Angrand, P.O.; Pinault, M.; Guimaraes, C.; Potier-Cartereau, M.; et al. Encapsulation of a TRPM8 Agonist, WS12, in Lipid Nanocapsules Potentiates PC3 Prostate Cancer Cell Migration Inhibition through Channel Activation. Sci. Rep. 2019, 9, 7926. [Google Scholar] [CrossRef]
- Yang, Z.H.; Wang, X.H.; Wang, H.P.; Hu, L.Q. Effects of TRPM8 on the proliferation and motility of prostate cancer PC-3 cells. Asian J. Androl. 2009, 11, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Genova, T.; Grolez, G.P.G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 inhibits endothelial cell migration via a nonchannel function by trapping the small GTPase Rap1. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, C.; Armache, J.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Nassar, N.; Horn, G.; Herrmann, C.A.; Scherer, A.; McCormick, F.; Wittinghofer, A. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with RaplA and a GTP analogue. Nature 1995, 375, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.; Xia, B.; Porter, K.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Quignot, C.; Rey, J.; Yu, J.; Tufféry, P.; Guerois, R.; Andreani, J. InterEvDock2: An expanded server for protein docking using evolutionary and biological information from homology models and multimeric inputs. Nucleic Acids Res. 2018, 46, W408. [Google Scholar] [CrossRef] [PubMed]
- Brysbaert, G.; Lorgouilloux, K.; Vranken, W.; Lensink, M. RINspector: A Cytoscape app for centrality analyses and DynaMine flexibility prediction. Bioinformatics 2018, 34, 294–296. [Google Scholar] [CrossRef]
- Brysbaert, G.; Mauri, T.; de Ruyck, J.; Lensink, M. Identification of Key Residues in Proteins Through Centrality Analysis and Flexibility Prediction with RINspector. Curr. Protoc. Bioinforma. 2019, 65, e66. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.; Demchak, B.; Bader, G. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinforma. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [Green Version]
- Amitai, G.; Shemesh, A.; Sitbon, E.; Shklar, M.; Netanely, D.; Venger, I.; Pietrokovski, S. Network analysis of protein structures identifies functional residues. J. Mol. Biol. 2004, 344, 1135–1146. [Google Scholar] [CrossRef]
- Del Sol, A.; Fujihashi, H.; Amoros, D.; Nussinov, R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006, 2, 2006.0019. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Wiener, H.H.; Ahearn, I.M.; Silletti, J.; Chiu, V.K.; Philips, M.R. Rap1 up-regulation and activation on plasma membrane regulates T cell adhesion. J. Cell Biol. 2004, 164, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Quatela, S.; Philips, M.R. Analysis of Ras Activation in Living Cells with GFP-RBD. Methods Enzymol. 2006, 407, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://xenabrowser.net/datapages/?dataset=GDC-%0APANCAN.mutect2_snv.tsv&host=https%3A%2F%2Fgdc.xenahubs.net&removeHub=https%0A%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443 (accessed on 25 February 2022).
- Edmund. Available online: https://github.com/MrHedmad/Edmund (accessed on 25 February 2022).
- Available online: https://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG000001164%0A73;r=1:111542218-111716691 (accessed on 25 February 2022).
- Available online: https://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG000001444%0A81;r=2:233917373-234019522 (accessed on 25 February 2022).
- ANOVA_merged.R. Available online: https://gist.github.com/MrHedmad/a53eb36ff1233e45bc638008912c0d35 (accessed on 4 March 2022).
