
The Cellular Tumor Immune Microenvironment of Childhood Solid Cancers: Informing More Effective Immunotherapies


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Pediatric Solid Cancers: Resistance to T-Cell Based Immunotherapeutics
1.2. Composition of the Tumor Microenvironment: Key to Effective Immunotherapies
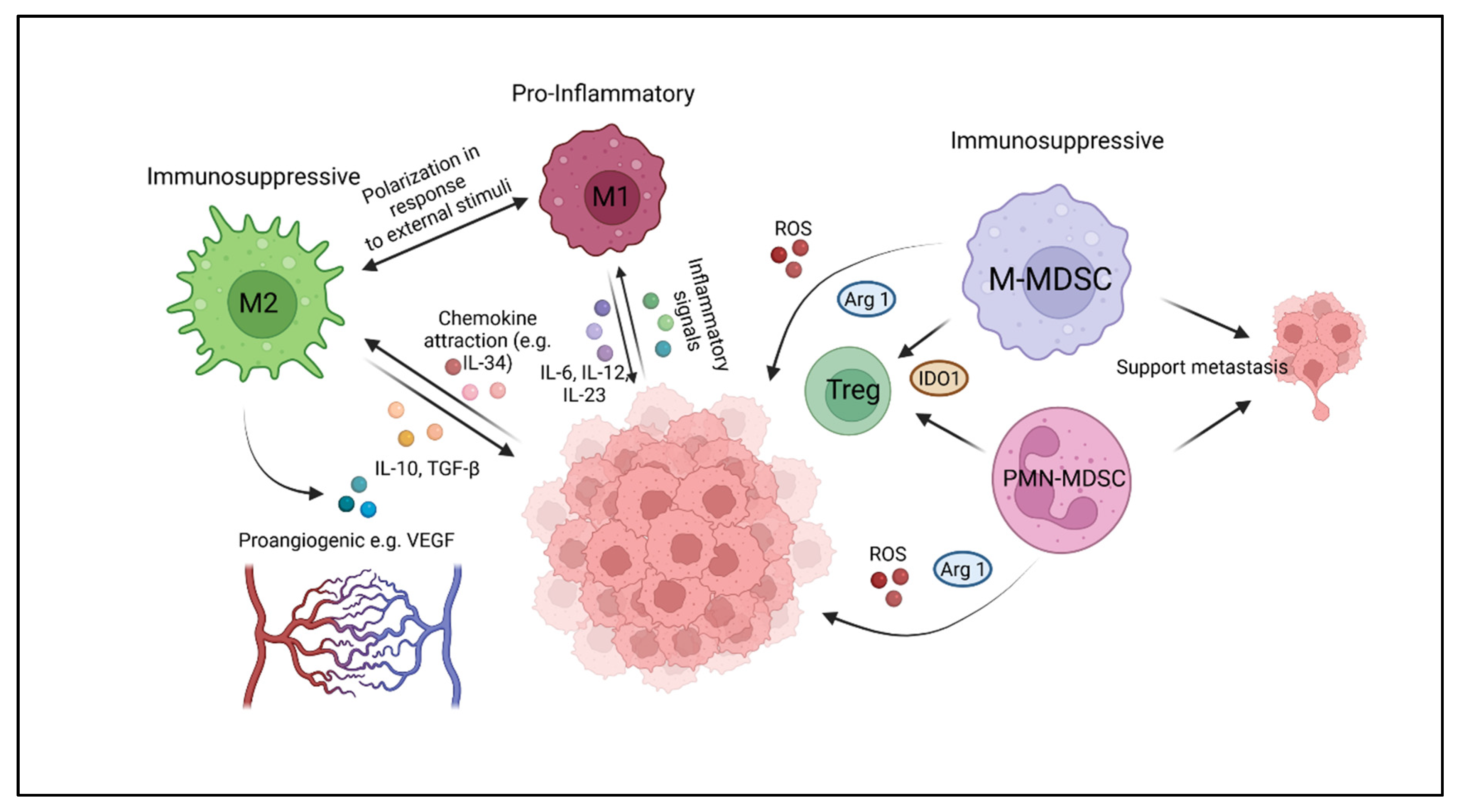
2. Myeloid Cell Components of the Immune Microenvironment of Solid Tumors
2.1. Tumor-Associated Macrophages (TAM)
2.2. Myeloid-Derived Suppressor Cells (MDSC)
3. Tools to Decipher Cellular Components of Tissues
3.1. Antibody-Based Analysis of Tissues
3.2. Gene Expression Analysis of the TME
3.3. Unbiased Molecular Profiling
4. Composition of the Immune Microenvironment of Pediatric Cancers
4.1. Neuroblastoma (NBL)
4.2. Osteosarcoma (OS)
4.3. Ewing Sarcoma (EwS)
4.4. Rhabdomyosarcoma

{kind=link}
{kind=link}
| Neuroblastoma | Patients | Samples | Method | Findings/Impact on Survival | Ref. | |
|---|---|---|---|---|---|---|
| Material | TAM | MDSC | ||||
| Human studies | ||||||
| pretreated tumor | 71 (IHC) 133 (RNA seq) | 133 (RNA seq) 71 (IHC) | RNA seq, IHC | M2 high in metastatic disease | [43] | |
| pretreated tumor | 41 | 41 | IHC | M2 associated with metastatic disease | [97] | |
| Mouse models | ||||||
| syngeneic | TAM accumulate during progression, shift from M1 to M2 | MDSC accumulate during disease progression | [98] | |||
| syngeneic | present in spleens, BM, blood, expressing Arg-1, ROS, TGF-β | [99] | ||||
| transgenic | Crosstalk with tumor cells polarizes macrophages for tumor progression | [100] | ||||
| Osteosarcoma | ||||||
| Human studies | ||||||
| primary tumors/ metastases | 53(GEP), 117(IHC) | 53(GEP), 174(IHC) | GEP/IHC | M1 and M2 associated with superior OS | [101] | |
| primary tumors | 124 | 124 | IHC | M1 associated with superior OS, MPFS | [102] | |
| primary tumor/ metastasis | 50 | 22 localized 28 metastasis | IHC | TAM associated with superior OS, M1 predominant in non-metastatic disease | [106] | |
| pre-/post treatment (matched) | 27 | 54 | IHC/IF | unchanged by chemotherapy | reduced after chemotherapy | [109] |
| Mouse models | ||||||
| syngeneic | IHC/IF | M2 associated with metastasis | [110] | |||
| xenograft | Fc | M2 associated with tumor growth | [111] | |||
| xenograft | M2 recruited by tumor | [112] | ||||
| Ewing sarcoma | ||||||
| Human studies | ||||||
| primary tumor pretreatment | 41 | 41 | IHC | high M1 associated with lower OS | [56] | |
| primary tumor pre-treatment | 197 | 197 | GEP | high M2 associated with poor outcome | [114] | |
| Mouse models | ||||||
| xenograft | IHC | TAM stimulate angiogenesis | [56] | |||
| xenograft | IHC | TAM inhibition reduces metastatic burden | [116] | |||
| xenograft | TAM negatively regulated by miRNA let-7a | [117] | ||||
| Rhabdomyosarcoma | ||||||
| Human studies | ||||||
| primary tumors | 39 | 20 aRMS 19 eRMS | IHC | high TAM infiltration | [118] | |
| primary tumors | 51 | 24 aRMS 27 eRMS | IHC/GEP | high M2 infiltration | [120] | |
| Mouse models | ||||||
| orthotopic | Fc | CXCR2-mediated tumor infiltration, promote local immunosuppression | [121] | |||
5. Strategies to Modify the TME for Effective Immunotherapies
5.1. Pharmacological Modification of the TME
5.1.1. Targeting the TME by Standard Anti-Proliferative Chemotherapeutic Agents
5.1.2. Targeting the TME by Molecularly Targeted Anticancer Agents
5.1.3. Targeting the TME by Epigenetic Anticancer Agents
5.2. Selective Targeting of Immunosuppressive Components of the TME
5.3. Inflammatory Reprogramming of the TME Using Locoregional Cytokine or Oncolytic Virus Deposits
5.4. Macrophage Immune Checkpoint Therapy: Unleashing TAM for Elimination of Tumor Cells
5.5. CAR-Engineered Macrophages for Tumor-Targeted Phagocytosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| ADCC | antibody dependent cell mediated cytotoxicity |
| ATRA | all-trans retinoic acid |
| c-FLIP | cellular FLICE-inhibitory protein |
| CAR | chimeric antigen receptor |
