
Head and Neck Cancer Susceptibility and Metabolism in Fanconi Anemia

 , , , , and
, , , , and
Abstract
Simple Summary
Abstract
1. Introduction
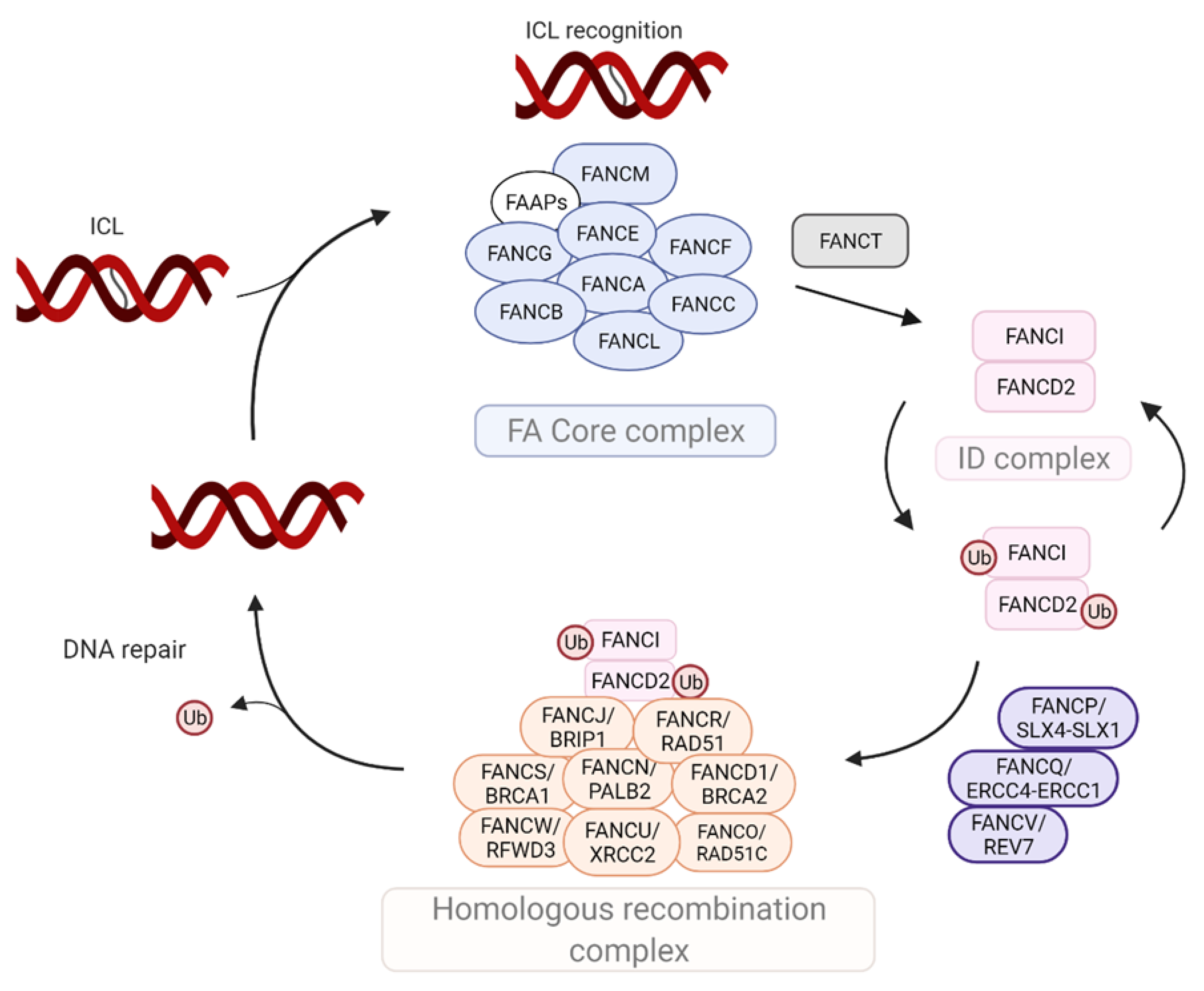
2. Molecular Components of the FA Machinery
3. Clinical Phenotypes in FA
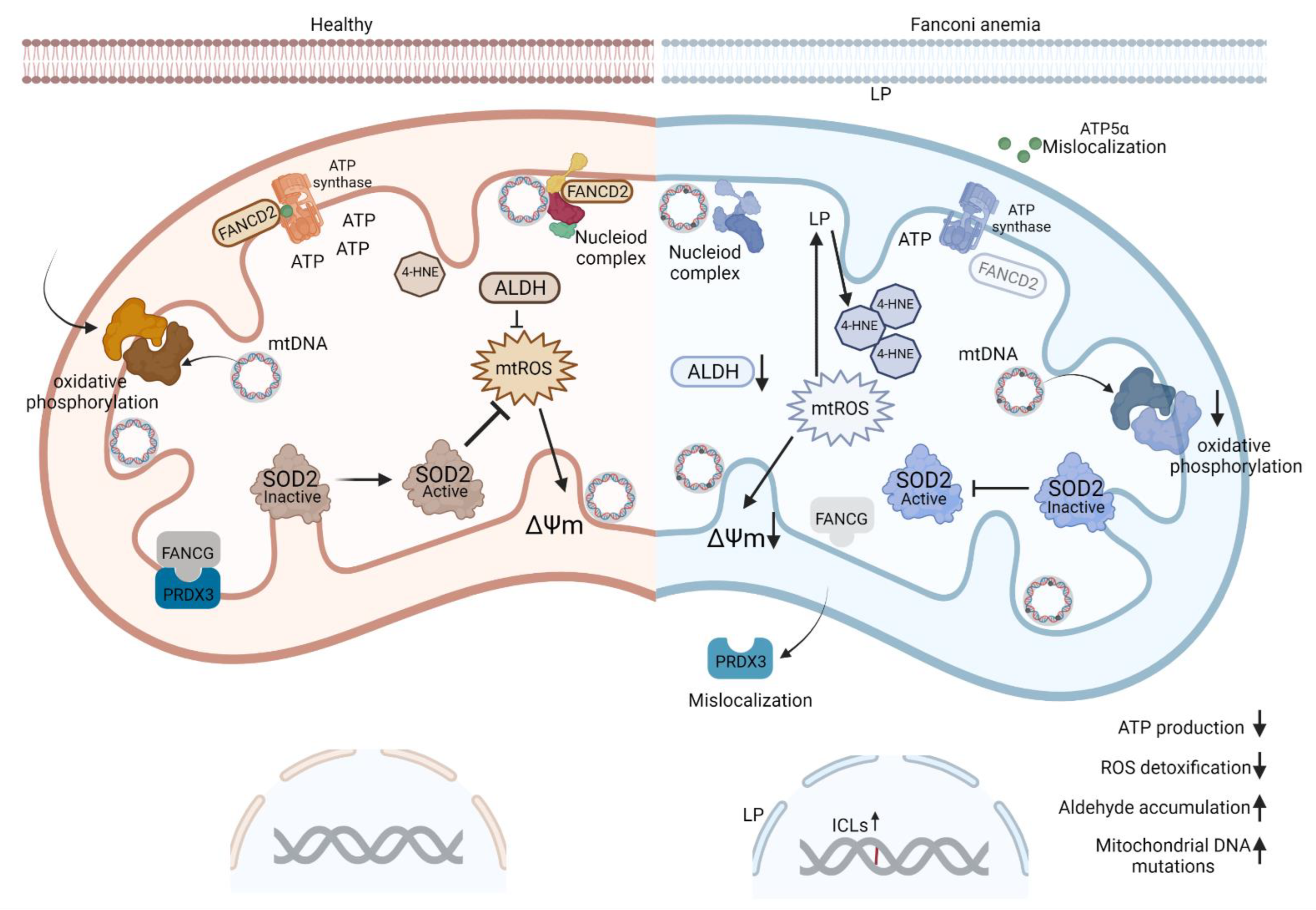
4. Dysregulation of Mitochondrial Metabolism in FA-Deficient Cells
4.1. Mitochondrial Activities Play a Role in Oncogenesis
4.2. FA Proteins Localize to Mitochondria
4.3. ROS May Be a Cause or Consequence of Mitochondrial Abnormalities in FA
4.4. Increased ROS Exacerbate DNA Damage in FA
5. Metabolic Dysregulation in FA
5.1. Aldehydes Are Relevant DNA Crosslinkers in Fanconi Anemia
5.2. Dysregulated Lipid Metabolism in FA Promotes Hyperproliferation and SCC Phenotypes
5.3. Tryptophan Metabolism Drives Serotonin Production in FA
5.4. Repurposing Therapies for Disease Prevention or Therapy in FA
6. Human Papillomavirus Is Tropic for Keratinocytes and Causes SCC
6.1. HPV Is Epidemiologically and Molecularly Linked to FA
6.2. HPV Positive and Negative SCCs May Harbor Differential Cellular Metabolism
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A.D. Diagnosis of Fanconi Anemia by Diepoxybutane Analysis. Curr. Protoc. Hum. Genet. 2015, 85, 8.7.1–8.7.17. [Google Scholar] [CrossRef] [PubMed]
- Bagby, G.C. Multifunctional Fanconi proteins, inflammation and the Fanconi phenotype. EBioMedicine 2016, 8, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; Ren, K. Fanconi Anemia Proteins, DNA Interstrand Crosslink Repair Pathways, and Cancer Therapy. Curr. Cancer Drug Targets 2009, 9, 101–117. [Google Scholar] [CrossRef]
- Ma, B.; Stepanov, I.; Hecht, S.S. Recent Studies on DNA Adducts Resulting from Human Exposure to Tobacco Smoke. Toxics 2019, 7, 16. [Google Scholar] [CrossRef]
- Rageul, J.; Kim, H. Fanconi anemia and the underlying causes of genomic instability. Environ. Mol. Mutagen. 2020, 61, 693–708. [Google Scholar] [CrossRef]
- Joenje, H.; Oostra, B. Effect of oxygen tension on chromosomal aberrations in Fanconi anaemia. Qual. Life Res. 1983, 65, 99–101. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef]
- Langevin, F.; Crossan, G.; Rosado, I.V.; Arends, M.J.; Patel, K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011, 475, 53–58. [Google Scholar] [CrossRef]
- Grompe, M.; D’Andrea, A. Fanconi anemia and DNA repair. Hum. Mol. Genet. 2001, 10, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Comar, M.; De Rocco, D.; Cappelli, E.; Zanotta, N.; Bottega, R.; Svahn, J.; Farruggia, P.; Misuraca, A.; Corsolini, F.; Dufour, C.; et al. Fanconi Anemia Patients Are More Susceptible to Infection with Tumor Virus SV40. PLoS ONE 2013, 8, e79683. [Google Scholar] [CrossRef] [PubMed]
- Sauter, S.L.; Wells, S.I.; Zhang, X.; Hoskins, E.E.; Davies, S.M.; Myers, K.C.; Mueller, R.; Panicker, G.; Unger, E.; Sivaprasad, U.; et al. Oral Human Papillomavirus Is Common in Individuals with Fanconi Anemia. Cancer Epidemiol. Biomark. Prev. 2015, 24, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Butturini AGale, R.P.; Verlander, P.C.; Adler-Brecher, B.; Gillio, A.P.; Auerbach, A.D. Hematologic abnormalities in Fanconi anemia: An International Fanconi Anemia Registry study. Blood 1994, 84, 1650–1655. [Google Scholar] [CrossRef]
- Hess, C.J.; Gale, R.P.; Verlander, P.C.; Adler-Brecher, B.; Gillio, A.P.; Auerbach, A.D. Hypermethylation of the FANCC and FANCL promoter regions in sporadic acute leukaemia. Anal. Cell. Pathol. 2008, 30, 299–306. [Google Scholar] [CrossRef]
- Li, J.; Sejas, D.P.; Zhang, X.; Qiu, Y.; Nattamai, K.J.; Rani, R.; Rathbun, K.R.; Geiger, H.; Williams, D.A.; Bagby, G.C.; et al. TNF-α induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J. Clin. Investig. 2007, 117, 3283–3295. [Google Scholar] [CrossRef]
- Savage, S.A.; Walsh, M.F. Myelodysplastic Syndrome, Acute Myeloid Leukemia, and Cancer Surveillance in Fanconi Anemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 657–668. [Google Scholar] [CrossRef]
- Tischkowitz, M.D.; Morgan, N.; Grimwade, D.; Eddy, C.; Ball, S.; Vorechovsky, I.; Langabeer, S.; Stöger, R.; Hodgson, S.V.; Mathew, C. Deletion and reduced expression of the Fanconi anemia FANCA gene in sporadic acute myeloid leukemia. Leukemia 2004, 18, 420–425. [Google Scholar] [CrossRef]
