
The Bone Marrow Microenvironment in B-Cell Development and Malignancy

 , and
, and 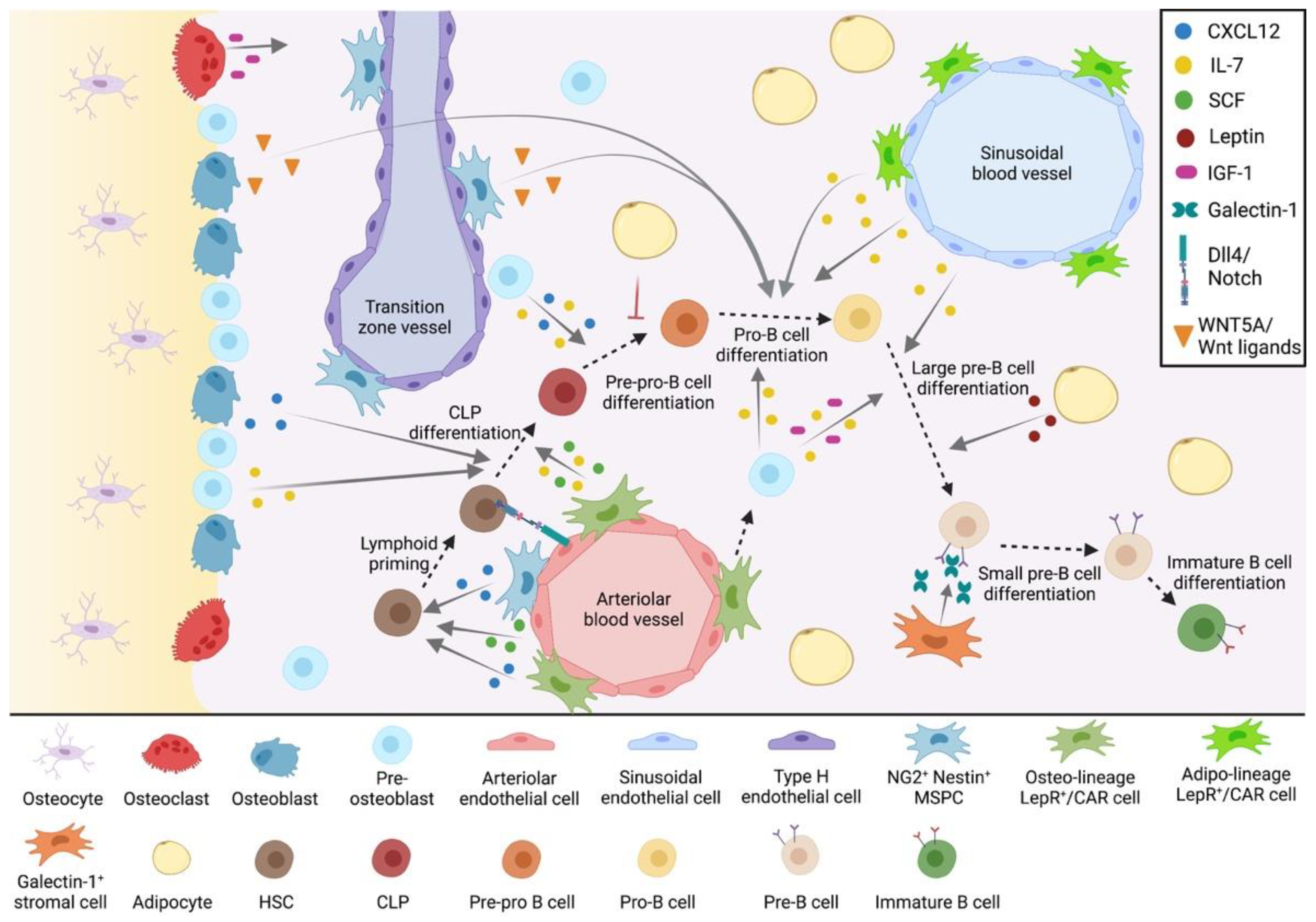{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. The Bone Marrow Microenvironment in B-Cell Development
2.1. The Endosteal B-Cell Niche
2.1.1. Osteoblasts
2.1.2. Osteocytes
2.1.3. Osteoclasts
2.2. The Central B-Cell Niche
2.2.1. Perivascular MSPCs
2.2.2. Galectin-1+ Stromal Cells
2.2.3. Endothelial Cells
2.2.4. Adipocytes
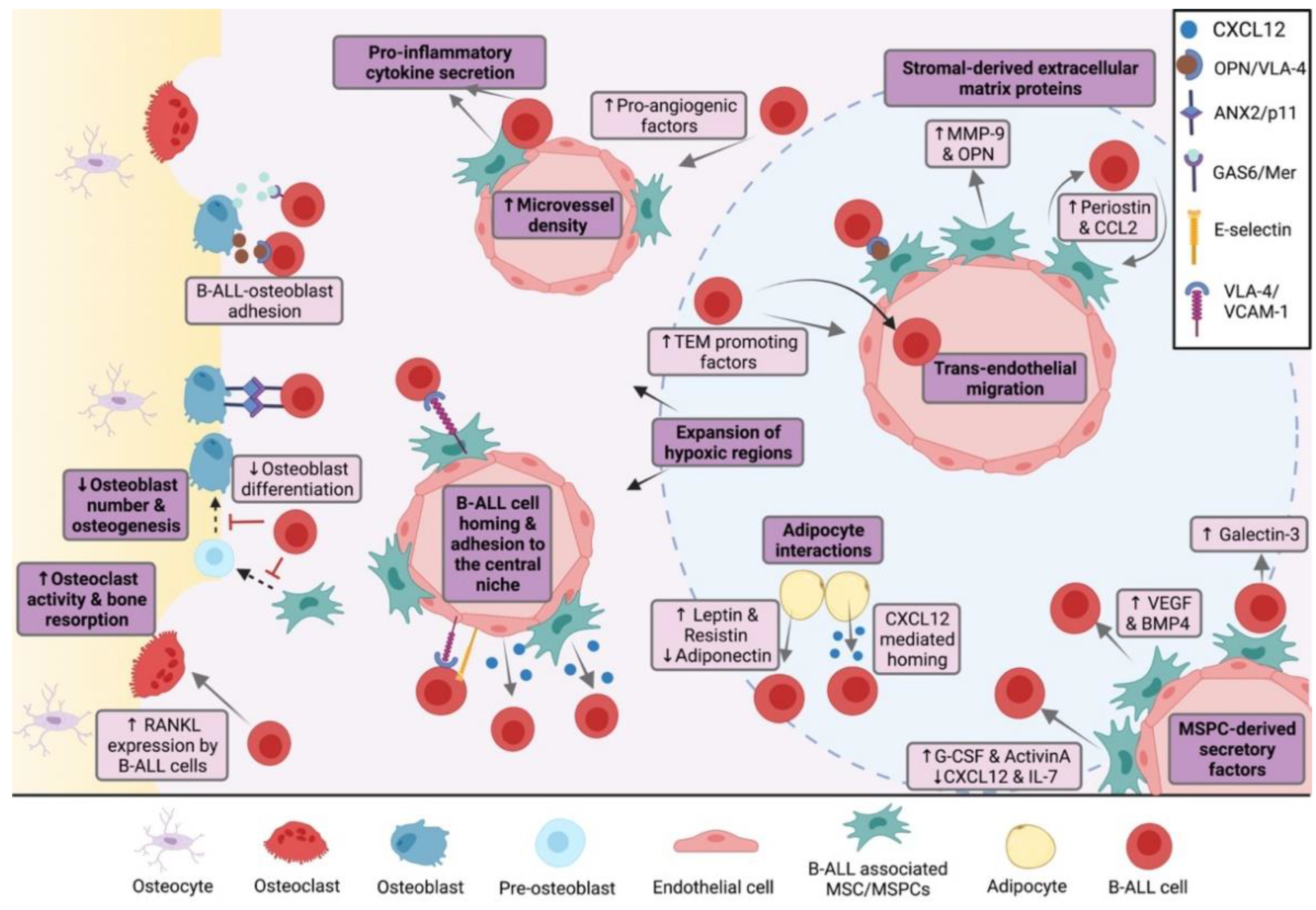
3. The Bone Marrow Microenvironment in B-Cell Malignancy
3.1. The Endosteal Niche in B-ALL
3.1.1. Osteoblasts
3.1.2. Osteoclasts
3.1.3. Endosteal Transition Zone Vessels
3.2. The Central Niche in B-ALL
3.2.1. CXCR4/CXCL12 and VLA-4/5-Mediated Mechanisms and Signaling Pathways
3.2.2. MSPC-Derived Secretory Factors
3.2.3. Stromal-Derived Extracellular Matrix Proteins
3.2.4. Pro-Inflammatory Cytokines
3.2.5. Hypoxia and Hypoxia-Related Mechanisms
3.2.6. Tunneling Nanotubes and Extracellular Vesicles
3.2.7. Endothelial Cells
3.2.8. Adipocytes
4. Emerging Techniques and Technologies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, J.; Mansson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.-J.; Thoren, L.A.; et al. Identification of Flt3+ Lympho-Myeloid Stem Cells Lacking Erythro-Megakaryocytic Potential: A Revised Road Map for Adult Blood Lineage Commitment. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Weissman, I.L.; Akashi, K. Identification of Clonogenic Common Lymphoid Progenitors in Mouse Bone Marrow. Cell 1997, 91, 661–672. [Google Scholar] [CrossRef]
- Mansson, R.; Zandi, S.; Anderson, K.; Martensson, I.-L.; Jacobsen, S.E.W.; Bryder, D.; Sigvardsson, M. B-lineage commitment prior to surface expression of B220 and CD19 on hematopoietic progenitor cells. Blood 2008, 112, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Inlay, M.A.; Bhattacharya, D.; Sahoo, D.; Serwold, T.; Seita, J.; Karsunky, H.; Plevritis, S.K.; Dill, D.L.; Weissman, I.L. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes Dev. 2009, 23, 2376–2381. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Carmack, C.E.; Shinton, S.A.; Kemp, J.D.; Hayakawa, K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991, 173, 1213–1225. [Google Scholar] [CrossRef]
- McLean, K.C.; Mandal, M. It Takes Three Receptors to Raise a B Cell. Trends Immunol. 2020, 41, 629–642. [Google Scholar] [CrossRef]
- Green, A.C.; Rudolph-Stringer, V.; Chantry, A.D.; Wu, J.Y.; Purton, L.E. Mesenchymal lineage cells and their importance in B lymphocyte niches. Bone 2019, 119, 42–56. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Lo Celso, C. The interplay of leukemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Tasdogan, A.; Ubellacker, J.M.; Zhang, J.; Nosyreva, E.D.; Du, L.; Murphy, M.M.; Hu, S.; Yi, Y.; Kara, N.; et al. A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature 2021, 591, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.; Osmond, D.G. Microenvironmental organization and stromal cell associations of B lymphocyte precursor cells in mouse bone marrow. Eur. J. Immunol. 1990, 20, 2395–2404. [Google Scholar] [CrossRef] [PubMed]
- Hermans, M.H.; Hartsuiker, H.; Opstelten, D. An in situ study of B-lymphocytopoiesis in rat bone marrow. Topographical arrangement of terminal deoxynucleotidyl transferase-positive cells and pre-B cells. J. Immunol. 1989, 142, 67–73. [Google Scholar] [PubMed]
- Cordeiro Gomes, A.; Hara, T.; Lim, V.Y.; Herndler-Brandstetter, D.; Nevius, E.; Sugiyama, T.; Tani-Ichi, S.; Schlenner, S.; Richie, E.; Rodewald, H.-R.; et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016, 45, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Garrett, R.; Jung, Y.; Zhang, Y.; Kim, N.; Wang, J.; Joe, G.J.; Hexner, E.; Choi, Y.; Taichman, R.S.; et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 2007, 109, 3706–3712. [Google Scholar] [CrossRef] [PubMed]
- Visnjic, D.; Kalajzic, Z.; Rowe, D.W.; Katavic, V.; Lorenzo, J.; Aguila, H.L. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004, 103, 3258–3264. [Google Scholar] [CrossRef]
- Terashima, A.; Okamoto, K.; Nakashima, T.; Akira, S.; Ikuta, K.; Takayanagi, H. Sepsis-Induced Osteoblast Ablation Causes Immunodeficiency. Immunity 2016, 44, 1434–1443. [Google Scholar] [CrossRef]
- Yu, V.W.; Lymperi, S.; Oki, T.; Jones, A.; Swiatek, P.; Vasic, R.; Ferraro, F.; Scadden, D.T. Distinctive Mesenchymal-Parenchymal Cell Pairings Govern B Cell Differentiation in the Bone Marrow. Stem Cell Rep. 2016, 7, 220–235. [Google Scholar] [CrossRef]
- Ponomaryov, T.; Peled, A.; Petit, I.; Taichman, R.S.; Habler, L.; Sandbank, J.; Arenzana-Seisdedos, F.; Magerus, A.; Caruz, A.; Fujii, N.; et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J. Clin. Investig. 2000, 106, 1331–1339. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, A.; Hsu, Y.-M.S.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.R.; Mandal, M.; Ochiai, K.; Singh, H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 2014, 14, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Fitter, S.; El Khawanky, N.; Grose, R.H.; Walkley, C.R.; Purton, L.E.; Ruegg, M.A.; Hall, M.N.; Gronthos, S.; Zannettino, A.C.W. mTORC1 plays an important role in osteoblastic regulation of B-lymphopoiesis. Sci. Rep. 2018, 8, 14501. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, M.; Tao, C.; Chen, J.; Wang, Z.; Yang, J.; Chen, Z.; Zou, Z.; Liu, A.; Cai, D.; et al. Inactivation of mTORC1 Signaling in Osterix-Expressing Cells Impairs B-cell Differentiation. J. Bone Miner. Res. 2018, 33, 732–742. [Google Scholar] [CrossRef]