| CCL | C-C motif chemokine ligand |
| CCR | C-C chemokine receptor |
| CD | cluster of differentiation |
| COX2 | cyclooxygenase 2 |
| CSF1 | colony stimulating factor 1 |
| CSF1R | colony stimulating factor 1 receptor |
| CXCL | C-X-C motif chemokine ligand |
| CXCR | C-X-C motif chemokine receptor |
| EGF | epidermal growth factor |
| EwS | Ewing sarcoma |
| EWSR1-FLI1 | Ewing sarcoma breakpoint region 1-friend leukemia integration 1 |
| EZH2 | enhancer of zeste homolog 2 |
| Fc | fragment crystallizable |
| FLICE | FADD-like IL1-β-converting enzyme |
| GMR4 | glutamate metabotropic receptor 4 |
| HER-2 | human epidermal growth factor receptor 2 |
| HDAC | histone deacetylase |
| HLA-DR | human leukocyte antigen DR isotype |
| HSPC | hematopoietic stem and progenitor cell |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| IF | immunofluorescence |
| IFN-γ | interferon-γ |
| IGF | insulin-like growth factor |
| IGF1R | insulin-like growth factor receptor 1 |
| IHC | immunohistochemistry |
| IL | interleukin |
| JAK | Janus kinase |
| LAG3 | lymphocyte activating gene 3 |
| MARCO | macrophage receptor with collagenous structure |
| MALDI-IMS | matrix-assisted laser desorption/ionization- imaging mass spectrometry |
| MCP1 | monocyte chemoattractant protein-1 |
| MCPcounter | microenvironment cell populations-counter |
| MDSC | myeloid-derived suppressor cells |
| MELC | multi-epitope-ligand cartography |
| MHC | major histocompatibility complex |
| M-MDSC | monocytic myeloid-derived suppressor cells |
| MMP9 | matrix metalloproteinase 9 |
| NA | non amplified |
| NBL | neuroblastoma |
| OS | osteosarcoma |
| P2 × 7R | P2X-Purinoreceptor 7 |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death protein ligand 1 |
| PI3Kγ | phosphatidyl-inositol 3 kinase γ isoform |
| PMN-MDSC | polymorphonuclear myeloid-derived suppressor cells |
| PRC2 | polycomb receptor complex 2 |
| RMS | rhabdomyosarcoma |
| RNAish | RNA in situ hybridization |
| ROS | reactive oxygen species |
| SIRPα | signal-regulatory protein alpha |
| STAT3 | signal transducer and activator of transcription 3 |
| TAM | tumor associated macrophages |
| TAN | tumor associated neutrophils |
| TCR | T-cell receptor |
| TGCT | tenosynovial giant cell tumor |
| TGF-β | transforming growth factor β |
| TLS | tertiary lymphoid structure |
| TME | tumor microenvironment |
| TNFα | tumor necrosis factor α |
| TRAIL | tumor necrosis factor-related apoptosis inducing ligand |
| TRUCK | T cells redirected for universal cytokine killing |
| UPS | undifferentiated polymorphic sarcoma |
| USP6 | ubiquitin-specific protease 6 |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
References
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- McKay, R.R.; McGregor, B.A.; Xie, W.; Braun, D.A.; Wei, X.; Kyriakopoulos, C.E.; Zakharia, Y.; Maughan, B.L.; Rose, T.L.; Stadler, W.M.; et al. Optimized Management of Nivolumab and Ipilimumab in Advanced Renal Cell Carcinoma: A Response-Based Phase II Study (OMNIVORE). J. Clin. Oncol. 2020, 38, 4240–4248. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): Interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 121–133. [Google Scholar] [CrossRef]
- Geoerger, B.; Zwaan, C.M.; Marshall, L.V.; Michon, J.; Bourdeaut, F.; Casanova, M.; Corradini, N.; Rossato, G.; Farid-Kapadia, M.; Shemesh, C.S.; et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): A multicentre phase 1-2 study. Lancet Oncol. 2020, 21, 134–144. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Pasqualini, C.; Rubino, J.; Brard, C.; Cassard, L.; Andre, N.; Rondof, W.; Scoazec, J.Y.; Marchais, A.; Nebchi, S.; Boselli, L.; et al. Phase II and biomarker study of programmed cell death protein 1 inhibitor nivolumab and metronomic cyclophosphamide in paediatric relapsed/refractory solid tumours: Arm G of AcSe-ESMART, a trial of the European Innovative Therapies for Children with Cancer Consortium. Eur. J. Cancer 2021, 150, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting from Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Pearson, A.D.J.; Rossig, C.; Lesa, G.; Diede, S.J.; Weiner, S.; Anderson, J.; Gray, J.; Geoerger, B.; Minard-Colin, V.; Marshall, L.V.; et al. ACCELERATE and European Medicines Agency Paediatric Strategy Forum for medicinal product development of checkpoint inhibitors for use in combination therapy in paediatric patients. Eur. J. Cancer 2020, 127, 52–66. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e854. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e844. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Mlecnik, B.; Bindea, G.; Pages, F.; Galon, J. Immunosurveillance in human non-viral cancers. Curr. Opin. Immunol. 2011, 23, 272–278. [Google Scholar] [CrossRef]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Pages, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e1010. [Google Scholar] [CrossRef] [Green Version]
- Spurny, C.; Kailayangiri, S.; Jamitzky, S.; Altvater, B.; Wardelmann, E.; Dirksen, U.; Hardes, J.; Hartmann, W.; Rossig, C. Programmed cell death ligand 1 (PD-L1) expression is not a predominant feature in Ewing sarcomas. Pediatric Blood Cancer 2018, 65, e26719. [Google Scholar] [CrossRef]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamora, A.E.; Crawford, J.C.; Allen, E.K.; Guo, X.J.; Bakke, J.; Carter, R.A.; Abdelsamed, H.A.; Moustaki, A.; Li, Y.; Chang, T.C.; et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8(+) T cell responses. Sci. Transl. Med. 2019, 11, eaat8549. [Google Scholar] [CrossRef]
- Walz, S.; Stickel, J.S.; Kowalewski, D.J.; Schuster, H.; Weisel, K.; Backert, L.; Kahn, S.; Nelde, A.; Stroh, T.; Handel, M.; et al. The antigenic landscape of multiple myeloma: Mass spectrometry (re)defines targets for T-cell-based immunotherapy. Blood 2015, 126, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Byron, S.A.; Hendricks, W.P.D.; Nagulapally, A.B.; Kraveka, J.M.; Ferguson, W.S.; Brown, V.I.; Eslin, D.E.; Mitchell, D.; Cornelius, A.; Roberts, W.; et al. Genomic and Transcriptomic Analysis of Relapsed and Refractory Childhood Solid Tumors Reveals a Diverse Molecular Landscape and Mechanisms of Immune Evasion. Cancer Res. 2021, 81, 5818–5832. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Vakkila, J.; Jaffe, R.; Michelow, M.; Lotze, M.T. Pediatric cancers are infiltrated predominantly by macrophages and contain a paucity of dendritic cells: A major nosologic difference with adult tumors. Clin. Cancer Res. 2006, 12, 2049–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgharzadeh, S.; Salo, J.A.; Ji, L.; Oberthuer, A.; Fischer, M.; Berthold, F.; Hadjidaniel, M.; Liu, C.W.; Metelitsa, L.S.; Pique-Regi, R.; et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 2012, 30, 3525–3532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013, 34, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163(+) TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef]
- Hu, Z.; Van Rooijen, N.; Yang, Y.G. Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood 2011, 118, 5938–5946. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Fukushi, J.; Yamamoto, S.; Matsumoto, Y.; Setsu, N.; Oda, Y.; Yamada, H.; Okada, S.; Watari, K.; Ono, M.; et al. Macrophage infiltration predicts a poor prognosis for human ewing sarcoma. Am. J. Pathol. 2011, 179, 1157–1170. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, S.M.; Lee, A.H.; Paish, E.C.; Macmillan, R.D.; Ellis, I.O.; Green, A.R. Tumour-infiltrating macrophages and clinical outcome in breast cancer. J. Clin. Pathol. 2012, 65, 159–163. [Google Scholar] [CrossRef]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef]
- Komohara, Y.; Hasita, H.; Ohnishi, K.; Fujiwara, Y.; Suzu, S.; Eto, M.; Takeya, M. Macrophage infiltration and its prognostic relevance in clear cell renal cell carcinoma. Cancer Sci. 2011, 102, 1424–1431. [Google Scholar] [CrossRef]