- Kutler, D.I.; Auerbach, A.D.; Satagopan, J.; Giampietro, P.F.; Batish, S.D.; Huvos, A.G.; Goberdhan, A.; Shah, J.P.; Singh, B. High Incidence of Head and Neck Squamous Cell Carcinoma in Patients with Fanconi Anemia. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 106–112. [Google Scholar] [CrossRef]
- Kutler, D.I.; Patel, K.R.; Auerbach, A.D.; Kennedy, J.; Lach, F.P.; Sanborn, E.; Cohen, M.A.; Kuhel, W.I.; Smogorzewska, A. Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. Laryngoscope 2016, 126, 870–879. [Google Scholar] [CrossRef]
- Kutler, D.I.; Singh, B.; Satagopan, J.; Batish, S.D.; Berwick, M.; Giampietro, P.F.; Hanenberg, H.; Auerbach, A. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003, 101, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Greene, M.H.; Alter, B.P. Cancer incidence in persons with Fanconi anemia. Blood 2003, 101, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Scheckenbach, K.; Wagenmann, M.; Freund, M.; Schipper, J.; Hanenberg, H.; Schethenbach, K. Squamous Cell Carcinomas of the Head and Neck in Fanconi Anemia: Risk, Prevention, Therapy, and the Need for Guidelines. Klin. Pädiatrie 2012, 224, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Velleuer, E.; Dietrich, R. Fanconi anemia: Young patients at high risk for squamous cell carcinoma. Mol. Cell. Pediatr. 2014, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Savage, S.; Rosenberg, P. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2017, 103, 30–39. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC pathway and mitosis: A replication legacy. Cell Cycle 2009, 8, 2907–2912. [Google Scholar] [CrossRef]
- Vinciguerra, P.; Godinho, S.A.; Parmar, K.; Pellman, D.; D’Andrea, A.D. Cytokinesis failure occurs in Fanconi anemia pathway–deficient murine and human bone marrow hematopoietic cells. J. Clin. Investig. 2010, 120, 3834–3842. [Google Scholar] [CrossRef]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing Nonhomologous End Joining Suppresses DNA Repair Defects of Fanconi Anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.H.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef]
- Myers, K.C.; Sauter, S.; Zhang, X.; Bleesing, J.J.; Davies, S.M.; Wells, S.I.; Mehta, P.A.; Kumar, A.; Marmer, D.; Marsh, R.; et al. Impaired immune function in children and adults with Fanconi anemia. Pediatr. Blood Cancer 2017, 64, e26599. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.M.; Mark, J.; DeMarcantonio, M.; Leino, D.; Sisson, R.; Geller, J.I. Pediatric laryngeal carcinoma in a heterozygous carrier of Fanconi anemia. Pediatr. Blood Cancer 2017, 64, e26463. [Google Scholar] [CrossRef] [PubMed]
- Khanal, S.; Galloway, D.A. High-risk human papillomavirus oncogenes disrupt the Fanconi anemia DNA repair pathway by impairing localization and de-ubiquitination of FancD2. PLoS Pathog. 2019, 15, e1007442. [Google Scholar] [CrossRef]
- Spardy, N.; Duensing, A.; Charles, D.; Haines, N.; Nakahara, T.; Lambert, P.F.; Duensing, S. The Human Papillomavirus Type 16 E7 Oncoprotein Activates the Fanconi Anemia (FA) Pathway and Causes Accelerated Chromosomal Instability in FA Cells. J. Virol. 2007, 81, 13265–13270. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Wreesmann, V.B.; Goberdhan, A.; Ben-Porat, L.; Satagopan, J.; Ngai, I.; Huvos, A.G.; Giampietro, P.; Levran, O.; Pujara, K.; et al. Human Papillomavirus DNA and p53 Polymorphisms in Squamous Cell Carcinomas from Fanconi Anemia Patients. JNCI J. Natl. Cancer Inst. 2003, 95, 1718–1721. [Google Scholar] [CrossRef]
- Ruppitsch, W.; Meißlitzer, C.; Weirich-Schwaiger, H.; Klocker, H.; Scheidereit, C.; Schweiger, M.; Hirsch-Kauffmann, M. The role of oxygen metabolism for the pathological phenotype of Fanconi anemia. Qual. Life Res. 1997, 99, 710–719. [Google Scholar] [CrossRef]
- Bartlett, A.L.; Romick-Rosendale, L.; Nelson, A.; Abdullah, S.; Luebbering, N.; Bartlett, J.; Brusadelli, M.; Palumbo, J.S.; Lake, K.; Litts, B.; et al. Tryptophan metabolism is dysregulated in individuals with Fanconi anemia. Blood Adv. 2021, 5, 250–261. [Google Scholar] [CrossRef]