- Wu, J.Y.; Purton, L.E.; Rodda, S.J.; Chen, M.; Weinstein, L.S.; McMahon, A.P.; Scadden, D.T.; Kronenberg, H.M. Osteoblastic regulation of B lymphopoiesis is mediated by Gsα-dependent signaling pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 16976–16981. [Google Scholar] [CrossRef]
- Panaroni, C.; Fulzele, K.; Saini, V.; Chubb, R.; Pajevic, P.D.; Wu, J.Y. PTH Signaling in Osteoprogenitors Is Essential for B-Lymphocyte Differentiation and Mobilization. J. Bone Miner. Res. 2015, 30, 2273–2286. [Google Scholar] [CrossRef]
- Taguchi, T.; Takenouchi, H.; Matsui, J.; Tang, W.-R.; Itagaki, M.; Shiozawa, Y.; Suzuki, K.; Sakaguchi, S.; Ktagiri, Y.U.; Takahashi, T.; et al. Involvement of insulin-like growth factor-I and insulin-like growth factor binding proteins in pro-B-cell development. Exp. Hematol. 2006, 34, 508–518. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Domínguez, Á.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Nemeth, M.J.; Topol, L.; Anderson, S.M.; Yang, Y.; Bodine, D.M. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proc. Natl. Acad. Sci. USA 2007, 104, 15436–15441. [Google Scholar] [CrossRef]
- Cao, J.; Zhang, L.; Wan, Y.; Li, H.; Zhou, R.; Ding, H.; Liu, Y.; Yao, Z.; Guo, X. Ablation of Wntless in endosteal niches impairs lymphopoiesis rather than HSCs maintenance. Eur. J. Immunol. 2015, 45, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Lu, C.; Cao, J.; Zhou, R.; Yao, Y.; Yu, J.; Zhang, L.; Zhao, H.; Li, H.; Zhao, J.; et al. Osteoblastic Wnts differentially regulate bone remodeling and the maintenance of bone marrow mesenchymal stem cells. Bone 2013, 55, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted Ablation of Osteocytes Induces Osteoporosis with Defective Mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, G.; Roberts, S.J.; Ke, H.Z. Novel actions of sclerostin on bone. J. Mol. Endocrinol. 2019, 62, R167–R185. [Google Scholar] [CrossRef]
- Li, X.; Ominsky, M.S.; Niu, Q.-T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted Deletion of the Sclerostin Gene in Mice Results in Increased Bone Formation and Bone Strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Cain, C.J.; Rueda, R.; McLelland, B.; Collette, N.M.; Loots, G.G.; Manilay, J.O. Absence of sclerostin adversely affects B-cell survival. J. Bone Miner. Res. 2012, 27, 1451–1461. [Google Scholar] [CrossRef]
- Sato, M.; Asada, N.; Kawano, Y.; Wakahashi, K.; Minagawa, K.; Kawano, H.; Sada, A.; Ikeda, K.; Matsui, T.; Katayama, Y. Osteocytes Regulate Primary Lymphoid Organs and Fat Metabolism. Cell Metab. 2013, 18, 749–758. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef]
- Kong, Y.-Y.; Yoshida, H.; Sarosi, I.; Tan, H.-L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Blin-Wakkach, C.; Wakkach, A.; Sexton, P.M.; Rochet, N.; Carle, G.F. Hematological defects in the oc/oc mouse, a model of infantile malignant osteopetrosis. Leukemia 2004, 18, 1505–1511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mansour, A.; Anginot, A.; Mancini, S.J.C.; Schiff, C.; Carle, G.F.; Wakkach, A.; Blin-Wakkach, C. Osteoclast activity modulates B-cell development in the bone marrow. Cell Res. 2011, 21, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef]
- Morikawa, S.; Mabuchi, Y.; Kubota, Y.; Nagai, Y.; Niibe, K.; Hiratsu, E.; Suzuki, S.; Miyauchi-Hara, C.; Nagoshi, N.; Sunabori, T.; et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med. 2009, 206, 2483–2496. [Google Scholar] [CrossRef]
- Mourcin, F.; Breton, C.; Tellier, J.; Narang, P.; Chasson, L.; Jorquera, A.; Coles, M.; Schiff, C.; Mancini, S.J.C. Galectin-1-expressing stromal cells constitute a specific niche for pre-BII cell development in mouse bone marrow. Blood 2011, 117, 6552–6561. [Google Scholar] [CrossRef]
- Cuminetti, V.; Arranz, L. Bone Marrow Adipocytes: The Enigmatic Components of the Hematopoietic Stem Cell Niche. J. Clin. Med. 2019, 8, 707. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-Renewing Osteoprogenitors in Bone Marrow Sinusoids Can Organize a Hematopoietic Microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef]
- Chan, C.K.F.; Chen, C.-C.; Luppen, C.A.; Kim, J.-B.; DeBoer, A.T.; Wei, K.; Helms, J.A.; Kuo, C.J.; Kraft, D.L.; Weissman, I.L. Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature 2009, 457, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Tokoyoda, K.; Egawa, T.; Sugiyama, T.; Choi, B.-I.; Nagasawa, T. Cellular Niches Controlling B Lymphocyte Behavior within Bone Marrow during Development. Immunity 2004, 20, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Balzano, M.; De Grandis, M.; Vu Manh, T.-P.; Chasson, L.; Bardin, F.; Farina, A.; Sergé, A.; Bidaut, G.; Charbord, P.; Hérault, L.; et al. Nidogen-1 Contributes to the Interaction Network Involved in Pro-B Cell Retention in the Peri-sinusoidal Hematopoietic Stem Cell Niche. Cell Rep. 2019, 26, 3257–3271.e8. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Okoreeh, M.K.; Kennedy, D.E.; Maienschein-Cline, M.; Ai, J.; McLean, K.C.; Kaverina, N.; Veselits, M.; Aifantis, I.; Gounari, F.; et al. CXCR4 signaling directs Igk recombination and the molecular mechanisms of late B lymphopoiesis. Nat. Immunol. 2019, 20, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The Essential Functions of Adipo-osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.O.; Yue, R.; Murphy, M.M.; Peyer, J.G.; Morrison, S.J. Leptin-Receptor-Expressing Mesenchymal Stromal Cells Represent the Main Source of Bone Formed by Adult Bone Marrow. Cell Stem Cell 2014, 15, 154–168. [Google Scholar] [CrossRef]
