
The Role of Fast-Cycling Atypical RHO GTPases in Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. The Origin of the RHO GTPases
3. The Concept of Atypical RHO GTPases
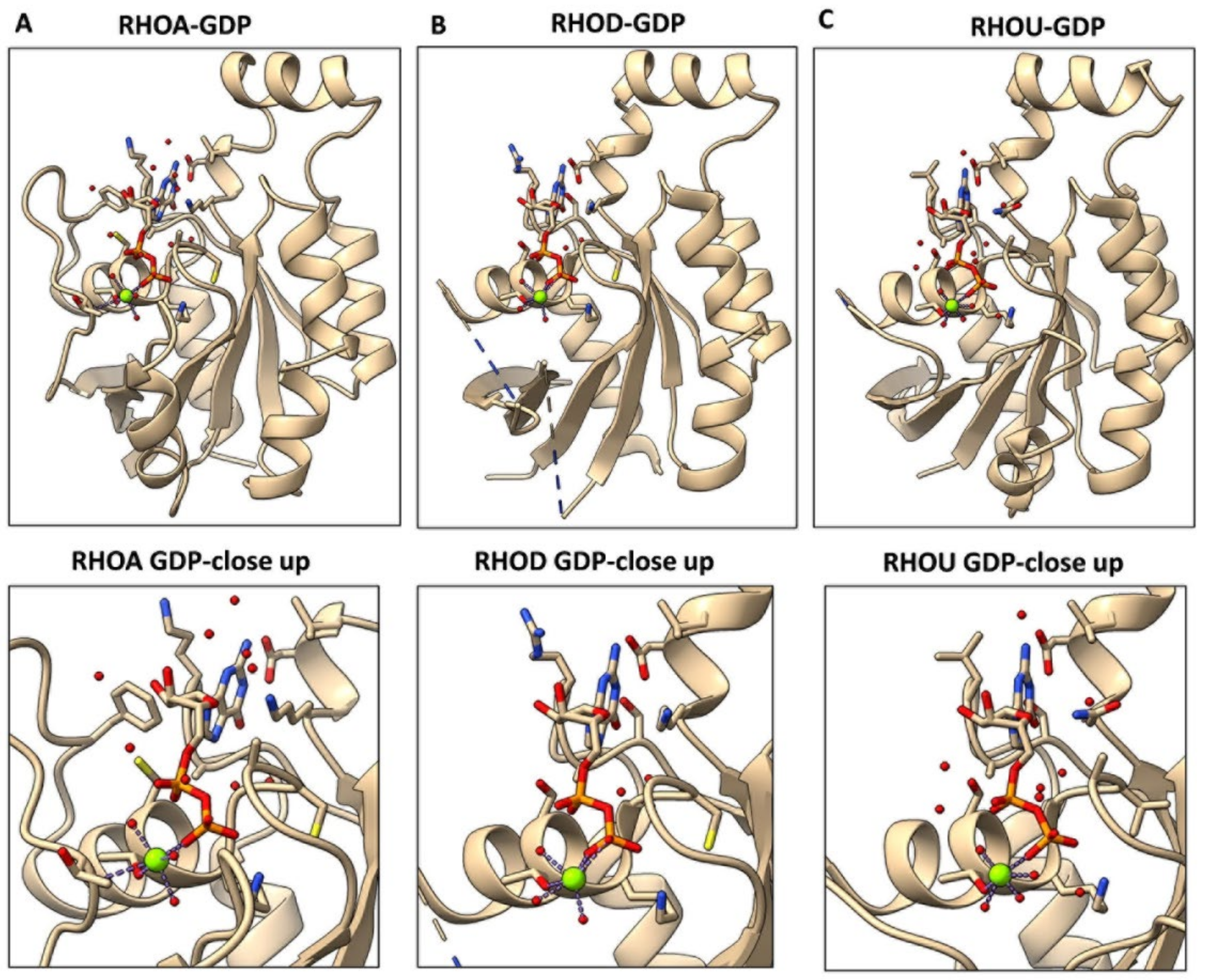
4. The Mechanisms Underlying an Increased Intrinsic Exchange Activity
5. RHOU and RHOV in Cancer
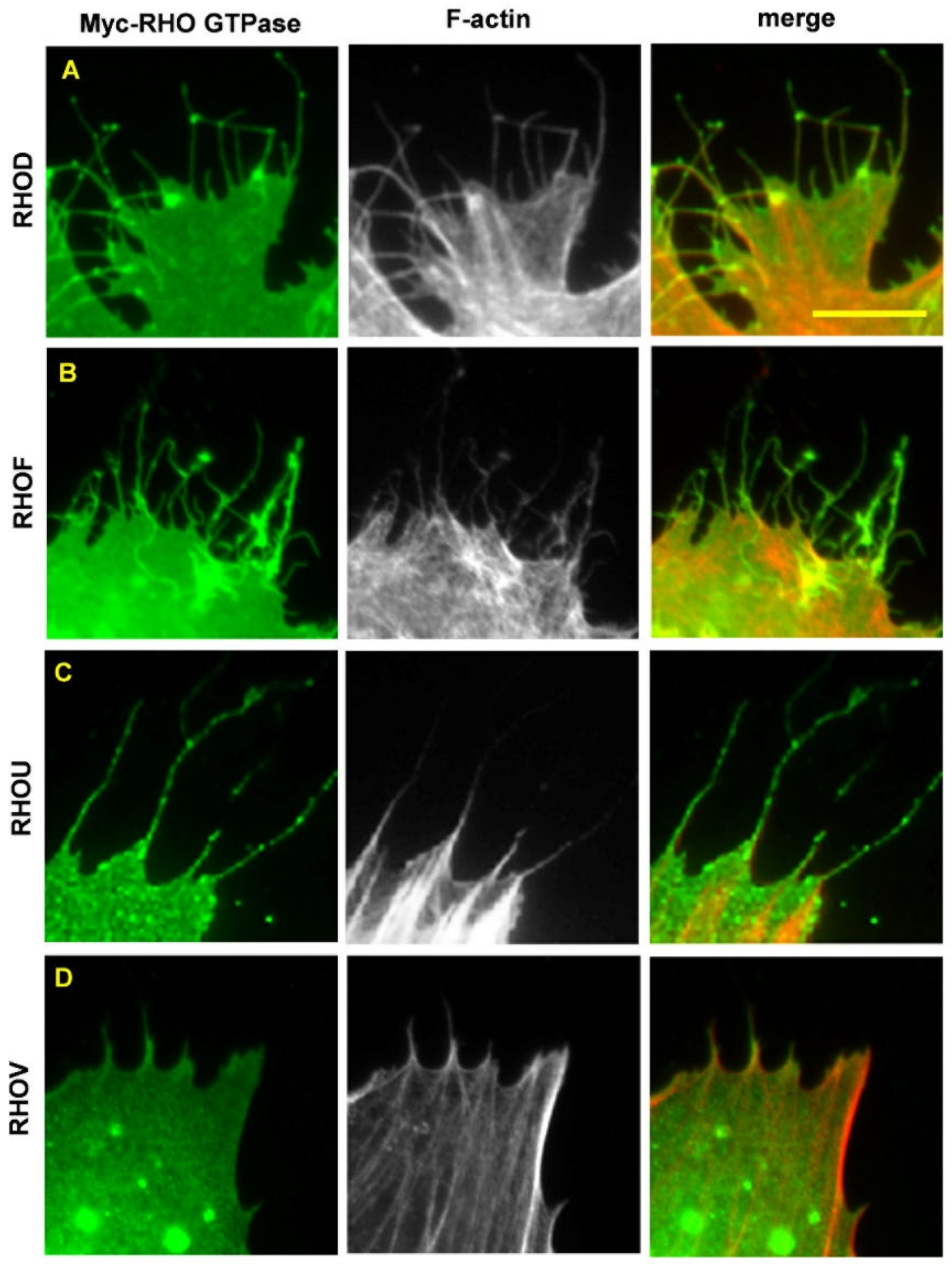
6. RHOD and RHOF in Cancer
7. Classical RHO GTPases as Proto-Oncogenes
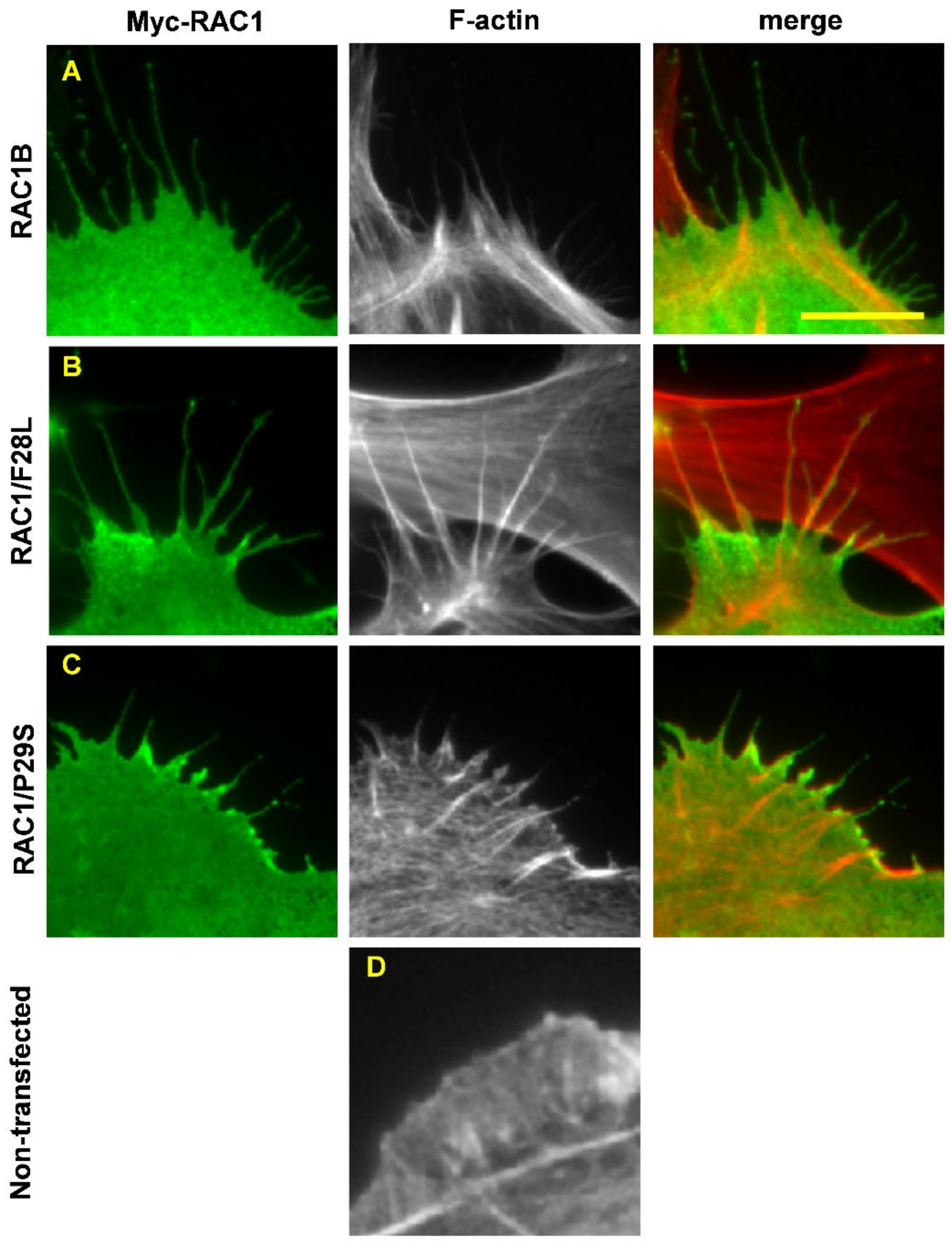
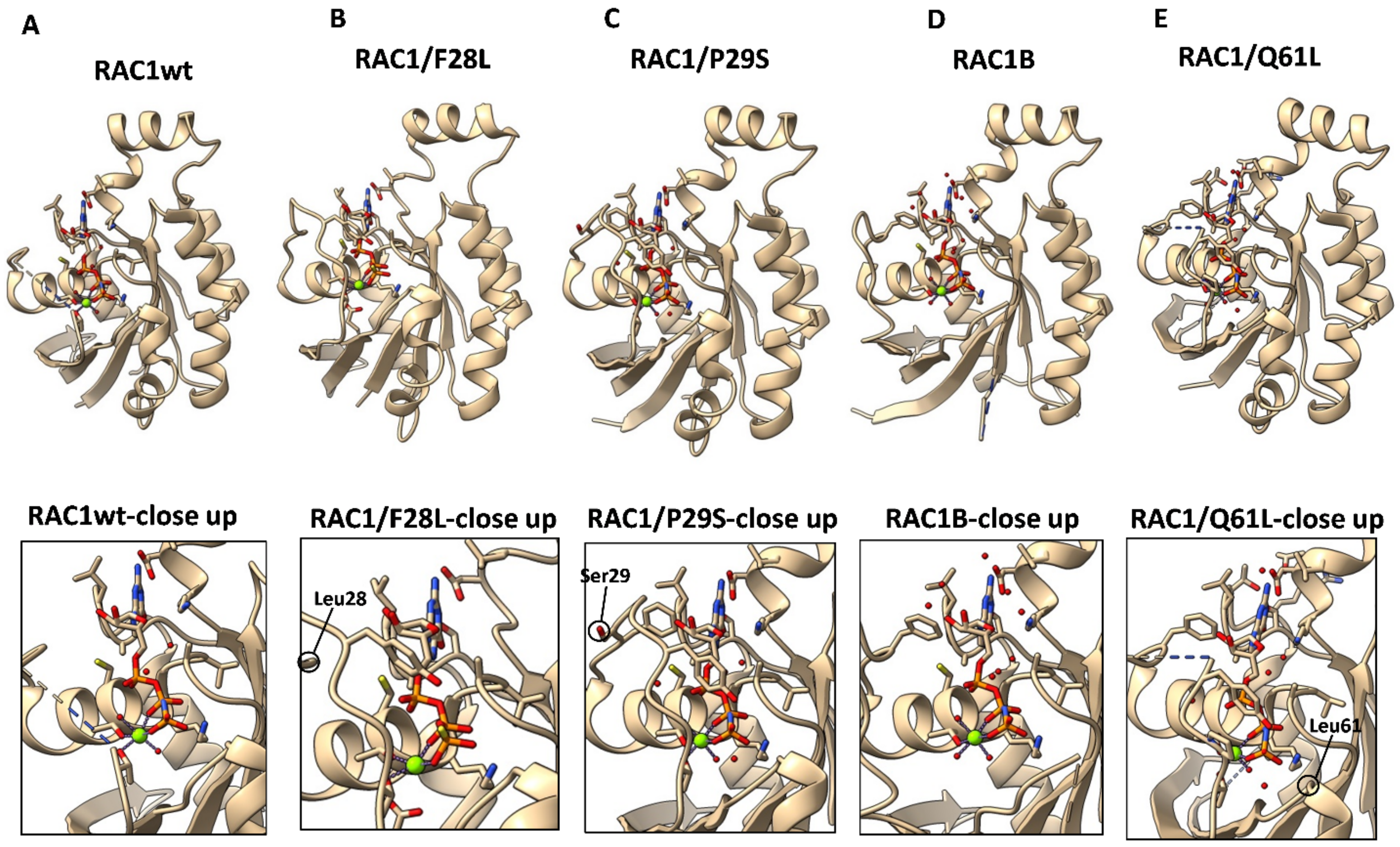
7.1. RAC1/P29S
7.2. RAC1B
8. RHO Mutants in Cancer
9. Additional Cancer-Associated Mutations in RHO GTPases
10. Summary
11. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Aspenström, P.; Fransson, Å.; Saras, J. The Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 2004, 377, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Madaule, P.; Axel, R. A novel Ras-related gene family. Cell 1985, 41, 31–40. [Google Scholar] [CrossRef]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Aspenström, P. Activated Rho GTPases in cancer-the beginning of a new paradigm. Int. J. Mol. Sci. 2018, 19, 3949. [Google Scholar] [CrossRef]
- Aspenström, P. Fast-cycling Rho GTPases. Small GTPases 2020, 11, 248–255. [Google Scholar] [CrossRef]
- Yeramian, P.; Chardin, P.; Madaule, P.; Tavitian, A. Nucleotide sequence of human rho cDNA clone 12. Nucleic Acids Res. 1987, 15, 1869. [Google Scholar] [CrossRef]
- Chardin, P.; Madaule, P.; Tavitian, A. Coding sequence of human rho cDNAs clone 6 and clone 9. Nucleic Acids Res. 1988, 16, 2717. [Google Scholar] [CrossRef]
- Evans, T.; Brown, M.L.; Fraser, E.D.; Northup, J.K. Purification of the major GTP-binding proteins from human placental membranes. J. Biol. Chem. 1986, 261, 7052–7059. [Google Scholar] [CrossRef]
- Didsbury, J.; Weber, R.F.; Bokoch, G.M.; Evans, T.; Snyderman, R. Rac, a novel ras-related family of proteins that are botulinum toxin substrates. J. Biol. Chem. 1989, 264, 16378–16382. [Google Scholar] [CrossRef]
- Munemitsu, S.; Innis, M.A.; Clark, R.; McCormick, F.; Ullrich, A.; Polakis, P. Molecular cloning and expression of a G25K cDNA, the human homolog of the yeast cell cyclegene CDC42. Mol. Cell. Biol. 1990, 10, 5977–5982. [Google Scholar] [CrossRef] [PubMed]