- Cappelli, E.; Cuccarolo, P.; Stroppiana, G.; Miano, M.; Bottega, R.; Cossu, V.; Degan, P.; Ravera, S. Defects in mitochondrial energetic function compels Fanconi Anaemia cells to glycolytic metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1214–1221. [Google Scholar] [CrossRef]
- Jayabal, P.; Manoj, N.; Nepal, M.; Shen, Y.; Che, R.; Turkson, J.; Fei, P. Involvement of FANCD2 in Energy Metabolism via ATP5α. Sci. Rep. 2017, 7, 4921. [Google Scholar] [CrossRef]
- Ciccia, A.; Ling, C.; Coulthard, R.; Yan, Z.; Xue, Y.; Meetei, A.R.; Laghmani, E.H.; Joenje, H.; McDonald, N.; de Winter, J.P.; et al. Identification of FAAP24, a Fanconi Anemia Core Complex Protein that Interacts with FANCM. Mol. Cell 2007, 25, 331–343. [Google Scholar] [CrossRef]
- Kaddar, T.; Carreau, M. Fanconi Anemia Proteins and Their Interacting Partners: A Molecular Puzzle. Anemia 2012, 2012, 425814. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.R.; Saro, D.; Ali, A.M.; Zheng, X.-F.; Du, C.-H.; Killen, M.W.; Sachpatzidis, A.; Wahengbam, K.; Pierce, A.J.; Xiong, Y.; et al. MHF1-MHF2, a Histone-Fold-Containing Protein Complex, Participates in the Fanconi Anemia Pathway via FANCM. Mol. Cell 2010, 37, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Delannoy, M.; Ling, C.; Daee, D.; Osman, F.; Muniandy, P.A.; Shen, X.; Oostra, A.B.; Du, H.; Steltenpool, J.; et al. A Histone-Fold Complex and FANCM Form a Conserved DNA-Remodeling Complex to Maintain Genome Stability. Mol. Cell 2010, 37, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, S.; Rajendra, E.; Alcón, P.; O’Reilly, F.J.; Chorev, D.; Maslen, S.; Degliesposti, G.; Russo, C.J.; He, S.; Hill, C.H.; et al. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 2019, 575, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef]
- Wang, X.; Kennedy, R.D.; Ray, K.; Stuckert, P.; Ellenberger, T.; D’Andrea, A.D. Chk1-Mediated Phosphorylation of FANCE Is Required for the Fanconi Anemia/BRCA Pathway. Mol. Cell. Biol. 2007, 27, 3098–3108. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Romick-Rosendale, L.E.; Lui, V.W.; Grandis, J.R.; Wells, S.I. The Fanconi anemia pathway: Repairing the link between DNA damage and squamous cell carcinoma. Mutat. Res. Mol. Mech. Mutagen. 2013, 743–744, 78–88. [Google Scholar] [CrossRef]
- Ishiai, M.; Kitao, H.; Smogorzewska, A.; Tomida, J.; Kinomura, A.; Uchida, E.; Saberi, A.; Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T.; et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2008, 15, 1138–1146. [Google Scholar] [CrossRef]
- Meetei, A.R.; De Winter, J.P.; Medhurst, A.L.; Wallisch, M.; Waisfisz, Q.; Van De Vrugt, H.J.; Oostra, A.B.; Yan, Z.; Ling, C.; Bishop, C.E.; et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003, 35, 165–170. [Google Scholar] [CrossRef]
- Sareen, A.; Chaudhury, I.; Adams, N.; Sobeck, A. Fanconi anemia proteins FANCD2 and FANCI exhibit different DNA damage responses during S-phase. Nucleic Acids Res. 2012, 40, 8425–8439. [Google Scholar] [CrossRef] [PubMed]
- Rickman, K.A.; Lach, F.; Abhyankar, A.; Donovan, F.; Sanborn, E.M.; Kennedy, J.A.; Sougnez, C.; Gabriel, S.B.; Elemento, O.; Chandrasekharappa, S.C.; et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase Necessary for FANCD2 and FANCI Ubiquitination, Causes FA-T Subtype of Fanconi Anemia. Cell Rep. 2015, 12, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Adachi, D.; Oda, T.; Yagasaki, H.; Nakasato, K.; Taniguchi, T.; D’Andrea, A.D.; Asano, S.; Yamashita, T. Heterogeneous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Hum. Mol. Genet. 2002, 11, 3125–3134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xue, Y.; Li, Y.; Guo, R.; Ling, C.; Wang, W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum. Mol. Genet. 2008, 17, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, N.J.; Knoll, A.; Puchta, H. The nuclease FAN1 is involved in DNA crosslink repair in Arabidopsis thaliana independently of the nuclease MUS81. Nucleic Acids Res. 2015, 43, 3653–3666. [Google Scholar] [CrossRef] [PubMed]