- Cheung, L.C.; Strickland, D.H.; Howlett, M.; Ford, J.; Charles, A.K.; Lyons, K.M.; Brigstock, D.R.; Goldschmeding, R.; Cole, C.H.; Alexander, W.S.; et al. Connective tissue growth factor is expressed in bone marrow stromal cells and promotes interleukin-7-dependent B lymphopoiesis. Haematologica 2014, 99, 1149–1156. [Google Scholar] [CrossRef]
- Comazzetto, S.; Murphy, M.M.; Berto, S.; Jeffery, E.; Zhao, Z.; Morrison, S.J. Restricted Hematopoietic Progenitors and Erythropoiesis Require SCF from Leptin Receptor+ Niche Cells in the Bone Marrow. Cell Stem Cell 2019, 24, 477–486.e6. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e16. [Google Scholar] [CrossRef]
- Baccin, C.; Al-Sabah, J.; Velten, L.; Helbling, P.M.; Grünschläger, F.; Hernández-Malmierca, P.; Nombela-Arrieta, C.; Steinmetz, L.M.; Trumpp, A.; Haas, S. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 2020, 22, 38–48. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Itkin, T.; Mäe, M.A.; Langen, U.H.; Betsholtz, C.; Lapidot, T.; Adams, R.H. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016, 532, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Marchand, T.; Yang, E.; Wei, Q.; Nerlov, C.; Frenette, P.S. Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev. Cell 2018, 44, 634–641.e4. [Google Scholar] [CrossRef] [PubMed]
- Espeli, M.; Mancini, S.J.C.; Breton, C.; Poirier, F.; Schiff, C. Impaired B-cell development at the pre-BII-cell stage in galectin-1-deficient mice due to inefficient pre-BII/stromal cell interactions. Blood 2009, 113, 5878–5886. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hendriks, M.; Chatzis, A.; Ramasamy, S.K.; Kusumbe, A.P. Bone Vasculature and Bone Marrow Vascular Niches in Health and Disease. J. Bone Miner. Res. 2020, 35, 2103–2120. [Google Scholar] [CrossRef]
- Ramasamy, S.K. Structure and Functions of Blood Vessels and Vascular Niches in Bone. Stem Cells Int. 2017, 2017, 5046953. [Google Scholar] [CrossRef]
- Dar, A.; Goichberg, P.; Shinder, V.; Kalinkovich, A.; Kollet, O.; Netzer, N.; Margalit, R.; Zsak, M.; Nagler, A.; Hardan, I.; et al. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat. Immunol. 2005, 6, 1038–1046. [Google Scholar] [CrossRef]
- Xu, C.; Gao, X.; Wei, Q.; Nakahara, F.; Zimmerman, S.E.; Mar, J.; Frenette, P.S. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat. Commun. 2018, 9, 2449. [Google Scholar] [CrossRef]
- Fiuza, U.-M.; Arias, A.M. Cell and molecular biology of Notch. J. Endocrinol. 2007, 194, 459–474. [Google Scholar] [CrossRef]
- Lecka-Czernik, B.; Baroi, S.; Stechschulte, L.A.; Chougule, A.S. Marrow Fat—A New Target to Treat Bone Diseases? Curr. Osteoporos. Rep. 2018, 16, 123–129. [Google Scholar] [CrossRef]
- Żelechowska, P.; Brzezińska-Błaszczyk, E.; Kusowska, A.; Kozłowska, E. The role of adipokines in the modulation of lymphoid lineage cell development and activity: An overview. Obes. Rev. 2020, 21, e13055. [Google Scholar] [CrossRef]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.J.; Green, D.E.; Pagnotti, G.M.; Chan, M.E.; Rubin, C.T. High Fat Diet Rapidly Suppresses B Lymphopoiesis by Disrupting the Supportive Capacity of the Bone Marrow Niche. PLoS ONE 2014, 9, e90639. [Google Scholar] [CrossRef]
- Bilwani, F.A.; Knight, K.L. Adipocyte-Derived Soluble Factor(s) Inhibits Early Stages of B Lymphopoiesis. J. Immunol. 2012, 189, 4379–4386. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.E.; Knight, K.L. Inflammatory Changes in Bone Marrow Microenvironment Associated with Declining B Lymphopoiesis. J. Immunol. 2017, 198, 3471–3479. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.E.; Knight, K.L. Inhibition of B Lymphopoiesis by Adipocytes and IL-1-Producing Myeloid-Derived Suppressor Cells. J. Immunol. 2015, 195, 2666–2674. [Google Scholar] [CrossRef]
- Bennett, B.D.; Solar, G.P.; Yuan, J.Q.; Mathias, J.; Thomas, G.R.; Matthews, W. A role for leptin and its cognate receptor in hematopoiesis. Curr. Biol. 1996, 6, 1170–1180. [Google Scholar] [CrossRef]
- Claycombe, K.; King, L.E.; Fraker, P.J. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 2017–2021. [Google Scholar] [CrossRef]
- Malouf, C.; Ottersbach, K. Molecular processes involved in B cell acute lymphoblastic leukaemia. Cell. Mol. Life Sci. 2018, 75, 417–446. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Roberts, K.G.; Morin, R.D.; Zhang, J.; Hirst, M.; Zhao, Y.; Su, X.; Chen, S.-C.; Payne-Turner, D.; Churchman, M.L.; Harvey, R.C.; et al. Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 153–166. [Google Scholar] [CrossRef]
- Okuyama, K.; Strid, T.; Kuruvilla, J.; Somasundaram, R.; Cristobal, S.; Smith, E.; Prasad, M.; Fioretos, T.; Lilljebjörn, H.; Soneji, S.; et al. PAX5 is part of a functional transcription factor network targeted in lymphoid leukemia. PLoS Genet. 2019, 15, e1008280. [Google Scholar] [CrossRef] [PubMed]