- Shinjo, K.; Koland, J.G.; Hart, M.J.; Narasimhan, V.; Johnson, D.I.; Evans, T.; Cerione, R.A. Molecular cloning of the gene for the human placental GTP-binding protein Gp (G25K): Identification of this GTP-binding protein as the human homolog of the yeast cell-division-cycle protein CDC42. Proc. Natl. Acad. Sci. USA 1990, 87, 9853–9857. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A.R. Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Tcherkezian, J.; Lamarche-Vane, N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell 2007, 99, 67–86. [Google Scholar] [CrossRef]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef]
- Xie, F.; Shao, S.; Aziz, A.U.R.; Zhang, B.; Wang, H.; Liu, B. Role of Rho-specific guanine nucleotide dissociation inhibitor α regulation in cell migration. Acta Histochem. 2017, 119, 183–189. [Google Scholar] [CrossRef]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Hu, K.Q.; Lu, Y.; Nolan, K.M.; Thissen, J.; Settleman, J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and In-Vivo farnesylation. Mol. Cell. Biol. 1996, 16, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Fiegen, D.; Blumenstein, L.; Stege, P.; Vetter, I.R.; Ahmadian, M.R. Crystal structure of Rnd3/RhoE: Functional implications. FEBS Lett. 2002, 525, 100–104. [Google Scholar] [CrossRef]
- Aspenström, P.; Ruusala, A.; Pacholsky, D. Taking Rho GTPases to the next level: The cellular functions of atypical Rho GTPases. Exp. Cell Res. 2007, 313, 3673–3679. [Google Scholar] [CrossRef] [PubMed]
- Chardin, P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 2006, 7, 54–62. [Google Scholar] [CrossRef]
- Saras, J.; Wollberg, P.; Aspenström, P. Wrch1 is a GTPase-deficient Cdc42-like protein with unusual binding characteristics and cellular effects. Exp. Cell Res. 2004, 299, 356–369. [Google Scholar] [CrossRef]
- Shutes, A.; Berzat, A.C.; Cox, A.D.; Der, C.J. Atypical mechanism of regulation of the Wrch-1 Rho family small GTPase. Curr. Biol. 2004, 14, 2052–2056. [Google Scholar] [CrossRef]