- Campbell, M.J.; Tonlaar, N.Y.; Garwood, E.R.; Huo, D.; Moore, D.H.; Khramtsov, A.I.; Au, A.; Baehner, F.; Chen, Y.; Malaka, D.O.; et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res. Treat. 2011, 128, 703–711. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvaro, T.; Lejeune, M.; Camacho, F.I.; Salvadó, M.T.; Sánchez, L.; García, J.F.; Lopez, C.; Jaén, J.; Bosch, R.; Pons, L.E.; et al. The presence of STAT1-positive tumor-associated macrophages and their relation to outcome in patients with follicular lymphoma. Haematologica 2006, 91, 1605–1612. [Google Scholar] [PubMed]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Kinouchi, M.; Miura, K.; Mizoi, T.; Ishida, K.; Fujibuchi, W.; Sasaki, H.; Ohnuma, S.; Saito, K.; Katayose, Y.; Naitoh, T.; et al. Infiltration of CD40-positive tumor-associated macrophages indicates a favorable prognosis in colorectal cancer patients. Hepatogastroenterology 2013, 60, 83–88. [Google Scholar] [CrossRef]
- He, Y.M.; Li, X.; Perego, M.; Nefedova, Y.; Kossenkov, A.V.; Jensen, E.A.; Kagan, V.; Liu, Y.F.; Fu, S.Y.; Ye, Q.J.; et al. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat. Med. 2018, 24, 224–231. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression within the Pre-metastatic Niche. Cancer Res. 2016, 76, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Maric, I.; DiPrima, M.J.; Khan, J.; Orentas, R.J.; Kaplan, R.N.; Mackall, C.L. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood 2013, 122, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seung, L.P.; Rowley, D.A.; Dubey, P.; Schreiber, H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc. Natl. Acad. Sci. USA 1995, 92, 6254–6258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [Green Version]
- Gorris, M.A.J.; Halilovic, A.; Rabold, K.; van Duffelen, A.; Wickramasinghe, I.N.; Verweij, D.; Wortel, I.M.N.; Textor, J.C.; de Vries, I.J.M.; Figdor, C.G. Eight-Color Multiplex Immunohistochemistry for Simultaneous Detection of Multiple Immune Checkpoint Molecules within the Tumor Microenvironment. J. Immunol. 2018, 200, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Parra, E.R.; Uraoka, N.; Jiang, M.; Cook, P.; Gibbons, D.; Forget, M.A.; Bernatchez, C.; Haymaker, C.; Wistuba, I.I., II; Rodriguez-Canales, J. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci. Rep. 2017, 7, 13380. [Google Scholar] [CrossRef] [Green Version]
- Melcher, V.; Graf, M.; Interlandi, M.; Moreno, N.; de Faria, F.W.; Kim, S.N.; Kastrati, D.; Korbanka, S.; Alfert, A.; Gerss, J.; et al. Macrophage-tumor cell interaction promotes ATRT progression and chemoresistance. Acta Neuropathol. 2020, 139, 913–936. [Google Scholar] [CrossRef] [Green Version]
- Schubert, W. Topological proteomics, toponomics, MELK-technology. Adv. Biochem. Eng. Biotechnol. 2003, 83, 189–209. [Google Scholar] [CrossRef]
- Schubert, W.; Bonnekoh, B.; Pommer, A.J.; Philipsen, L.; Bockelmann, R.; Malykh, Y.; Gollnick, H.; Friedenberger, M.; Bode, M.; Dress, A.W. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006, 24, 1270–1278. [Google Scholar] [CrossRef]
- Schafer, D.; Tomiuk, S.; Kuster, L.N.; Rawashdeh, W.A.; Henze, J.; Tischler-Hohle, G.; Agorku, D.J.; Brauner, J.; Linnartz, C.; Lock, D.; et al. Identification of CD318, TSPAN8 and CD66c as target candidates for CAR T cell based immunotherapy of pancreatic adenocarcinoma. Nat. Commun. 2021, 12, 1453. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Deisseroth, K. CLARITY for mapping the nervous system. Nat. Methods 2013, 10, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shen, Q.; White, S.L.; Gokmen-Polar, Y.; Badve, S.; Goodman, L.J. Three-dimensional imaging and quantitative analysis in CLARITY processed breast cancer tissues. Sci. Rep. 2019, 9, 5624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perfetto, S.P.; Chattopadhyay, P.K.; Roederer, M. Seventeen-colour flow cytometry: Unravelling the immune system. Nat. Rev. Immunol. 2004, 4, 648–655. [Google Scholar] [CrossRef]