- Dander, E.; Palmi, C.; D’amico, G.; Cazzaniga, G. The Bone Marrow Niche in B-Cell Acute Lymphoblastic Leukemia: The Role of Microenvironment from Pre-Leukemia to Overt Leukemia. Int. J. Mol. Sci. 2021, 22, 4426. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.G.P. Niche contributions to oncogenesis: Emerging concepts and implications for the hematopoietic system. Haematologica. 2011, 96, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Kokkaliaris, K.D.; Scadden, D.T. Cell interactions in the bone marrow microenvironment affecting myeloid malignancies. Blood Adv. 2020, 4, 3795–3803. [Google Scholar] [CrossRef]
- Halton, J.M.; Atkinson, S.A.; Fraher, L.; Webber, C.; Gill, G.J.; Dawson, S.; Barr, R.D. Altered mineral metabolism and bone mass in children during treatment for acute lymphoblastic leukemia. J. Bone Miner. Res. 1996, 11, 1774–1783. [Google Scholar] [CrossRef]
- Leeuw, J.A.; Koudstaal, J.; Wiersema-Buist, J.; Kamps, W.A.; Timens, W. Bone Histomorphometry in Children with Newly Diagnosed Acute Lymphoblastic Leukemia. Pediatr. Res. 2003, 54, 814–818. [Google Scholar] [CrossRef][Green Version]
- Davies, J.H.; Evans, B.A.J.; Jenney, M.E.M.; Gregory, J.W. Effects of Chemotherapeutic Agents on the Function of Primary Human Osteoblast-Like Cells Derived from Children. J. Clin. Endocrinol. Metab. 2003, 88, 6088–6097. [Google Scholar] [CrossRef][Green Version]
- Kroschwald, L.M.; Tauer, J.T.; Kroschwald, S.I.; Suttorp, M.; Wiedenfeld, A.; Beissert, S.; Bauer, A.; Rauner, M. Imatinib mesylate and nilotinib decrease synthesis of bone matrix in vitro. Oncol. Lett. 2019, 18, 2102–2108. [Google Scholar] [CrossRef]
- Nguyen, T.-V.; Melville, A.; Nath, S.; Story, C.; Howell, S.; Sutton, R.; Zannettino, A.; Revesz, T. Bone Marrow Recovery by Morphometry during Induction Chemotherapy for Acute Lymphoblastic Leukemia in Children. PLoS ONE 2015, 10, e0126233. [Google Scholar] [CrossRef]
- Le, P.M.; Andreeff, M.; Battula, V.L. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 2018, 103, 1945–1955. [Google Scholar] [CrossRef]
- Cheung, L.C.; Tickner, J.; Hughes, A.M.; Skut, P.; Howlett, M.; Foley, B.; Oommen, J.; Wells, J.E.; He, B.; Singh, S.; et al. New therapeutic opportunities from dissecting the pre-B leukemia bone marrow microenvironment. Leukemia 2018, 32, 2326–2338. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-C.; Xu, Y.-H.; Chen, H.-X.; Wang, X.-J. Acute Lymphoblastic Leukemia Cells Inhibit the Differentiation of Bone Mesenchymal Stem Cells into Osteoblasts In Vitro by Activating Notch Signaling. Stem Cells Int. 2015, 2015, 162410. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Zafrir, M.; Yesilkanal, A.E.; Price, T.T.; Hyjek, E.M.; Sipkins, D.A. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013, 121, 4821–4831. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Witkowski, M.T.; Harris, J.; Dolgalev, I.; Sreeram, S.; Qian, W.; Tong, J.; Chen, X.; Aifantis, I.; Chen, W. Leukemia-on-a-chip: Dissecting the chemoresistance mechanisms in B cell acute lymphoblastic leukemia bone marrow niche. Sci. Adv. 2020, 6, eaba5536. [Google Scholar] [CrossRef]
- Gopalakrishnapillai, A.; Kolb, E.A.; Dhanan, P.; Mason, R.W.; Napper, A.; Barwe, S.P. Disruption of Annexin II /p11 Interaction Suppresses Leukemia Cell Binding, Homing and Engraftment, and Sensitizes the Leukemia Cells to Chemotherapy. PLoS ONE 2015, 10, e0140564. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Taichman, R.S. GAS6/Mer axis regulates the homing and survival of the E2A/PBX1-positive B-cell precursor acute lymphoblastic leukemia in the bone marrow niche. Exp. Hematol. 2010, 38, 132–140. [Google Scholar] [CrossRef]
- Moses, B.S.; Slone, W.L.; Thomas, P.; Evans, R.; Piktel, D.; Angel, P.M.; Walsh, C.M.; Cantrell, P.S.; Rellick, S.L.; Martin, K.H.; et al. Bone marrow microenvironment modulation of acute lymphoblastic leukemia phenotype. Exp. Hematol. 2016, 44, 50–59.e2. [Google Scholar] [CrossRef]
- Moses, B.S.; Evans, R.; Slone, W.L.; Piktel, D.; Martinez, I.; Craig, M.D.; Gibson, L.F. Bone Marrow Microenvironment Niche Regulates miR-221/222 in Acute Lymphoblastic Leukemia. Mol. Cancer Res. 2016, 14, 909–919. [Google Scholar] [CrossRef]
- Slone, W.L.; Moses, B.S.; Hare, I.; Evans, R.; Piktel, D.; Gibson, L.F. BCL6 modulation of acute lymphoblastic leukemia response to chemotherapy. Oncotarget 2016, 7, 23439–23453. [Google Scholar] [CrossRef]
- Rajakumar, S.A.; Papp, E.; Lee, K.K.; Grandal, I.; Merico, D.; Liu, C.C.; Allo, B.; Zhang, L.; Grynpas, M.D.; Minden, M.D.; et al. B cell acute lymphoblastic leukemia cells mediate RANK-RANKL-dependent bone destruction. Sci. Transl. Med. 2020, 12, eaba5942. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Blair, H.J.; Elder, A.; Dormon, K.; Rennie, K.J.; Coleman, D.J.L.; Weiland, J.; Rankin, K.S.; Filby, A.; Heidenreich, O.; et al. Long-term in vitro maintenance of clonal abundance and leukaemia-initiating potential in acute lymphoblastic leukaemia. Leukemia 2016, 30, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Manabe, A.; Coustan-Smith, E.; Behm, F.G.; Raimondi, S.C.; Campana, D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood 1992, 79, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, M.; Manabe, A.; Pui, C.H.; Behm, F.G.; Raimondi, S.C.; Hancock, M.L.; Mahmoud, H.; Crist, W.M.; Campana, D. Stroma-supported culture in childhood B-lineage acute lymphoblastic leukemia cells predicts treatment outcome. J. Clin. Investig. 1996, 97, 755–760. [Google Scholar] [CrossRef] [PubMed]