- Jaiswal, M.; Fansa, E.K.; Dvorsky, R.; Ahmadian, M.R. New insight into the molecular switch mechanism of human Rho family proteins: Shifting a paradigm. Biol. Chem. 2013, 394, 89–95. [Google Scholar] [CrossRef]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Tao, W.; Pennica, D.; Xu, L.; Kalejta, R.F.; Levine, A.J. Wrch-1, a novel member of the Rho gene family that is regulated by Wnt-1. Genes Dev. 2001, 15, 1796–1807. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulation and function of RhoU and RhoV. Small GTPases 2020, 11, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, R.; Ueda, H.; Honda, R. Structural and functional characterization of fast-cycling RhoF GTPase. Biochem. Biophys. Res. Commun. 2019, 513, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, Q.; Ge, X.; Shi, Y.; Ye, T.; Mi, Y.; Xie, T.; Li, Q.; Ye, Q. RHOV promotes lung adenocarcinoma cell growth and metastasis through JNK/c-Jun pathway. Int. J. Biol. Sci. 2021, 17, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xia, R.; Jiang, L.; Zhou, Y.; Xu, H.; Peng, W.; Yao, C.; Zhou, G.; Zhang, Y.; Xia, H.; et al. Overexpression of RhoV promotes the progression and EGFR-TKI resistance of lung adenocarcinoma. Front. Oncol. 2021, 11, 619013. [Google Scholar] [CrossRef] [PubMed]
- Shepelev, M.V.; Korobko, I.V. The RHOV gene is overexpressed in human non-small cell lung cancer. Cancer Genet. 2013, 206, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho family of Ras-like GTPases in eukaryotes. Mol. Biol. Evol. 2007, 24, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, P.J.; Infante, E.; Khwaja, A.; Ridley, A.J. Analysis of Rho GTPase expression in T-ALL identifies RhoU as a target for Notch involved in T-ALL cell migration. Oncogene 2013, 32, 198–208. [Google Scholar] [CrossRef]
- Piano, D.M.; Manuelli, V.; Zadra, G.; Otte, J.; Edqvist, P.-H.D.; Pontén, F.; Nowinski, S.; Niaouris, A.; Grigoriadis, A.; Loda, M.; et al. Lipogenic signalling modulates prostate cancer cell adhesion and migration via modification of Rho GTPases. Oncogene 2020, 39, 3666–3679. [Google Scholar] [CrossRef]
- Yamada, Y.; Nishikawa, R.; Kato, M.; Okato, A.; Arai, T.; Kojima, S.; Yamazaki, K.; Naya, Y.; Ichikawa, T.; Seki, N. Regulation of HMGB3 by antitumor miR-205-5p inhibits cancer cell aggressiveness and is involved in prostate cancer pathogenesis. J. Hum. Genet. 2018, 63, 195–205. [Google Scholar] [CrossRef]
- Alinezhad, S.; Väänänen, R.-M.; Mattsson, J.; Li, Y.; Tallgrén, T.; Tong Ochoa, N.; Bjartell, A.; Åkerfelt, M.; Taimen, P.; Boström, P.J.; et al. Validation of novel biomarkers for prostate cancer progression by the combination of bioinformatics, clinical and functional studies. PLoS ONE 2016, 11, e0155901. [Google Scholar] [CrossRef]