- Douwel, D.K.; Boonen, R.A.C.M.; Long, D.T.; Szypowska, A.A.; Räschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 Acts in Unhooking DNA Interstrand Crosslinks in Cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef]
- Murina, O.; von Aesch, C.; Karakus, U.; Ferretti, L.P.; Bolck, H.A.; Hänggi, K.; Sartori, A.A. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014, 7, 1030–1038. [Google Scholar] [CrossRef]
- Yamamoto, K.N.; Kobayashi, S.; Tsuda, M.; Kurumizaka, H.; Takata, M.; Kono, K.; Jiricny, J.; Takeda, S.; Hirota, K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 6492–6496. [Google Scholar] [CrossRef]
- Mason, J.M.; Sekiguchi, J.M. Snm1B/Apollo functions in the Fanconi anemia pathway in response to DNA interstrand crosslinks. Hum. Mol. Genet. 2011, 20, 2549–2559. [Google Scholar] [CrossRef]
- Benitez, A.; Yuan, F.; Nakajima, S.; Wei, L.; Qian, L.; Myers, R.; Hu, J.J.; Lan, L.; Zhang, Y. Damage-dependent regulation of MUS81-EME1 by Fanconi anemia complementation group A protein. Nucleic Acids Res. 2013, 42, 1671–1683. [Google Scholar] [CrossRef]
- Bluteau, D.; Masliah-Planchon, J.; Clairmont, C.; Rousseau, A.; Ceccaldi, R.; D’Enghien, C.D.; Bluteau, O.; Cuccuini, W.; Gachet, S.; De Latour, R.P.; et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J. Clin. Investig. 2016, 126, 3580–3584. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Dudimah, F.; Zhang, J.; Pickering, A.; Paneerselvam, J.; Palrasu, M.; Wang, H.; Fei, P. Recruitment of DNA polymerase eta by FANCD2 in the early response to DNA damage. Cell Cycle 2013, 12, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; D’Andrea, A.; Niedernhofer, L.J. Mouse models of Fanconi anemia. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 668, 133–140. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.-S.; Pondarre, C.; et al. Bone Marrow Failure in Fanconi Anemia Is Triggered by an Exacerbated p53/p21 DNA Damage Response that Impairs Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Ameziane, N.; Waisfisz, Q.; De Winter, J.P.; Harris, R.; Taniguchi, T.; D’Andrea, A.; Hodgson, S.V.; Mathew, C.; Joenje, H. Bi-allelic silencing of the Fanconi anaemia gene FANCF in acute myeloid leukaemia. Br. J. Haematol. 2003, 123, 469–471. [Google Scholar] [CrossRef]
- Alter, B.P. Cancer in Fanconi anemia, 1927–2001. Cancer 2003, 97, 425–440. [Google Scholar] [CrossRef]
- Rosenberg, P.S.; Alter, B.P.; Ebell, W. Cancer risks in Fanconi anemia: Findings from the German Fanconi Anemia Registry. Haematologica 2008, 93, 511–517. [Google Scholar] [CrossRef]
- Ruiz-Torres, S.; Brusadelli, M.G.; Witte, D.P.; Wikenheiser-Brokamp, K.A.; Sauter, S.; Nelson, A.S.; Sertorio, M.; Chlon, T.M.; Lane, A.; Mehta, P.A.; et al. Inherited DNA Repair Defects Disrupt the Structure and Function of Human Skin. Cell Stem Cell 2021, 28, 424–435.e6. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Savage, S.A.; Quint, W.G.; De Koning, M.N.; Schiffman, M. Squamous cell carcinomas in patients with Fanconi anemia and dyskeratosis congenita: A search for human papillomavirus. Int. J. Cancer 2013, 133, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Sauter, S.; Kovacic, M.B.; Nelson, A.S.; Myers, K.C.; Mehta, P.A.; Davies, S.M.; Wells, S.I. Risk of Human Papillomavirus Infection in Cancer-Prone Individuals: What We Know. Viruses 2018, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Lane, A.N.; Higashi, R.M.; Fan, T.W.M. Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. Genes Dis. 2019, 7, 185–198. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef]
- Abad, E.; Samino, S.; Grodzicki, R.L.; Pagano, G.; Trifuoggi, M.; Graifer, D.; Potesil, D.; Zdrahal, Z.; Yanes, O.; Lyakhovich, A. Identification of metabolic changes leading to cancer susceptibility in Fanconi anemia cells. Cancer Lett. 2021, 503, 185–196. [Google Scholar] [CrossRef]
- VanderWerf, S.M.; Svahn, J.; Olson, S.; Rathbun, R.K.; Harrington, C.; Yates, J.; Keeble, W.; Anderson, D.C.; Anur, P.; Pereira, N.F.; et al. TLR8-dependent TNF-α overexpression in Fanconi anemia group C cells. Blood 2009, 114, 5290–5298. [Google Scholar] [CrossRef]