- van den Berk, L.C.J.; van der Veer, A.; Willemse, M.E.; Theeuwes, M.J.G.A.; Luijendijk, M.W.; Tong, W.H.; van der Sluis, I.M.; Pieters, R.; den Boer, M.L. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2014, 166, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Balandrán, J.C.; Purizaca, J.; Enciso, J.; Dozal, D.; Sandoval, A.; Jiménez-Hernández, E.; Alemán-Lazarini, L.; Perez-Koldenkova, V.; Quintela-Nunez Del Prado, H.; Ríos de Los Ríos, J.; et al. Pro-inflammatory-Related Loss of CXCL12 Niche Promotes Acute Lymphoblastic Leukemic Progression at the Expense of Normal Lymphopoiesis. Front. Immunol. 2017, 7, 666. [Google Scholar] [CrossRef] [PubMed]
- Konoplev, S.; Jorgensen, J.L.; Thomas, D.A.; Lin, E.; Burger, J.; Kantarjian, H.M.; Andreeff, M.; Medeiros, L.J.; Konopleva, M. Phosphorylated CXCR4 is associated with poor survival in adults with B-acute lymphoblastic leukemia. Cancer 2011, 117, 4689–4695. [Google Scholar] [CrossRef]
- Nishii, K.; Katayama, N.; Miwa, H.; Shikami, M.; Masuya, M.; Shiku, H.; Kita, K. Survival of human leukaemic B-cell precursors is supported by stromal cells and cytokines: Association with the expression of bcl-2 protein. Br. J. Haematol. 1999, 105, 701–710. [Google Scholar] [CrossRef]
- Juarez, J.; Bradstock, K.F.; Gottlieb, D.J.; Bendall, L.J. Effects of inhibitors of the chemokine receptor CXCR4 on acute lymphoblastic leukemia cells in vitro. Leukemia 2003, 17, 1294–1300. [Google Scholar] [CrossRef]
- Juarez, J.; Baraz, R.; Gaundar, S.; Bradstock, K.; Bendall, L. Interaction of interleukin-7 and interleukin-3 with the CXCL12-induced proliferation of B-cell progenitor acute lymphoblastic leukemia. Haematologica 2007, 92, 450–459. [Google Scholar] [CrossRef]
- Juárez, J.; Dela Pena, A.; Baraz, R.; Hewson, J.; Khoo, M.; Cisterne, A.; Fricker, S.; Fujii, N.; Bradstock, K.F.; Bendall, L.J. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia 2007, 21, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Sison, E.A.R.; Magoon, D.; Li, L.; Annesley, C.E.; Rau, R.E.; Small, D.; Brown, P. Plerixafor as a chemosensitizing agent in pediatric acute lymphoblastic leukemia: Efficacy and potential mechanisms of resistance to CXCR4 inhibition. Oncotarget 2014, 5, 8947–8958. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Vanegas, N.-D.P.; Vernot, J.P. Acute Leukemia Induces Senescence and Impaired Osteogenic Differentiation in Mesenchymal Stem Cells Endowing Leukemic Cells with Functional Advantages. Stem Cells Int. 2019, 2019, 3864948. [Google Scholar] [CrossRef]
- Shalapour, S.; Hof, J.; Kirschner-Schwabe, R.; Bastian, L.; Eckert, C.; Prada, J.; Henze, G.; von Stackelberg, A.; Seeger, K. High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica 2011, 96, 1627–1635. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, D.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef]
- Hu, Z.; Slayton, W.B. Integrin VLA-5 and FAK are Good Targets to Improve Treatment Response in the Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia. Front. Oncol. 2014, 4, 112. [Google Scholar] [CrossRef]
- Hou, J.; Yan, D.; Liu, Y.; Huang, P.; Cui, H. The Roles of Integrin α5β1 in Human Cancer. Onco. Targets Ther. 2020, 13, 13329–13344. [Google Scholar] [CrossRef]
- Fistonich, C.; Zehentmeier, S.; Bednarski, J.J.; Miao, R.; Schjerven, H.; Sleckman, B.P.; Pereira, J.P. Cell circuits between B cell progenitors and IL-7+ mesenchymal progenitor cells control B cell development. J. Exp. Med. 2018, 215, 2586–2599. [Google Scholar] [CrossRef] [PubMed]
- Portale, F.; Cricrì, G.; Bresolin, S.; Lupi, M.; Gaspari, S.; Silvestri, D.; Russo, B.; Marino, N.; Ubezio, P.; Pagni, F.; et al. ActivinA: A new leukemia-promoting factor conferring migratory advantage to B-cell precursor-acute lymphoblastic leukemic cells. Haematologica 2019, 104, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Wang, J.; Lu, T.; Ma, D.; Wei, D.; Guo, Y.; Cheng, B.; Wang, W.; Fang, Q. Overexpression of heme oxygenase-1 in microenvironment mediates vincristine resistance of B-cell acute lymphoblastic leukemia by promoting vascular endothelial growth factor secretion. J. Cell. Biochem. 2019, 120, 17791–17810. [Google Scholar] [CrossRef]
- Vincente López, Á.; Vázquez Garcia, M.N.; Melen, G.J.; Entrena Martínez, A.; Cubillo Moreno, I.; Garcia-Castro, J.; Orellana, M.R.; González, A.G.Z. Mesenchymal Stromal Cells Derived from the Bone Marrow of Acute Lymphoblastic Leukemia Patients Show Altered BMP4 Production: Correlations with the Course of Disease. PLoS ONE 2014, 9, e84496. [Google Scholar] [CrossRef] [PubMed]
- Voeltzel, T.; Flores-Violante, M.; Zylbersztejn, F.; Lefort, S.; Billandon, M.; Jeanpierre, S.; Joly, S.; Fossard, G.; Milenkov, M.; Mazurier, F.; et al. A new signaling cascade linking BMP4, BMPR1A, ΔNp73 and NANOG impacts on stem-like human cell properties and patient outcome. Cell Death Dis. 2018, 9, 1011. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mallampati, S.; Sun, B.; Zhang, J.; Kim, S.-B.; Lee, J.-S.; Gong, Y.; Cai, Z.; Sun, X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 2013, 333, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Gu, Y.; Lou, L.; Liu, L.; Hu, Y.; Wang, B.; Luo, Y.; Shi, J.; Yu, X.; Huang, H. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/β-catenin signaling pathway. J. Hematol. Oncol. 2015, 8, 1. [Google Scholar] [CrossRef]