- Canovas Nunes, S.; Manzoni, M.; Pizzi, M.; Mandato, E.; Carrino, M.; Quotti Tubi, L.; Zambello, R.; Adami, F.; Visentin, A.; Barilà, G.; et al. The small GTPase RhoU lays downstream of JAK/STAT signaling and mediates cell migration in multiple myeloma. Blood Cancer J. 2018, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, E.; Orecchia, V.; Corrieri, P.; Schiavone, D.; Avalle, L.; Moiso, E.; Savino, A.; Molineris, I.; Provero, P.; Poli, V. SP1 and STAT3 functionally synergize to induce the RhoU small GTPase and a subclass of non-canonical WNT-responsive genes correlating with poor prognosis in breast cancer. Cancers 2019, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Slaymi, C.; Vignal, E.; Crès, G.; Roux, P.; Blangy, A.; Raynaud, P.; Fort, P. The atypical RhoU/Wrch1 Rho GTPase controls cell proliferation and apoptosis in the gut epithelium. Biol. Cell 2019, 111, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zi, J.; Ge, Z. High expression of RhoF predicts worse overall survival: A potential therapeutic target for non-M3 acute myeloid leukemia. J. Cancer 2021, 12, 5530–5542. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, Y.; Bai, Y.; Chen, M.; Cheng, D.; Wu, M.; Xia, J. Ras homolog family member F, filopodia associated promotes hepatocellular carcinoma metastasis by altering the metabolic status of cancer cells through RAB3D. Hepatology 2021, 73, 2361–2379. [Google Scholar] [CrossRef]
- Eva, A.; Aaronson, S.A. Isolation of a new human oncogene from a diffuse B-cell lymphoma. Nature 1985, 316, 273–275. [Google Scholar] [CrossRef]
- Ron, D.; Tronick, S.R.; Aaronson, S.A.; Eva, A. Molecular cloning and characterization of the human dbl proto-oncogene: Evidence that its overexpression is sufficient to transform NIH/3T3 cells. EMBO J. 1988, 7, 2465–2473. [Google Scholar] [CrossRef]
- Qiu, R.G.; Chen, J.; McCormick, F.; Symons, M. A role for Rho in Ras transformation. Proc. Natl. Acad. Sci. USA 1995, 92, 11781–11785. [Google Scholar] [CrossRef]
- Khosravi-Far, R.; Solski, P.A.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell Biol. 1995, 15, 6443–6453. [Google Scholar] [CrossRef]