- Zhao, X.; Brusadelli, M.G.; Sauter, S.L.; Kovacic, M.B.; Zhang, W.; Romick-Rosendale, L.E.; Lambert, P.F.; Setchell, K.D.; Wells, S.I. Lipidomic Profiling Links the Fanconi Anemia Pathway to Glycosphingolipid Metabolism in Head and Neck Cancer Cells. Clin. Cancer Res. 2018, 24, 2700–2709. [Google Scholar] [CrossRef]
- Nepal, M.; Ma, C.; Xie, G.; Jia, W.; Fei, P. Fanconi Anemia complementation group C protein in metabolic disorders. Aging 2018, 10, 1506–1522. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Uber den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Tseng, L.-M.; Lee, H.-C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef]
- Lee, H.-C. Somatic alterations in mitochondrial DNA and mitochondrial dysfunction in gastric cancer progression. World J. Gastroenterol. 2014, 20, 3950–3959. [Google Scholar] [CrossRef]
- Cuccarolo, P.; Viaggi, S.; Degan, P. New insights into redox response modulation in Fanconi’s anemia cells by hydrogen peroxide and glutathione depletors. FEBS J. 2012, 279, 2479–2494. [Google Scholar] [CrossRef]
- Kumari, U.; Jun, W.Y.; Bay, B.H.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi Anemia cells. Oncogene 2013, 33, 165–172. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.S.; Leung, K.S.; Hicks, M.J.; Hastings, P.J.; Youssoufian, H.; Plon, S.E. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J. Cell Biol. 2006, 175, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Pallardó, F.V.; Lloret, A.; Lebel, M.; D’Ischia, M.; Cogger, V.C.; Le Couteur, D.; Gadaleta, M.N.; Castello, G.; Pagano, G. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: Ataxia-Telangiectasia, Down Syndrome, Fanconi Anaemia and Werner Syndrome. Biogerontology 2010, 11, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.; Rajendran, A.; Mohan, S.; Raj, R.; Vundinti, B.R. Mitochondrial DNA variations and mitochondrial dysfunction in Fanconi anemia. PLoS ONE 2020, 15, e0227603. [Google Scholar] [CrossRef] [PubMed]
- Milletti, G.; Strocchio, L.; Pagliara, D.; Girardi, K.; Carta, R.; Mastronuzzi, A.; Locatelli, F.; Nazio, F. Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition. Cancers 2020, 12, 2684. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Lyakhovich, A. Damaged mitochondria and overproduction of ROS in Fanconi anemia cells. Rare Dis. 2013, 1, e24048. [Google Scholar] [CrossRef]
- Tu, V.; Ayari, A.; O’Connor, R. Beyond the Lactate Paradox: How Lactate and Acidity Impact T Cell Therapies against Cancer. Antibodies 2021, 10, 25. [Google Scholar] [CrossRef]
- Pagano, G.; Tiano, L.; Pallardó, F.V.; Lyakhovich, A.; Mukhopadhyay, S.S.; Di Bartolomeo, P.; Zatterale, A.; Trifuoggi, M. Re-definition and supporting evidence toward Fanconi Anemia as a mitochondrial disease: Prospects for new design in clinical management. Redox Biol. 2021, 40, 101860. [Google Scholar] [CrossRef]
- DiMauro, S.; Hirano, M. Pathogenesis and Treatment of Mitochondrial Disorders. Adv. Exp. Med. Biol. 2009, 652, 139–170. [Google Scholar] [CrossRef]
- Chatla, S.; Du, W.; Wilson, A.F.; Meetei, A.R.; Pang, Q. Fancd2-deficient hematopoietic stem and progenitor cells depend on augmented mitochondrial translation for survival and proliferation. Stem Cell Res. 2019, 40, 101550. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.R.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A.; et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P.; Miotto, B.; Saint-Ruf, C.; Said, M.; Barra, V.; Nähse, V.; Ravera, S.; Cappelli, E.; Naim, V. FANCD2 modulates the mitochondrial stress response to prevent common fragile site instability. Commun. Biol. 2021, 4, 127. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.S.; Daugherity, E.K.; Karambizi, D.I.; Cummings, B.P.; Behling-Kelly, E.; Schaefer, D.M.W.; Southard, T.L.; McFadden, J.W.; Weiss, R.S. Sex-specific hepatic lipid and bile acid metabolism alterations in Fancd2-deficient mice following dietary challenge. J. Biol. Chem. 2019, 294, 15623–15637. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Wang, Y. Bulky DNA Lesions Induced by Reactive Oxygen Species. Chem. Res. Toxicol. 2008, 21, 276–281. [Google Scholar] [CrossRef]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.-C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxid. Med. Cell. Longev. 2021, 2021, 9965916. [Google Scholar] [CrossRef]