- Grabinski, T.M.; Kneynsberg, A.; Manfredsson, F.P.; Kanaan, N.M. A method for combining RNAscope in situ hybridization with immunohistochemistry in thick free-floating brain sections and primary neuronal cultures. PLoS ONE 2015, 10, e0120120. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Sanchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765.e716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.O.; Oehler, M.K.; Ruszkiewicz, A.; McColl, S.R.; Hoffmann, P. MALDI Imaging Mass Spectrometry (MALDI-IMS)-application of spatial proteomics for ovarian cancer classification and diagnosis. Int. J. Mol. Sci. 2011, 12, 773–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghmans, E.; Van Raemdonck, G.; Schildermans, K.; Willems, H.; Boonen, K.; Maes, E.; Mertens, I.; Pauwels, P.; Baggerman, G. MALDI Mass Spectrometry Imaging Linked with Top-Down Proteomics as a Tool to Study the Non-Small-Cell Lung Cancer Tumor Microenvironment. Methods Protoc. 2019, 2, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wienke, J.; Dierselhuis, M.P.; Tytgat, G.A.M.; Kunkele, A.; Nierkens, S.; Molenaar, J.J. The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer 2021, 144, 123–150. [Google Scholar] [CrossRef]
- Wei, J.S.; Kuznetsov, I.B.; Zhang, S.; Song, Y.K.; Asgharzadeh, S.; Sindiri, S.; Wen, X.; Patidar, R.; Najaraj, S.; Walton, A.; et al. Clinically Relevant Cytotoxic Immune Cell Signatures and Clonal Expansion of T-Cell Receptors in High-Risk MYCN-Not-Amplified Human Neuroblastoma. Clin. Cancer Res. 2018, 24, 5673–5684. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, O.; Yoshida, M.; Koma, Y.; Yanai, T.; Hasegawa, D.; Kosaka, Y.; Nishimura, N.; Yokozaki, H. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J. Pathol. 2016, 240, 211–223. [Google Scholar] [CrossRef]
- Carlson, L.M.; Rasmuson, A.; Idborg, H.; Segerström, L.; Jakobsson, P.J.; Sveinbjörnsson, B.; Kogner, P. Low-dose aspirin delays an inflammatory tumor progression in vivo in a transgenic mouse model of neuroblastoma. Carcinogenesis 2013, 34, 1081–1088. [Google Scholar] [CrossRef]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef] [Green Version]
- Fultang, L.; Gamble, L.D.; Gneo, L.; Berry, A.M.; Egan, S.A.; De Bie, F.; Yogev, O.; Eden, G.L.; Booth, S.; Brownhill, S.; et al. Macrophage-Derived IL1beta and TNFalpha Regulate Arginine Metabolism in Neuroblastoma. Cancer Res. 2019, 79, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Burger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; Larousserie, F.; Entz-Werlé, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Han, Y.; Guo, W.; Ren, T.; Huang, Y.; Wang, S.; Liu, K.; Zheng, B.; Yang, K.; Zhang, H.; Liang, X. Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis. Cancer Lett. 2019, 440–441, 116–125. [Google Scholar] [CrossRef]
- Han, Q.; Shi, H.; Liu, F. CD163(+) M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int. Immunopharmacol. 2016, 34, 101–106. [Google Scholar] [CrossRef]
- Dumars, C.; Ngyuen, J.M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef] [Green Version]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Brouchet, A.; Gilhodes, J.; Acker, N.V.; Brion, R.; Bouvier, C.; Assemat, P.; Gaspar, N.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; et al. Characterization of Macrophages and Osteoclasts in the Osteosarcoma Tumor Microenvironment at Diagnosis: New Perspective for Osteosarcoma Treatment? Cancers 2021, 13, 423. [Google Scholar] [CrossRef]
- Deng, C.; Xu, Y.; Fu, J.; Zhu, X.; Chen, H.; Xu, H.; Wang, G.; Song, Y.; Song, G.; Lu, J.; et al. Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci. 2020, 111, 1899–1909. [Google Scholar] [CrossRef]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-Trans Retinoic Acid Prevents Osteosarcoma Metastasis by Inhibiting M2 Polarization of Tumor-Associated Macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Zhang, X.; Wu, Y.; Yang, Y. Inhibition of macrophage polarization prohibits growth of human osteosarcoma. Tumour. Biol. 2014, 35, 7611–7616. [Google Scholar] [CrossRef] [PubMed]