- del Peso, L.; Hernández-Alcoceba, R.; Embade, N.; Carnero, A.; Esteve, P.; Paje, C.; Lacal, J.C. Rho proteins induce metastatic properties In Vivo. Oncogene 1997, 15, 3047–3057. [Google Scholar] [CrossRef]
- Jordan, P.; Brazåo, R.; Boavida, M.G.; Gespach, C.; Chastre, E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999, 18, 6835–6839. [Google Scholar] [CrossRef] [PubMed]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Aspenström, P. The Intrinsic GDP/GTP exchange activities of Cdc42 and Rac1 are critical determinants for their specific effects on mobilization of the actin filament system. Cells 2019, 8, 759. [Google Scholar] [CrossRef]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. Molecular dynamic simulation reveals damaging impact of RAC1 F28L mutation in the switch I region. PLoS ONE 2013, 8, e77453. [Google Scholar] [CrossRef]
- Henriques, A.F.; Barros, P.; Moyer, M.P.; Matos, P.; Jordan, P. Expression of tumor-related Rac1b antagonizes B-Raf-induced senescence in colorectal cells. Cancer Lett. 2015, 369, 368–375. [Google Scholar] [CrossRef]
- Zhou, C.; Licciulli, S.; Avila, J.L.; Cho, M.; Troutman, S.; Jiang, P.; Kossenkov, A.V.; Showe, L.C.; Liu, Q.; Vachani, A.; et al. The Rac1 splice form Rac1b promotes K-Ras-induced lung tumorigenesis. Oncogene 2013, 32, 903–909. [Google Scholar] [CrossRef]
- Seiz, J.R.; Klinke, J.; Scharlibbe, L.; Lohfink, D.; Heipel, M.; Ungefroren, H.; Giehl, K.; Menke, A. Different signaling and functionality of Rac1 and Rac1b in the progression of lung adenocarcinoma. Biol. Chem. 2020, 401, 517–531. [Google Scholar] [CrossRef]
- Mehner, C.; Miller, E.; Khauv, D.; Nassar, A.; Oberg, A.L.; Bamlet, W.R.; Zhang, L.; Waldmann, J.; Radisky, E.S.; Crawford, H.C.; et al. Tumor cell-derived MMP3 orchestrates Rac1b and tissue alterations that promote pancreatic adenocarcinoma. Mol. Cancer Res. 2014, 12, 1430–1439. [Google Scholar] [CrossRef]
- Faria, M.; Matos, P.; Pereira, T.; Cabrera, R.; Cardoso, B.A.; Bugalho, M.J.; Silva, A.L. RAC1b overexpression stimulates proliferation and NF-κB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS ONE 2017, 12, e0172689. [Google Scholar] [CrossRef]