- Park, S.-J.; Ciccone, S.L.M.; Beck, B.D.; Hwang, B.; Freie, B.; Clapp, D.W.; Lee, S.-H. Oxidative Stress/Damage Induces Multimerization and Interaction of Fanconi Anemia Proteins. J. Biol. Chem. 2004, 279, 30053–30059. [Google Scholar] [CrossRef]
- Beswick, R.A.; Dorrance, A.M.; Leite, R.; Webb, R.C. NADH/NADPH Oxidase and Enhanced Superoxide Production in the Mineralocorticoid Hypertensive Rat. Hypertension 2001, 38, 1107–1111. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Xing, C.; Jiang, F.; Wang, X.; Guo, H.; Nan, J.; Qian, L.; Yang, P.; Lin, J.; Li, M.; et al. Benzo[a]pyrene induces oxidative stress and endothelial progenitor cell dysfunction via the activation of the NF-κB pathway. Int. J. Mol. Med. 2013, 31, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Columbaro, M.; Capanni, C.; Bartolucci, M.; Panfoli, I.; Morelli, A.; Dufour, C.; Cappelli, E.; et al. Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie 2013, 95, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Adam, Z.; Rani, R.; Zhang, X.; Pang, Q. Oxidative Stress in Fanconi Anemia Hematopoiesis and Disease Progression. Antioxid. Redox Signal. 2008, 10, 1909–1921. [Google Scholar] [CrossRef]
- Degan, P.; Bonassi, S.; De Caterina, M.; Korkina, L.G.; Pinto, L.; Scopacasa, F.; Zatterale, A.; Calzone, R.; Pagano, G. In vivo accumulation of 8-hydroxy-2’-deoxyguanosine in DNA correlates with release of reactive oxygen species in Fanconi’s anaemia families. Carcinogenesis 1995, 16, 735–742. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Shen, X.; Li, L. Mutagenic repair of DNA interstrand crosslinks. Environ. Mol. Mutagen. 2010, 51, 493–499. [Google Scholar] [CrossRef]
- Leto, T.L.; Morand, S.; Hurt, D.; Ueyama, T. Targeting and Regulation of Reactive Oxygen Species Generation by Nox Family NADPH Oxidases. Antioxid. Redox Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef]
- Yang, H.-C.; Cheng, M.-L.; Ho, H.-Y.; Chiu, D.T.-Y. The microbicidal and cytoregulatory roles of NADPH oxidases. Microbes Infect. 2011, 13, 109–120. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Komori, A.; Yatsunami, J.; Suganuma, M.; Okabe, S.; Abe, S.; Sakai, A.; Sasaki, K.; Fujiki, H. Tumor necrosis factor acts as a tumor promoter in BALB/3T3 cell transformation. Cancer Res. 1993, 53, 1982–1985. [Google Scholar] [PubMed]
- Hudson, J.D.; Shoaibi, M.A.; Maestro, R.; Carnero, A.; Hannon, G.J.; Beach, D.H. A Proinflammatory Cytokine Inhibits P53 Tumor Suppressor Activity. J. Exp. Med. 1999, 190, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.; Strieter, R. The Role of CXC Chemokines in the Regulation of Angiogenesis. Chem. Immunol. Allergy 1999, 72, 86–101. [Google Scholar] [CrossRef]
- Raman, D.; Baugher, P.J.; Thu, Y.M.; Richmond, A. Role of chemokines in tumor growth. Cancer Lett. 2007, 256, 137–165. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000, 60, 184. [Google Scholar]
- Yau, T.M. Mutagenicity and cytotoxicity of malonaldehyde in mammalian cells. Mech. Ageing Dev. 1979, 11, 137–144. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, X.S.; Wei, Y.; Polyanskaya, S.A.; Iyer, S.V.; Jung, M.; Lach, F.P.; Adelman, E.R.; Klingbeil, O.; Milazzo, J.P.; et al. Transcriptional Silencing of ALDH2 Confers a Dependency on Fanconi Anemia Proteins in Acute Myeloid Leukemia. Cancer Discov. 2021, 11, 2300–2315. [Google Scholar] [CrossRef]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 668, 4–10. [Google Scholar] [CrossRef]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Hadjur, S.; Ung, K.; Wadsworth, L.; Dimmick, J.; Rajcan-Separovic, E.; Scott, R.W.; Buchwald, M.; Jirik, F.R. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood 2001, 98, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Hira, A.; Yabe, H.; Yoshida, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Nakamura, J.; Kojima, S.; et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 2013, 122, 3206–3209. [Google Scholar] [CrossRef] [PubMed]