- Bidaux, G.; Flourakis, M.; Thebault, S.; Zholos, A.; Beck, B.; Gkika, D.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; et al. Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. J. Clin. Investig. 2007, 117, 1647–1657. [Google Scholar] [CrossRef] [Green Version]
- Pillozzi, S.; Brizzi, M.F.; Bernabei, P.A.; Bartolozzi, B.; Caporale, R.; Basile, V.; Boddi, V.; Pegoraro, L.; Becchetti, A.; Arcangeli, A. VEGFR-1 (FLT-1), β1 integrin, and hERG K+ channel for a macromolecular signaling complex in acute myeloid leukemia: Role in cell migration and clinical outcome. Blood 2007, 110, 1238–1250. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef]
- Desai, B.N.; Krapivinsky, G.; Navarro, B.; Krapivinsky, L.; Carter, B.C.; Febvay, S.; Delling, M.; Penumaka, A.; Ramsey, I.S.; Manasian, Y.; et al. Cleavage of TRPM7 Releases the Kinase Domain from the Ion Channel and Regulates Its Participation in Fas-Induced Apoptosis. Dev. Cell 2012, 22, 1149–1162. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 2017, 595, 3165–3180. [Google Scholar] [CrossRef] [Green Version]
- Joly, D.; Ishibe, S.; Nickel, C.; Yu, Z.; Somlo, S.; Cantley, L. The polycystin 1-C-terminal fragment stimulates ERK-dependent spreading of renal epithelial cells. J. Biol. Chem. 2006, 281, 26329–26339. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L. Linking Rap to cell adhesion. Curr. Opin. Cell Biol. 2005, 17, 123–128. [Google Scholar] [CrossRef]
- Boettner, B.; Van Aelst, L. Control of cell adhesion dynamics by Rap1 signaling. Curr. Opin. Cell Biol. 2009, 21, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moissoglu, K.; Schwartz, M.A. Spatial and temporal control of Rho GTPase functions. Cell. Logist. 2014, 4, e943618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarwood, S.; Cullen, P.J.; Kupzig, S. The GAP1 family of GTPase-activating proteins: Spatial and temporal regulators of small GTPase signalling. Biochem. Soc. Trans. 2006, 34, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [Green Version]
- Reedquist, K.A.; Ross, E.; Koop, E.A.; Wolthuis, R.M.F.; Zwartkruis, F.J.T.; Van Kooyk, Y.; Salmon, M.; Buckley, C.D.; Bos, J.L. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J. Cell Biol. 2000, 148, 1151–1158. [Google Scholar] [CrossRef]
- Carmona, G.; Go, S.; Orlandi, A.; Ba, T.; Jugold, M.; Kiessling, F.; Henschler, R.; Zeiher, A.M.; Dimmeler, S.; Chavakis, E. Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood J. Am. Soc. Hematol. 2009, 113, 488–497. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Zhang, L.; Barritt, G. TRP channels in cancer. Biochim. Biophys. Acta—Mol. Basis Dis. 2007, 1772, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Skryma, R.; Mariot, P.; Bourhis, X.; Coppenolle, F.; Shuba, Y.; Abeele, F.V.; Legrand, G.; Humez, S.; Boilly, B.; Prevarskaya, N. Store depletion and store-operated Ca2+ current in human prostate cancer LNCaP cells: Involvement in apoptosis. J. Physiol. 2000, 527 Pt 1, 71–83. [Google Scholar] [CrossRef]