- Ségaliny, A.I.; Mohamadi, A.; Dizier, B.; Lokajczyk, A.; Brion, R.; Lanel, R.; Amiaud, J.; Charrier, C.; Boisson-Vidal, C.; Heymann, D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 2015, 137, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Thomson, K.; Pang, P.; Dutour, A.; Mirabello, L.; Acher, F.; Pin, J.P.; Demicco, E.G.; Yan, J.; Teng, M.W.L.; et al. Infiltrating Myeloid Cells Drive Osteosarcoma Progression via GRM4 Regulation of IL23. Cancer Discov. 2019, 9, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, D.; Gentles, A.J.; Thiele, R.; Gutgemann, I. Prognostic profiling of the immune cell microenvironment in Ewing s Sarcoma Family of Tumors. Oncoimmunology 2019, 8, e1674113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.J.; Han, J.; Han, Z.Y.; Sax, B.; Kream, B.E.; et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2017, 8, 34141–34163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, A.J.; Maloney, C.; Behr, C.A.; Edelman, M.C.; Glick, R.D.; Al-Abed, Y.; Symons, M.; Soffer, S.Z.; Steinberg, B.M. The Macrophage Inhibitor CNI-1493 Blocks Metastasis in a Mouse Model of Ewing Sarcoma through Inhibition of Extravasation. PLoS ONE 2015, 10, e0145197. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Huang, L.; Xiao, Q.; Chen, X.; Zhong, J.; Chen, Y.; Yang, D.; Han, Z.; Shu, Y.; et al. Let-7a suppresses macrophage infiltrations and malignant phenotype of Ewing sarcoma via STAT3/NF-kappaB positive regulatory circuit. Cancer Lett. 2016, 374, 192–201. [Google Scholar] [CrossRef]
- Kather, J.N.; Hörner, C.; Weis, C.A.; Aung, T.; Vokuhl, C.; Weiss, C.; Scheer, M.; Marx, A.; Simon-Keller, K. CD163+ immune cell infiltrates and presence of CD54+ microvessels are prognostic markers for patients with embryonal rhabdomyosarcoma. Sci. Rep. 2019, 9, 9211. [Google Scholar] [CrossRef]
- Nakajima, K.; Raz, A. T-cell infiltration profile in musculoskeletal tumors. J. Orthop. Res. 2021, 39, 536–542. [Google Scholar] [CrossRef]
- Chen, L.; Oke, T.; Siegel, N.; Cojocaru, G.; Tam, A.J.; Blosser, R.L.; Swailes, J.; Ligon, J.A.; Lebid, A.; Morris, C.; et al. The Immunosuppressive Niche of Soft-Tissue Sarcomas is Sustained by Tumor-Associated Macrophages and Characterized by Intratumoral Tertiary Lymphoid Structures. Clin. Cancer Res. 2020, 26, 4018–4030. [Google Scholar] [CrossRef] [Green Version]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rebe, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.J.; Kim, Y.J.; Kim, Y.S.; Chang, W.S.; Ko, S.Y.; Chang, S.Y.; Sakaguchi, S.; Kang, C.Y. A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res. 2007, 67, 7477–7486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugel, S.; Peranzoni, E.; Desantis, G.; Chioda, M.; Walter, S.; Weinschenk, T.; Ochando, J.C.; Cabrelle, A.; Mandruzzato, S.; Bronte, V. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep. 2012, 2, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Welters, M.J.; van der Sluis, T.C.; van Meir, H.; Loof, N.M.; van Ham, V.J.; van Duikeren, S.; Santegoets, S.J.; Arens, R.; de Kam, M.L.; Cohen, A.F.; et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci. Transl. Med. 2016, 8, 334ra352. [Google Scholar] [CrossRef]
- Ratti, C.; Botti, L.; Cancila, V.; Galvan, S.; Torselli, I.; Garofalo, C.; Manara, M.C.; Bongiovanni, L.; Valenti, C.F.; Burocchi, A.; et al. Trabectedin Overrides Osteosarcoma Differentiative Block and Reprograms the Tumor Immune Environment Enabling Effective Combination with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 5149–5161. [Google Scholar] [CrossRef] [Green Version]
- Fiore, A.; Ugel, S.; De Sanctis, F.; Sandri, S.; Fracasso, G.; Trovato, R.; Sartoris, S.; Solito, S.; Mandruzzato, S.; Vascotto, F.; et al. Induction of immunosuppressive functions and NF-kappaB by FLIP in monocytes. Nat. Commun. 2018, 9, 5193. [Google Scholar] [CrossRef]