- Ridpath, J.R.; Nakamura, A.; Tano, K.; Luke, A.M.; Sonoda, E.; Arakawa, H.; Buerstedde, J.-M.; Gillespie, D.; Sale, J.; Yamazoe, M.; et al. Cells Deficient in the FANC/BRCA Pathway Are Hypersensitive to Plasma Levels of Formaldehyde. Cancer Res. 2007, 67, 11117–11122. [Google Scholar] [CrossRef]
- Hodskinson, M.R.; Bolner, A.; Sato, K.; Kamimae-Lanning, A.; Rooijers, K.; Witte, M.; Mahesh, M.; Silhan, J.; Petek, M.; Williams, D.M.; et al. Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature 2020, 579, 603–608. [Google Scholar] [CrossRef]
- Morellato, A.E.; Umansky, C.; Pontel, L.B. The toxic side of one-carbon metabolism and epigenetics. Redox Biol. 2020, 40, 101850. [Google Scholar] [CrossRef] [PubMed]
- Pontel, B.L.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Heit, C.; Dong, H.; Chen, Y.; Thompson, D.C.; Deitrich, R.A.; Vasiliou, V.K. The Role of CYP2E1 in Alcohol Metabolism and Sensitivity in the Central Nervous System. In Cytochrome P450 2E1: Its Role in Disease and Drug Metabolism; Springer: Berlin/Heidelberg, Germany, 2013; Volume 67, pp. 235–247. [Google Scholar] [CrossRef]
- Muñoz-Clares, R.A.; González-Segura, L.; Riveros-Rosas, H.; Julián-Sánchez, A. Amino acid residues that affect the basicity of the catalytic glutamate of the hydrolytic aldehyde dehydrogenases. Chem. Interact. 2015, 234, 45–58. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines 2020, 8, 50. [Google Scholar] [CrossRef]
- Lindahl, R.; Petersen, D.R. Lipid aldehyde oxidation as a physiological role for class 3 aldehyde dehydrogenases. Biochem. Pharmacol. 1991, 41, 1583–1587. [Google Scholar] [CrossRef]
- Labrecque, J.; Dumas, F.; Lacroix, A.; Bhat, P.V. A novel isoenzyme of aldehyde dehydrogenase specifically involved in the biosynthesis of 9-cis and all-trans retinoic acid. Biochem. J. 1995, 305, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, O.; Parkkila, S.; Juvonen, R.O.; Viitala, K.; Gelboin, H.V.; Pasanen, M. Cytochromes P450 2A6, 2E1, and 3A and production of protein-aldehyde adducts in the liver of patients with alcoholic and non-alcoholic liver diseases. J. Hepatol. 2000, 33, 893–901. [Google Scholar] [CrossRef]
- Jia, J.; Parikh, H.; Xiao, W.; Hoskins, J.W.; Pflicke, H.; Liu, X.; Collins, I.; Zhou, W.; Wang, Z.; Powell, J.; et al. An integrated transcriptome and epigenome analysis identifies a novel candidate gene for pancreatic cancer. BMC Med. Genom. 2013, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Saw, Y.-T.; Yang, J.; Ng, S.-K.; Liu, S.; Singh, S.; Singh, M.; Welch, W.R.; Tsuda, H.; Fong, W.-P.; Thompson, D.; et al. Characterization of aldehyde dehydrogenase isozymes in ovarian cancer tissues and sphere cultures. BMC Cancer 2012, 12, 329. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-H.; Wang, Y.-C.; Chou, Y.-C.; Yu, M.-H.; Chao, T.-K. The combination of aldehyde dehydrogenase 1 (ALDH1) and CD44 is associated with poor outcomes in endometrial cancer. PLoS ONE 2018, 13, e0206685. [Google Scholar] [CrossRef]
- Yao, T.; Wu, Z.; Liu, Y.; Rao, Q.; Lin, Z. Aldehyde dehydrogenase 1 (ALDH1) positivity correlates with poor prognosis in cervical cancer. J. Int. Med. Res. 2014, 42, 1038–1042. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.-H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef]
- van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.T.; Lippitt, J.M.; Guzmán-Ramírez, N.; Hamdy, F.C.; Eaton, C.L.; Thalmann, G.N.; Cecchini, M.G.; et al. High Aldehyde Dehydrogenase Activity Identifies Tumor-Initiating and Metastasis-Initiating Cells in Human Prostate Cancer. Cancer Res. 2010, 70, 5163–5173. [Google Scholar] [CrossRef]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck 2010, 32, 1195–1201. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res. Treat. 2011, 133, 75–87. [Google Scholar] [CrossRef]