- Fei, F.; Abdel-Azim, H.; Lim, M.; Arutyunyan, A.; von Itzstein, M.; Groffen, J.; Heisterkamp, N. Galectin-3 in pre-B acute lymphoblastic leukemia. Leukemia 2013, 27, 2385–2388. [Google Scholar] [CrossRef]
- Iwamoto, S.; Mihara, K.; Downing, J.R.; Pui, C.-H.; Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Investig. 2007, 117, 1049–1057. [Google Scholar] [CrossRef]
- Laranjeira, A.B.A.; de Vasconcellos, J.F.; Sodek, L.; Spago, M.C.; Fornazim, M.C.; Tone, L.G.; Brandalise, S.R.; Nowill, A.E.; Yunes, J.A. IGFBP7 participates in the reciprocal interaction between acute lymphoblastic leukemia and BM stromal cells and in leukemia resistance to asparaginase. Leukemia 2012, 26, 1001–1011. [Google Scholar] [CrossRef]
- Ma, Z.; Zhao, X.; Huang, J.; Jia, X.; Deng, M.; Cui, D.; Du, Z.; Fu, G.; Ouyang, G.; Xiao, C. A critical role of periostin in B-cell acute lymphoblastic leukemia. Leukemia 2017, 31, 1835–1837. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein with Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Ma, Z.; Zhao, X.; Deng, M.; Huang, Z.; Wang, J.; Wu, Y.; Cui, D.; Liu, Y.; Liu, R.; Ouyang, G. Bone Marrow Mesenchymal Stromal Cell-Derived Periostin Promotes B-ALL Progression by Modulating CCL2 in Leukemia Cells. Cell Rep. 2019, 26, 1533–1543.e4. [Google Scholar] [CrossRef]
- Verma, D.; Zanetti, C.; Godavarthy, P.S.; Kumar, R.; Minciacchi, V.R.; Pfeiffer, J.; Metzler, M.; Lefort, S.; Maguer-Satta, V.; Nicolini, F.E.; et al. Bone marrow niche-derived extracellular matrix-degrading enzymes influence the progression of B-cell acute lymphoblastic leukemia. Leukemia 2020, 34, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Figueroa, E.; Sánchez-Cuaxospa, M.; Martínez-Soto, K.A.; Sanchez-Zauco, N.; Medina-Sansón, A.; Jiménez-Hernández, E.; Torres-Nava, J.R.; Félix-Castro, J.M.; Gómez, A.; Ortega, E.; et al. Strong inflammatory response and Th1-polarization profile in children with acute lymphoblastic leukemia without apparent infection. Oncol. Rep. 2016, 35, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcellos, J.F.; Laranjeira, A.B.A.; Zanchin, N.I.T.; Otubo, R.; Vaz, T.H.; Cardoso, A.A.; Brandalise, S.R.; Yunes, J.A. Increased CCL2 and IL-8 in the bone marrow microenvironment in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2010, 56, 568–577. [Google Scholar] [CrossRef]
- Vilchis-Ordoñez, A.; Contreras-Quiroz, A.; Vadillo, E.; Dorantes-Acosta, E.; Reyes-López, A.; Quintela-Nunez del Prado, H.M.; Venegas-Vázquez, J.; Mayani, H.; Ortiz-Navarrete, V.; López-Martínez, B.; et al. Bone Marrow Cells in Acute Lymphoblastic Leukemia Create a Proinflammatory Microenvironment Influencing Normal Hematopoietic Differentiation Fates. BioMed Res. Int. 2015, 2015, 386165. [Google Scholar] [CrossRef]
- Isidro-Hernández, M.; Mayado, A.; Casado-García, A.; Martínez-Cano, J.; Palmi, C.; Fazio, G.; Orfao, A.; Ribera, J.; Ribera, J.M.; Zamora, L.; et al. Inhibition of inflammatory signaling in Pax5 mutant cells mitigates B-cell leukemogenesis. Sci. Rep. 2020, 10, 19189. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Cao, W.; Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef]
- Niedźwiecki, M.; Budziło, O.; Adamkiewicz-Drożyńska, E.; Pawlik-Gwozdecka, D.; Zieliński, M.; Maciejka-Kembłowska, L.; Szczepanski, T.; Trzonkowski, P. CD4+CD25highCD127low/-FoxP3+ Regulatory T-Cell Population in Acute Leukemias: A Review of the Literature. J. Immunol. Res. 2019, 2019, 2816498. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Benito, J.; Shi, Y.; Szymanska, B.; Carol, H.; Boehm, I.; Lu, H.; Konoplev, S.; Fang, W.; Zweidler-McKay, P.A.; Campana, D.; et al. Pronounced Hypoxia in Models of Murine and Human Leukemia: High Efficacy of Hypoxia-Activated Prodrug PR-104. PLoS ONE 2011, 6, e23108. [Google Scholar] [CrossRef] [PubMed]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol. Ther. 2012, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, B.; Polak, R.; Stalpers, F.; Pieters, R.; den Boer, M.L. Tunneling nanotubes facilitate autophagosome transfer in the leukemic niche. Leukemia 2017, 31, 1651–1654. [Google Scholar] [CrossRef]
- Johnson, S.M.; Dempsey, C.; Parker, C.; Mironov, A.; Bradley, H.; Saha, V. Acute lymphoblastic leukaemia cells produce large extracellular vesicles containing organelles and an active cytoskeleton. J. Extracell. Vesicles 2017, 6, 1294339. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Dempsey, C.; Chadwick, A.; Harrison, S.; Liu, J.; Di, Y.; McGinn, O.J.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P.; et al. Metabolic reprogramming of bone marrow stromal cells by leukemic extracellular vesicles in acute lymphoblastic leukemia. Blood 2016, 128, 453–456. [Google Scholar] [CrossRef]
- Schneider, P.; Dubus, I.; Gouel, F.; Legrand, E.; Vannier, J.P.; Vasse, M. What Role for Angiogenesis in Childhood Acute Lymphoblastic Leukaemia? Adv. Hematol. 2011, 2011, 27462. [Google Scholar] [CrossRef]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Côté, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef]