- Haverkamp, J.M.; Smith, A.M.; Weinlich, R.; Dillon, C.P.; Qualls, J.E.; Neale, G.; Koss, B.; Kim, Y.; Bronte, V.; Herold, M.J.; et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 2014, 41, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e488. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Cheng, F.; Yu, B.; Nefedova, Y.; Sotomayor, E.; Lush, R.; Gabrilovich, D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003, 63, 4441–4449. [Google Scholar] [PubMed]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, X.J.; Xiang, S.F.; Chen, Y.Q.; Zhang, N.; Cao, J.; Zhu, H.; Yang, B.; Zhou, Q.; Ying, M.D.; He, Q.J. Inhibition of M2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta Pharmacol. Sin. 2019, 40, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Irey, E.A.; Lassiter, C.M.; Brady, N.J.; Chuntova, P.; Wang, Y.; Knutson, T.P.; Henzler, C.; Chaffee, T.S.; Vogel, R.I.; Nelson, A.C.; et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proc. Natl. Acad. Sci. USA 2019, 116, 12442–12451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [Green Version]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, N.; Campbell-Hewson, Q.; Gallego Melcon, S.; Locatelli, F.; Venkatramani, R.; Hecker-Nolting, S.; Gambart, M.; Bautista, F.; Thebaud, E.; Aerts, I.; et al. Phase I/II study of single-agent lenvatinib in children and adolescents with refractory or relapsed solid malignancies and young adults with osteosarcoma (ITCC-050). ESMO Open 2021, 6, 100250. [Google Scholar] [CrossRef]
- Gaspar, N.; Venkatramani, R.; Hecker-Nolting, S.; Melcon, S.G.; Locatelli, F.; Bautista, F.; Longhi, A.; Lervat, C.; Entz-Werle, N.; Casanova, M.; et al. Lenvatinib with etoposide plus ifosfamide in patients with refractory or relapsed osteosarcoma (ITCC-050): A multicentre, open-label, multicohort, phase 1/2 study. Lancet Oncol. 2021, 22, 1312–1321. [Google Scholar] [CrossRef]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, F.; Quintana, F.J.; Cohen, I.R. Angiogenesis-inflammation cross-talk: Vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J. Immunol. 2004, 172, 4618–4623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Huang, H.; Coleman, M.; Ziemys, A.; Gopal, P.; Kazmi, S.M.; Brekken, R.A. VEGFR2 activity on myeloid cells mediates immune suppression in the tumor microenvironment. JCI Insight 2021, 6, e150735. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, D.; Tran, E.; Yu, Z.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013, 73, 3371–3380. [Google Scholar] [CrossRef] [Green Version]
- Chinnasamy, D.; Yu, Z.; Theoret, M.R.; Zhao, Y.; Shrimali, R.K.; Morgan, R.A.; Feldman, S.A.; Restifo, N.P.; Rosenberg, S.A. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Investig. 2010, 120, 3953–3968. [Google Scholar] [CrossRef] [Green Version]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Gottlich, C.; Kuhnemundt, J.; Mikesch, J.H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol. Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Yuan, J.; Li, N.; Pei, S.; Xu, J.; Luo, X.; Mao, C.; Liu, J.; Yu, T.; et al. Macrophage/microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3. J. Exp. Med. 2018, 215, 1365–1382. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, Y.; Pienta, K.J. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst. 2010, 102, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tille, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e655. [Google Scholar] [CrossRef] [Green Version]
- Park, J.A.; Wang, L.; Cheung, N.V. Modulating tumor infiltrating myeloid cells to enhance bispecific antibody-driven T cell infiltration and anti-tumor response. J. Hematol. Oncol. 2021, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.Y.; Alcindor, T.; Ganjoo, K.; Martin-Broto, J.; Ryan, C.W.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Tresckow, B.; Morschhauser, F.; Ribrag, V.; Topp, M.S.; Chien, C.; Seetharam, S.; Aquino, R.; Kotoulek, S.; de Boer, C.J.; Engert, A. An Open-Label, Multicenter, Phase I/II Study of JNJ-40346527, a CSF-1R Inhibitor, in Patients with Relapsed or Refractory Hodgkin Lymphoma. Clin. Cancer Res. 2015, 21, 1843–1850. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.J.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S.; et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef] [Green Version]