- Thebault, S.; Lemonnier, L.; Bidaux, G.; Flourakis, M.; Bavencoffe, A.; Gordienko, D.; Roudbaraki, M.; Delcourt, P.; Panchin, Y.; Shuba, Y.; et al. Novel role of cold/menthol-sensitive transient receptor potential melastatine family member 8 (TRPM8) in the activation of store-operated channels in LNCaP human prostate cancer epithelial cells. J. Biol. Chem. 2005, 280, 39423–39435. [Google Scholar] [CrossRef] [Green Version]
- Abeele, F.V.; Skryma, R.; Shuba, Y.; Van Coppenolle, F.; Slomianny, C.; Roudbaraki, M.; Mauroy, B.; Wuytack, F.; Prevarskaya, N. Bcl-2-dependent modulation of Ca2+ homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 2002, 1, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Vanoverberghe, K.; Abeele, F.V.; Mariot, P.; Lepage, G.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004, 11, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Mak, S.; Li, L.; Parra, A.; Denlinger, B.; Belmonte, C.; McNaughton, P.A. Direct inhibition of the cold-Activated TRPM8 ion channel by Gα q. Nat. Cell Biol. 2012, 14, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klasen, K.; Hollatz, D.; Zielke, S.; Gisselmann, G.; Hatt, H.; Wetzel, C.H. The TRPM8 ion channel comprises direct Gq protein-activating capacity. Eur. J. Physiol. 2012, 463, 779–797. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Miyano, N.; Baba, S.; Liao, J.; Kawamura, T.; Tsuda, C.; Takeda, A.; Yamamoto, M.; Kumasaka, T.; Kataoka, T.; et al. Molecular Mechanism for Conformational Dynamics of Ras·GTP Elucidated from In-Situ Structural Transition in Crystal. Sci. Rep. 2016, 6, 25931. [Google Scholar] [CrossRef]
- Khaled, M.; Gorfe, A.; Sayyed-Ahmad, A. Conformational and Dynamical Effects of Tyr32 Phosphorylation in K-Ras: Molecular Dynamics Simulation and Markov State Models Analysis. J. Phys. Chem. B 2019, 123, 7667–7675. [Google Scholar] [CrossRef]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.K.; St-Germain, J.; Cook, J.D.; Valencia-Sama, I.; Grant, B.M.M.; Herrera, S.G.; et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 2019, 10, 224. [Google Scholar] [CrossRef]
- Bunda, S.; Heir, P.; Srikumar, T.; Cook, J.D.; Burrell, K.; Kano, Y.; Lee, J.E.; Zadeh, G.; Raught, B.; Ohh, M. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3785–E3794. [Google Scholar] [CrossRef] [Green Version]
- Van de Graaf, S.F.J.; Chang, Q.; Mensenkamp, A.R.; Hoenderop, J.G.J.; Bindels, R.J.M. Direct Interaction with Rab11a Targets the Epithelial Ca 2+ Channels TRPV5 and TRPV6 to the Plasma Membrane. Mol. Cell. Biol. 2006, 26, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334. [Google Scholar] [CrossRef]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habault, J.; Poyet, J.L. Recent advances in cell penetrating peptide-based anticancer therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Kang, R.H.; Jang, J.E.; Huh, E.; Kang, S.J.; Ahn, D.R.; Kang, J.S.; Sailor, M.J.; Yeo, S.G.; Oh, M.S.; Kim, D.; et al. A brain tumor-homing tetra-peptide delivers a nano-therapeutic for more effective treatment of a mouse model of glioblastoma. Nanoscale Horizons 2020, 5, 1213–1225. [Google Scholar] [CrossRef]