- Spertini, C.; Baïsse, B.; Bellone, M.; Gikic, M.; Smirnova, T.; Spertini, O. Acute Myeloid and Lymphoblastic Leukemia Cell Interactions with Endothelial Selectins: Critical Role of PSGL-1, CD44 and CD43. Cancers 2019, 11, 1253. [Google Scholar] [CrossRef]
- Veiga, J.P.; Costa, L.F.; Sallan, S.E.; Nadler, L.M.; Cardoso, A.A. Leukemia-stimulated bone marrow endothelium promotes leukemia cell survival. Exp. Hematol. 2006, 34, 610–621. [Google Scholar] [CrossRef]
- Perez-Atayde, A.R.; Sallan, S.E.; Tedrow, U.; Connors, S.; Allred, E.; Folkman, J. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am. J. Pathol. 1997, 150, 815–821. [Google Scholar] [PubMed]
- Pule, M.A.; Gullmann, C.; Dennis, D.; McMahon, C.; Jeffers, M.; Smith, O.P. Increased angiogenesis in bone marrow of children with acute lymphoblastic leukaemia has no prognostic significance. Br. J. Haematol. 2002, 118, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Ahsberg, J.; Xiao, P.; Okuyama, K.; Somasundaram, R.; Strid, T.; Qian, H.; Sigvardsson, M. Progression of progenitor B-cell leukemia is associated with alterations of the bone marrow micro-environment. Haematologica 2020, 105, e102–e106. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Vasse, M.; Sbaa-Ketata, E.; Lenormand, B.; Hong, L.; Soria, C.; Vannier, J.P. The growth of highly proliferative acute lymphoblastic leukemia may be independent of stroma and/or angiogenesis. Leukemia 2001, 15, 1143–1145. [Google Scholar] [CrossRef][Green Version]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Velázquez-Avila, M.; Balandrán, J.C.; Ramírez-Ramírez, D.; Velázquez-Avila, M.; Sandoval, A.; Felipe-López, A.; Nava, P.; Alvarado-Moreno, J.A.; Dozal, D.; Prieto-Chávez, J.L.; et al. High cortactin expression in B-cell acute lymphoblastic leukemia is associated with increased transendothelial migration and bone marrow relapse. Leukemia 2019, 33, 1337–1348. [Google Scholar] [CrossRef]
- Thompson, S.B.; Wigton, E.J.; Krovi, S.H.; Chung, J.W.; Long, R.A.; Jacobelli, J. The Formin mDia1 Regulates Acute Lymphoblastic Leukemia Engraftment, Migration, and Progression in vivo. Front. Oncol. 2018, 8, 389. [Google Scholar] [CrossRef]
- Münch, V.; Trentin, L.; Herzig, J.; Demir, S.; Seyfried, F.; Kraus, J.M.; Kestler, H.A.; Köhler, R.; Barth, T.F.E.; Te Kronnie, G.; et al. Central nervous system involvement in acute lymphoblastic leukemia is mediated by vascular endothelial growth factor. Blood 2017, 130, 643–654. [Google Scholar] [CrossRef]
- Samimi, A.; Ghanavat, M.; Shahrabi, S.; Azizidoost, S.; Saki, N. Role of bone marrow adipocytes in leukemia and chemotherapy challenges. Cell. Mol. Life Sci. 2019, 76, 2489–2497. [Google Scholar] [CrossRef]
- Zinngrebe, J.; Debatin, K.-M.; Fischer-Posovszky, P. Adipocytes in hematopoiesis and acute leukemia: Friends, enemies, or innocent bystanders? Leukemia 2020, 34, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, R.; Sheng, X.; Ichihara, B.; Heisterkamp, N.; Mittelman, S.D. Adipose tissue attracts and protects acute lymphoblastic leukemia cells from chemotherapy. Leuk. Res. 2013, 37, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Chen, Y.; Cabreira Mda, G.; Ruvolo, V.; Wang, Z.; Ma, W.; Konoplev, S.; Shpall, E.; Lyons, K.; Strunk, D.; et al. Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood 2013, 122, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Sala-Torra, O.; Gundacker, H.M.; Stirewalt, D.L.; Ladne, P.A.; Pogosova-Agadjanyan, E.L.; Slovak, M.L.; Willman, C.L.; Heimfeld, S.; Boldt, D.H.; Radich, J.P. Connective tissue growth factor (CTGF) expression and outcome in adult patients with acute lymphoblastic leukemia. Blood 2007, 109, 3080–3083. [Google Scholar] [CrossRef]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Brigstock, D.R.; de Klerk, N.H.; Kees, U.R. High expression of connective tissue growth factor in pre-B acute lymphoblastic leukaemia. Br. J. Haematol. 2007, 138, 740–748. [Google Scholar] [CrossRef]
- Tucci, J.; Chen, T.; Margulis, K.; Orgel, E.; Paszkiewicz, R.L.; Cohen, M.D.; Oberley, M.J.; Wahhab, R.; Jones, A.E.; Divakaruni, A.S.; et al. Adipocytes Provide Fatty Acids to Acute Lymphoblastic Leukemia Cells. Front. Oncol. 2021, 11, 665763. [Google Scholar] [CrossRef]
- Attané, C.; Estève, D.; Chaoui, K.; Iacovoni, J.S.; Corre, J.; Moutahir, M.; Valet, P.; Schiltz, O.; Reina, N.; Muller, C. Human Bone Marrow Is Comprised of Adipocytes with Specific Lipid Metabolism. Cell Rep. 2020, 30, 949–958.e6. [Google Scholar] [CrossRef]
- Shin, E.; Koo, J.S. The Role of Adipokines and Bone Marrow Adipocytes in Breast Cancer Bone Metastasis. Int. J. Mol. Sci. 2020, 21, 4967. [Google Scholar] [CrossRef]
- Moschovi, M.; Trimis, G.; Vounatsou, M.; Katsibardi, K.; Margeli, A.; Damianos, A.; Chrousos, G.; Papassotiriou, I. Serial Plasma Concentrations of Adiponectin, Leptin, and Resistin during Therapy in Children with Acute Lymphoblastic Leukemia. J. Pediatr. Hematol. Oncol. 2010, 32, e8–e13. [Google Scholar] [CrossRef]
- Konopleva, M.; Mikhail, A.; Estrov, Z.; Zhao, S.; Harris, D.; Sanchez-Williams, G.; Kornblau, S.M.; Dong, J.; Kliche, K.O.; Jiang, S.; et al. Expression and function of leptin receptor isoforms in myeloid leukemia and myelodysplastic syndromes: Proliferative and anti-apoptotic activities. Blood 1999, 93, 1668–1676. [Google Scholar] [CrossRef]