- Kawazu, M.; Ueno, T.; Kontani, K.; Ogita, Y.; Ando, M.; Fukumura, K.; Yamato, A.; Soda, M.; Takeuchi, K.; Miki, Y.; et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.E.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T-cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Kontani, K.; Enami, T.; Kataoka, K.; Ishii, R.; Totoki, Y.; Kataoka, T.R.; Hirata, M.; Aoki, K.; Nakano, K.; et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood 2016, 127, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T-cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef]
- Stevers, M.; Rabban, J.T.; Garg, K.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Zaloudek, C.; Solomon, D.A. Well-differentiated papillary mesothelioma of the peritoneum is genetically defined by mutually exclusive mutations in TRAF7 and CDC42. Mod. Pathol. 2019, 32, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Shakir, M.A.; Gill, J.S.; Lundquist, E.A. Interactions of UNC-34 enabled with Rac GTPases and the NIK kinase MIG-15 in Caenorhabditis elegans axon pathfinding and neuronal migration. Genetics 2006, 172, 893–913. [Google Scholar] [CrossRef][Green Version]
- Senyuz, S.; Jang, H.; Nussinov, R.; Keskin, O.; Gursoy, A. Mechanistic differences of activation of Rac1(P29S) and Rac1(A159V). J. Phys. Chem. B 2021, 125, 3790–3802. [Google Scholar] [CrossRef]
- Mohan, A.S.; Dean, K.M.; Isogai, T.; Kasitinon, S.Y.; Murali, V.S.; Roudot, P.; Groisman, A.; Reed, D.K.; Welf, E.S.; Han, S.J.; et al. Enhanced dendritic actin network formation in extended lamellipodia drives proliferation in growth-challenged Rac1(P29S) melanoma cells. Dev. Cell 2019, 49, 444–460. [Google Scholar] [CrossRef]
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. Rac1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Overcoming immune evasion in melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar] [CrossRef] [PubMed]
- Mar, V.J.; Wong, S.Q.; Logan, A.; Nguyen, T.; Cebon, J.; Kelly, J.W.; Wolfe, R.; Dobrovic, A.; McLean, C.; McArthur, G.A. Clinical and pathological associations of the activating RAC1 P29S mutation in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2014, 27, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1(P29S) induces a mesenchymal phenotypic switch via serum response factor to promote melanoma development and therapy resistance. Cancer Cell 2019, 36, 68–83.e9. [Google Scholar] [CrossRef] [PubMed]