- Bledzka, K.; Smyth, S.S.; Plow, E.F. Integrin alphaIIbbeta3: From discovery to efficacious therapeutic target. Circ. Res. 2013, 112, 1189–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, M.; Btesh, J.; McNaughton, P. Disrupting Sensitization of Transient Receptor Potential Vanilloid Subtype 1 Inhibits Inflammatory Hyperalgesia. J. Neurosci. 2013, 33, 7407–7414. [Google Scholar] [CrossRef] [Green Version]
- Schulie, A.J.; Yeh, C.Y.; Orang, B.N.; Pa, O.J.; Hopkin, M.P.; Moutal, A.; Khanna, R.; Sun, D.; Justic, J.A.; Aizenman, E. Targeted disruption of Kv2.1-VAPA association provides neuroprotection against ischemic stroke in mice by declustering Kv2.1 channels. Sci. Adv. 2020, 6, eaaz8110. [Google Scholar] [CrossRef]
- Brittain, J.M.; Duarte, D.B.; Wilson, S.M.; Zhu, W.; Ballard, C.; Johnson, P.L.; Liu, N.; Xiong, W.; Ripsch, M.S.; Wang, Y.; et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 2011, 17, 822–829. [Google Scholar] [CrossRef]
- Fischer, A.; Rosen, A.C.; Ensslin, C.J.; Wu, S.; Lacouture, M.E. Pruritus to anticancer agents targeting the EGFR, BRAF, and CTLA-4. Dermatol. Ther. 2013, 26, 135–148. [Google Scholar] [CrossRef]
- Tu, C.L.; Chang, W.; Bikle, D.D. Phospholipase Cγ1 is required for activation of store-operated channels in human keratinocytes. J. Investig. Dermatol. 2005, 124, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Katterle, Y.; Brandt, B.H.; Dowdy, S.F.; Niggemann, B.; Zänker, K.S.; Dittmar, T. Antitumour effects of PLC-γ1-(SH2)2-TAT fusion proteins on EGFR/c-erbB-2-positive breast cancer cells. Br. J. Cancer 2004, 90, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laínez, S.; Valente, P.; Ontoria-Oviedo, I.; Estévez-Herrera, J.; Camprubí-Robles, M.; Ferrer-Montiel, A.; Planells-Cases, R. GABA A receptor associated protein (GABARAP) modulates TRPV1 expression and channel function and desensitization. FASEB J. 2010, 24, 1958–1970. [Google Scholar] [CrossRef] [PubMed]
- Saldías, M.P.; Maureira, D.; Orellana-Serradell, O.; Silva, I.; Lavanderos, B.; Cruz, P.; Torres, C.; Cáceres, M.; Cerda, O. TRP Channels Interactome as a Novel Therapeutic Target in Breast Cancer. Front. Oncol. 2021, 11, 621614. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.J.; Patel, K.N.; Jeske, N.A.; Bierbower, S.M.; Zou, W.; Tiwari, V.; Zheng, Q.; Tang, Z.; Mo, G.C.H.; Wang, Y.; et al. Tmem100 Is a Regulator of TRPA1-TRPV1 Complex and Contributes to Persistent Pain. Neuron 2015, 85, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabonga, L.; Kappo, A.P. Peptidomimetics: A Synthetic Tool for Inhibiting Protein–Protein Interactions in Cancer. Int. J. Pept. Res. Ther. 2020, 26, 225–241. [Google Scholar] [CrossRef] [Green Version]
- Corbi-Verge, C.; Kim, P.M. Motif mediated protein-protein interactions as drug targets. Cell Commun. Signal. 2016, 14, 8. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vector | Mutation | Fw Primer | Rv Primer | Kit Used |
|---|---|---|---|---|
| pGEX-TRPM8 Nt | E207A | caatggccacaatattcgcctctgaactcctgctg | cagcaggagttcagaggcgaatattgtggccattg | Agilent kit |
| pGEX-TRPM8 Nt | Y240A | cttgtgaagtcatccataagggcctgggctaaaaaatagccctc | gagggctattttttagcccaggcccttatggatgacttcacaag | Agilent kit |
| pCDNA4-TRPM8 | E207A | caatggccacaatattcgcctctgaactcctgctg | cagcaggagttcagaggcgaatattgtggccattg | Agilent kit |