- Lu, Z.; Xie, J.; Wu, G.; Shen, J.; Collins, R.; Chen, W.; Kang, X.; Luo, M.; Zou, Y.; Huang, L.J.-S.; et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat. Med. 2017, 23, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Clifton, K.K.; Ma, C.X.; Fontana, L.; Peterson, L.L. Intermittent fasting in the prevention and treatment of cancer. CA Cancer J. Clin. 2021, 71, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Cawthorn, W.P.; Scheller, E.L.; Parlee, S.D.; Pham, H.A.; Learman, B.S.; Redshaw, C.M.H.; Sulston, R.J.; Burr, A.A.; Das, A.K.; Simon, B.R.; et al. Expansion of Bone Marrow Adipose Tissue During Caloric Restriction Is Associated with Increased Circulating Glucocorticoids and Not With Hypoleptinemia. Endocrinology 2016, 157, 508–521. [Google Scholar] [CrossRef]
- Orgel, E.; Tucci, J.; Alhushki, W.; Malvar, J.; Sposto, R.; Fu, C.H.; Freyer, D.R.; Abdel-Azim, H.; Mittelman, S.D. Obesity is associated with residual leukemia following induction therapy for childhood B-precursor acute lymphoblastic leukemia. Blood 2014, 124, 3932–3938. [Google Scholar] [CrossRef] [PubMed]
- Behan, J.W.; Yun, J.P.; Proektor, M.P.; Ehsanipour, E.A.; Arutyunyan, A.; Moses, A.S.; Avramis, V.I.; Louie, S.G.; Butturini, A.; Heisterkamp, N.; et al. Adipocytes Impair Leukemia Treatment in Mice. Cancer Res. 2009, 69, 7867–7874. [Google Scholar] [CrossRef] [PubMed]
- Ehsanipour, E.A.; Sheng, X.; Behan, J.W.; Wang, X.; Butturini, A.; Avramis, V.I.; Mittelman, S.D. Adipocytes Cause Leukemia Cell Resistance to L-Asparaginase via Release of Glutamine. Cancer Res. 2013, 73, 2998–3006. [Google Scholar] [CrossRef]
- Sheng, X.; Tucci, J.; Parmentier, J.-H.; Ji, L.; Behan, J.W.; Heisterkamp, N.; Mittelman, S.D. Adipocytes cause leukemia cell resistance to daunorubicin via oxidative stress response. Oncotarget 2016, 7, 73147–73159. [Google Scholar] [CrossRef]
- Sheng, X.; Parmentier, J.-H.; Tucci, J.; Pei, H.; Cortez-Toledo, O.; Dieli-Conwright, C.M.; Oberley, M.J.; Neely, M.; Orgel, E.; Louie, S.G.; et al. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol. Cancer Res. 2017, 15, 1704–1713. [Google Scholar] [CrossRef]
- Tjin, G.; Flores-Figueroa, E.; Duarte, D.; Straszkowski, L.; Scott, M.; Khorshed, R.A.; Purton, L.E.; Lo Celso, C. Imaging methods used to study mouse and human HSC niches: Current and emerging technologies. Bone 2019, 119, 19–35. [Google Scholar] [CrossRef]
- Al-Sabah, J.; Baccin, C.; Haas, S. Single-cell and spatial transcriptomics approaches of the bone marrow microenvironment. Curr. Opin. Oncol. 2020, 32, 146–153. [Google Scholar] [CrossRef]
- Baharlou, H.; Canete, N.P.; Cunningham, A.L.; Harman, A.N.; Patrick, E. Mass Cytometry Imaging for the Study of Human Diseases—Applications and Data Analysis Strategies. Front. Immunol. 2019, 10, 2657. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.; Skut, P.; Hughes, A.M.; Ferrari, E.; Tickner, J.; Xu, J.; Mullin, B.H.; Tang, D.; Malinge, S.; Kees, U.R.; et al. The bone marrow microenvironment of pre-B acute lymphoblastic leukemia at single-cell resolution. Sci. Rep. 2020, 10, 19173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Link, D.C. Targeting of Mesenchymal Stromal Cells by Cre-Recombinase Transgenes Commonly Used to Target Osteoblast Lineage Cells. J. Bone Miner. Res. 2016, 31, 2001–2007. [Google Scholar] [CrossRef]
- Joseph, C.; Quach, J.M.; Walkley, C.R.; Lane, S.W.; Lo Celso, C.; Purton, L.E. Deciphering Hematopoietic Stem Cells in Their Niches: A Critical Appraisal of Genetic Models, Lineage Tracing, and Imaging Strategies. Cell Stem Cell 2013, 13, 520–533. [Google Scholar] [CrossRef] [PubMed]
- Lampreht Tratar, U.; Horvat, S.; Cemazar, M. Transgenic Mouse Models in Cancer Research. Front. Oncol. 2018, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef]
- Breese, E.H.; Kotecha, R.S.; Guest, E.M. Acute Lymphoblastic Leukemia in Infants: A Distinctive, High-Risk Subtype of Childhood Acute Lymphoblastic Leukemia. In Clinical Management of Acute Lymphoblastic Leukemia; Litzow, M.R., Raetz, E.A., Eds.; Springer Nature: Cham, Switzerland, 2022; pp. 135–148. [Google Scholar]
- Kotecha, R.S.; Gottardo, N.G.; Kees, U.R.; Cole, C.H. The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e200. [Google Scholar] [CrossRef]
- Kuek, V.; Hughes, A.M.; Kotecha, R.S.; Cheung, L.C. Therapeutic Targeting of the Leukaemia Microenvironment. Int. J. Mol. Sci. 2021, 22, 6888. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, A.M.; Kuek, V.; Kotecha, R.S.; Cheung, L.C. The Bone Marrow Microenvironment in B-Cell Development and Malignancy. Cancers 2022, 14, 2089. https://doi.org/10.3390/cancers14092089
Hughes AM, Kuek V, Kotecha RS, Cheung LC. The Bone Marrow Microenvironment in B-Cell Development and Malignancy. Cancers. 2022; 14(9):2089. https://doi.org/10.3390/cancers14092089
Chicago/Turabian StyleHughes, Anastasia M., Vincent Kuek, Rishi S. Kotecha, and Laurence C. Cheung. 2022. "The Bone Marrow Microenvironment in B-Cell Development and Malignancy" Cancers 14, no. 9: 2089. https://doi.org/10.3390/cancers14092089
APA StyleHughes, A. M., Kuek, V., Kotecha, R. S., & Cheung, L. C. (2022). The Bone Marrow Microenvironment in B-Cell Development and Malignancy. Cancers, 14(9), 2089. https://doi.org/10.3390/cancers14092089