- Fiegen, D.; Haeusler, L.C.; Blumenstein, L.; Herbrand, U.; Dvorsky, R.; Vetter, I.R.; Ahmadian, M.R. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 2004, 279, 4743–4749. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Collard, J.G.; Jordan, P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J. Biol. Chem. 2003, 278, 50442–50448. [Google Scholar] [CrossRef]
- Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380. [Google Scholar] [CrossRef]
- Nimnual, A.S.; Taylor, L.J.; Nyako, M.; Jeng, H.-H.; Bar-Sagi, D. Perturbation of cytoskeleton dynamics by the opposing effects of Rac1 and Rac1b. Small GTPases 2010, 1, 89–97. [Google Scholar] [CrossRef]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef]
- Gudiño, V.; Pohl, S.Ö.; Billard, C.V.; Cammareri, P.; Bolado, A.; Aitken, S.; Stevenson, D.; Hall, A.E.; Agostino, M.; Cassidy, J. RAC1B modulates intestinal tumourigenesis via modulation of WNT and EGFR signalling pathways. Nat. Commun. 2021, 12, 2335. [Google Scholar] [CrossRef]
- Ungefroren, H.; Kumarasinghe, A.; Musfeldt, M.; Fiedler, C.; Lehnert, H.; Marquardt, J.U. RAC1B induces SMAD7 via USP26 to suppress TGFβ1-dependent cell migration in mesenchymal-subtype carcinoma cells. Cancers 2020, 12, 1545. [Google Scholar] [CrossRef]
- Ungefroren, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.U. The small GTPase RAC1B: A potent negative regulator of-and useful tool to study-TGFβ signaling. Cancers 2020, 12, 3475. [Google Scholar] [CrossRef] [PubMed]
- Witte, D.; Otterbein, H.; Förster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Schaefer, A.; Howard, S.V.; Der, C.J. RAS and RHO family GTPase mutations in cancer: Twin sons of different mothers? Crit. Rev. Biochem. Mol. Biol. 2020, 55, 386–407. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Alberts, A.S.; Treisman, R. RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO J. 1998, 17, 1350–1361. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RHO Subfamily | Member | Catalytic Type | Kinetics and Enzymatic Activities |
|---|---|---|---|
| RAC | RAC1 | Classical | Classical RHO GTPases have intact enzymatic activities and can cycle between GTP-bound and GDP-bound conformations |
| RAC2 | |||
| RAC3 | |||