| pCDNA4-TRPM8 | Y240A | cttgtgaagtcatccataagggcctgggctaaaaaatagccctc | gagggctattttttagcccaggcccttatggatgacttcacaag | Agilent kit |
| pCMV-TNT-Rap1A | K31A | gaatcttctatcgttgggtcatatgcttcaacaaaaattccctgaacaaac | gtttgttcagggaatttttgttgaagcatatgacccaacgatagaagattc | Agilent kit |
| pCMV-TNT-Rap1A | Y32A | cttctatcgttgggtcagctttttcaacaaaaattccctgaacaaactgaac | gttcagtttgttcagggaatttttgttgaaaaagctgacccaacgatagaag | Agilent kit |
| pCMV-TNT-Rap1A | K31A | gaatcttctatcgttgggtcatatgcttcaacaaaaattccctgaacaaac | gtttgttcagggaatttttgttgaagcatatgacccaacgatagaagattc | Agilent kit |
| pCMV-TNT-Rap1A | Y32A | cttctatcgttgggtcagctttttcaacaaaaattccctgaacaaactgaac | gttcagtttgttcagggaatttttgttgaaaaagctgacccaacgatagaag | Agilent kit |
| peGFP-Rap1A-N17 | K31A | agttcagaggcgaatattgtgg | cctgctgatggtgttatc | NEB |
| peGFP-Rap1A-N17 | Y32A | tttagcccaggcccttatggatgac | aaatagccctcagcatcg | NEB |
| pMT2SM-HA-Rap1A | K31A | gaatcttctatcgttgggtcatatgcttcaacaaaaattccctgaacaaac | gtttgttcagggaatttttgttgaagcatatgacccaacgatagaagattc | Agilent kit |
| pMT2SM-HA-Rap1A | Y32A | cttctatcgttgggtcagctttttcaacaaaaattccctgaacaaactgaac | gttcagtttgttcagggaatttttgttgaaaaagctgacccaacgatagaag | Agilent kit |
| pMT2SM-HA-Rap1A N17 | K31A | gaatcttctatcgttgggtcatatgcttcaacaaaaattccctgaacaaac | gtttgttcagggaatttttgttgaagcatatgacccaacgatagaagattc | Agilent kit |
| pMT2SM-HA-Rap1A N17 | Y32A | cttctatcgttgggtcagctttttcaacaaaaattccctgaacaaactgaac | gttcagtttgttcagggaatttttgttgaaaaagctgacccaacgatagaag | Agilent kit |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinigò, G.; Grolez, G.P.; Audero, M.; Bokhobza, A.; Bernardini, M.; Cicero, J.; Toillon, R.-A.; Bailleul, Q.; Visentin, L.; Ruffinatti, F.A.; et al. TRPM8-Rap1A Interaction Sites as Critical Determinants for Adhesion and Migration of Prostate and Other Epithelial Cancer Cells. Cancers 2022, 14, 2261. https://doi.org/10.3390/cancers14092261
Chinigò G, Grolez GP, Audero M, Bokhobza A, Bernardini M, Cicero J, Toillon R-A, Bailleul Q, Visentin L, Ruffinatti FA, et al. TRPM8-Rap1A Interaction Sites as Critical Determinants for Adhesion and Migration of Prostate and Other Epithelial Cancer Cells. Cancers. 2022; 14(9):2261. https://doi.org/10.3390/cancers14092261
Chicago/Turabian StyleChinigò, Giorgia, Guillaume P. Grolez, Madelaine Audero, Alexandre Bokhobza, Michela Bernardini, Julien Cicero, Robert-Alain Toillon, Quentin Bailleul, Luca Visentin, Federico Alessandro Ruffinatti, and et al. 2022. "TRPM8-Rap1A Interaction Sites as Critical Determinants for Adhesion and Migration of Prostate and Other Epithelial Cancer Cells" Cancers 14, no. 9: 2261. https://doi.org/10.3390/cancers14092261
APA StyleChinigò, G., Grolez, G. P., Audero, M., Bokhobza, A., Bernardini, M., Cicero, J., Toillon, R.-A., Bailleul, Q., Visentin, L., Ruffinatti, F. A., Brysbaert, G., Lensink, M. F., De Ruyck, J., Cantelmo, A. R., Fiorio Pla, A., & Gkika, D. (2022). TRPM8-Rap1A Interaction Sites as Critical Determinants for Adhesion and Migration of Prostate and Other Epithelial Cancer Cells. Cancers, 14(9), 2261. https://doi.org/10.3390/cancers14092261