- Razak, A.R.; Cleary, J.M.; Moreno, V.; Boyer, M.; Calvo Aller, E.; Edenfield, W.; Tie, J.; Harvey, R.D.; Rutten, A.; Shah, M.A.; et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J. Immunother. Cancer 2020, 8, e001006. [Google Scholar] [CrossRef]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol./ESMO 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- La Fleur, L.; Boura, V.F.; Alexeyenko, A.; Berglund, A.; Ponten, V.; Mattsson, J.S.M.; Djureinovic, D.; Persson, J.; Brunnstrom, H.; Isaksson, J.; et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int. J. Cancer 2018, 143, 1741–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020, 12, eaax6337. [Google Scholar] [CrossRef] [PubMed]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin. Cancer Res. 2012, 18, 1672–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Drakes, D.J.; Rafiq, S.; Purdon, T.J.; Lopez, A.V.; Chandran, S.S.; Klebanoff, C.A.; Brentjens, R.J. Optimization of T-cell Receptor-Modified T Cells for Cancer Therapy. Cancer Immunol. Res. 2020, 8, 743–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, K.; Kuehle, J.; Dragon, A.C.; Galla, M.; Kloth, C.; Rudek, L.S.; Sandalcioglu, I.E.; Neyazi, B.; Moritz, T.; Meyer, J.; et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines. Cancers 2020, 12, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masemann, D.; Meissner, R.; Schied, T.; Lichty, B.D.; Rapp, U.R.; Wixler, V.; Ludwig, S. Synergistic anti-tumor efficacy of oncolytic influenza viruses and B7-H3 immune- checkpoint inhibitors against IC-resistant lung cancers. Oncoimmunology 2021, 10, 1885778. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheth, V.S.; Gauthier, J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transpl. 2021, 56, 552–566. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Xu, J.F.; Pan, X.H.; Zhang, S.J.; Zhao, C.; Qiu, B.S.; Gu, H.F.; Hong, J.F.; Cao, L.; Chen, Y.; Xia, B.; et al. CD47 blockade inhibits tumor progression human osteosarcoma in xenograft models. Oncotarget 2015, 6, 23662–23670. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.; Aghighi, M.; Yerneni, K.; Theruvath, J.L.; Daldrup-Link, H.E. Improving the efficacy of osteosarcoma therapy: Combining drugs that turn cancer cell ‘don’t eat me’ signals off and ‘eat me’ signals on. Mol. Oncol. 2019, 13, 2049–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahri, M.; Kailayangiri, S.; Vermeulen, S.; Galopin, N.; Rossig, C.; Paris, F.; Fougeray, S.; Birkle, S. SIRPalpha-specific monoclonal antibody enables antibody-dependent phagocytosis of neuroblastoma cells. Cancer Immunol. Immunother. 2022, 71, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Theruvath, J.; Menard, M.; Smith, B.A.H.; Linde, M.H.; Coles, G.L.; Dalton, G.N.; Wu, W.; Kiru, L.; Delaidelli, A.; Sotillo, E.; et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 2022, 28, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holterhus, M.; Altvater, B.; Kailayangiri, S.; Rossig, C. The Cellular Tumor Immune Microenvironment of Childhood Solid Cancers: Informing More Effective Immunotherapies. Cancers 2022, 14, 2177. https://doi.org/10.3390/cancers14092177
Holterhus M, Altvater B, Kailayangiri S, Rossig C. The Cellular Tumor Immune Microenvironment of Childhood Solid Cancers: Informing More Effective Immunotherapies. Cancers. 2022; 14(9):2177. https://doi.org/10.3390/cancers14092177
Chicago/Turabian StyleHolterhus, Malcolm, Bianca Altvater, Sareetha Kailayangiri, and Claudia Rossig. 2022. "The Cellular Tumor Immune Microenvironment of Childhood Solid Cancers: Informing More Effective Immunotherapies" Cancers 14, no. 9: 2177. https://doi.org/10.3390/cancers14092177
APA StyleHolterhus, M., Altvater, B., Kailayangiri, S., & Rossig, C. (2022). The Cellular Tumor Immune Microenvironment of Childhood Solid Cancers: Informing More Effective Immunotherapies. Cancers, 14(9), 2177. https://doi.org/10.3390/cancers14092177