| RHOG | |||
| RHO | RHOA | ||
| RHOB | |||
| RHOC | |||
| CDC42 | CDC42 | ||
| RHOJ (TCL) | |||
| RHOQ (TC10) | |||
| RND | RND1 | GTPase deficient | GTPase deficient RHO GTPases cannot hydrolyze GTP |
| RND2 | |||
| RND3/RHOE | |||
| RHOH | RHOH | ||
| RHOBTB | RHOBTB1 | ||
| RHOBTB2/DBC | |||
| RHOD/F | RHOD | Fast-cycling | Fast-cycling RHO GTPases can freely cycle between GTP-bound and GDP-bound conformations without the involvement of RHOGEFs |
| RHOF | |||
| RHOU/V | RHOU(WRCH1) | ||
| RHOV(CHP) | Fast-cycling (?) | RHOV has not been confirmed to function as a fast-cycling RHO GTPase; however, its similarity to RHOU suggests that it is. |
| Mutant Protein | Catalytic Type | Cancer Type | Ref |
|---|---|---|---|
| RAC/P29S | Fast-cycling | Sun-exposed melanoma | [5] |
| RAC1/F28L | Fast-cycling | Laboratory-generated mutant | [55] |
| RAC1B | Fast-cycling | Colorectal cancer | [51,56] |
| Breast cancer | [52] | ||
| Lung cancer | [57,58] | ||
| Pancreatic cancer | [59] | ||
| Thyroid cancer | [60] | ||
| RAC1/N92I | Fast-cycling | Fibrosarcoma cell-line HT1080 | [61] |
| RAC1/C157Y | Fast-cycling | Lung cancer | [61] |
| RHOA/G17V | Decreased affinity for guanosine nucleotides | Angioimmunoblastic T-cell-lymphoma | [62,63] |
| Adult T-cell leukemia/lymphoma (ATLL) | [64] | ||
| Peripheral T-cell lymphoma | [65] | ||
| Diffuse-type gastric carcinoma | [66] | ||
| RHOA/C16R | Fast-cycling | ATLL | [64] |
| RHOA/A161P | Fast-cycling | ATLL | [64] |
| CDC42/P34Q | Effector-binding? | Well-differentiated papillary mesothelioma | [67] |
| CDC42/Q61R | GTPase-deficient? | Well-differentiated papillary mesothelioma | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspenström, P. The Role of Fast-Cycling Atypical RHO GTPases in Cancer. Cancers 2022, 14, 1961. https://doi.org/10.3390/cancers14081961
Aspenström P. The Role of Fast-Cycling Atypical RHO GTPases in Cancer. Cancers. 2022; 14(8):1961. https://doi.org/10.3390/cancers14081961
Chicago/Turabian StyleAspenström, Pontus. 2022. "The Role of Fast-Cycling Atypical RHO GTPases in Cancer" Cancers 14, no. 8: 1961. https://doi.org/10.3390/cancers14081961
APA StyleAspenström, P. (2022). The Role of Fast-Cycling Atypical RHO GTPases in Cancer. Cancers, 14(8), 1961. https://doi.org/10.3390/cancers14081961