- Dingler, F.A.; Wang, M.; Mu, A.; Millington, C.L.; Oberbeck, N.; Watcham, S.; Pontel, L.B.; Kamimae-Lanning, A.N.; Langevin, F.; Nadler, C.; et al. Two Aldehyde Clearance Systems Are Essential to Prevent Lethal Formaldehyde Accumulation in Mice and Humans. Mol. Cell 2020, 80, 996–1012.e9. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118.e15. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2009, 5, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Yabe, M.; Koike, T.; Ohtsubo, K.; Imai, E.; Morimoto, T.; Takakura, H.; Koh, K.; Yoshida, K.; Ogawa, S.; Ito, E.; et al. Associations of complementation group, ALDH2 genotype, and clonal abnormalities with hematological outcome in Japanese patients with Fanconi anemia. Ann. Hematol. 2018, 98, 271–280. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2019, 122, 4–22. [Google Scholar] [CrossRef]
- Birklé, S.; Zeng, G.; Gao, L.; Yu, R.; Aubry, J. Role of tumor-associated gangliosides in cancer progression. Biochimie 2003, 85, 455–463. [Google Scholar] [CrossRef]
- Rauter, A.P.; Lindhorst, T.K. Carbohydrate Chemistry; Royal Society of Chemistry: Cambridge, UK, 2011; Volume 37. [Google Scholar]
- Krengel, U.; Bousquet, P.A. Molecular Recognition of Gangliosides and Their Potential for Cancer Immunotherapies. Front. Immunol. 2014, 5, 325. [Google Scholar] [CrossRef]
- Kim, S.-J.; Chung, T.-W.; Choi, H.-J.; Kwak, C.-H.; Song, K.-H.; Suh, S.-J.; Kwon, K.-M.; Chang, Y.-C.; Park, Y.-G.; Chang, H.W.; et al. Ganglioside GM3 participates in the TGF-β1-induced epithelial–mesenchymal transition of human lens epithelial cells. Biochem. J. 2012, 449, 241–251. [Google Scholar] [CrossRef]
- Guan, F.; Handa, K.; Hakomori, S.-I. Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc. Natl. Acad. Sci. USA 2009, 106, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.L.H.; Sanders, M.A.; Patel, K.; Dietrich, R.; Noonan, R.J.; Lach, F.P.; White, R.R.; Goldfarb, A.; Hadi, K.; Edwards, M.M.; et al. Fanconi Anemia Pathway Deficiency Drives Copy Number Variation in Squamous Cell Carcinomas. bioRxiv 2021. [Google Scholar] [CrossRef]
- Boros, F.A.; Vécsei, L. Immunomodulatory Effects of Genetic Alterations Affecting the Kynurenine Pathway. Front. Immunol. 2019, 10, 2570. [Google Scholar] [CrossRef] [PubMed]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.-Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [PubMed]
- van der Goot, A.T.; Nollen, E.A. Tryptophan metabolism: Entering the field of aging and age-related pathologies. Trends Mol. Med. 2013, 19, 336–344. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Liu, X.-H.; Zhai, X.-Y. Role of tryptophan metabolism in cancers and therapeutic implications. Biochimie 2021, 182, 131–139. [Google Scholar] [CrossRef]
- Economopoulou, P.; Kladi-Skandali, A.; Strati, A.; Koytsodontis, G.; Kyrodimos, E.; Giotakis, E.; Maragoudakis, P.; Gagari, E.; Maratou, E.; Dimitriadis, G.; et al. Prognostic impact of indoleamine 2,3-dioxygenase 1 (IDO1) mRNA expression on circulating tumour cells of patients with head and neck squamous cell carcinoma. ESMO Open 2020, 5, e000646. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients with Advanced Solid Tumors: Phase I Results from a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Ponte, F.; Sousa, R.; Fernandes, A.P.; Gonçalves, C.; Barbot, J.; Carvalho, F.; Porto, B. Improvement of genetic stability in lymphocytes from Fanconi anemia patients through the combined effect of α-lipoic acid and N-acetylcysteine. Orphanet J. Rare Dis. 2012, 7, 28. [Google Scholar] [CrossRef]
- Zhang, Q.-S.; Eaton, L.; Snyder, E.R.; Houghtaling, S.; Mitchell, J.B.; Finegold, M.; Van Waes, C.; Grompe, M. Tempol Protects against Oxidative Damage and Delays Epithelial Tumor Onset in Fanconi Anemia Mice. Cancer Res. 2008, 68, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi Anemia Links Reactive Oxygen Species to Insulin Resistance and Obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.A.; Braune, A.; Hölzlwimmer, G.; Quintanilla-Fend, L.; Haller, D. Quercetin Inhibits TNF-Induced NF-κB Transcription Factor Recruitment to Proinflammatory Gene Promoters in Murine Intestinal Epithelial Cells. J. Nutr. 2007, 137, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, L.; Li, S.; Ye, D.; Yang, L.; Liu, Q.; Zhao, Z.; Cai, Q.; Tan, J.; Li, X. Quercetin Inhibits Breast Cancer Stem Cells via Downregulation of Aldehyde Dehydrogenase 1A1 (ALDH1A1), Chemokine Receptor Type 4 (CXCR4), Mucin 1 (MUC1), and Epithelial Cell Adhesion Molecule (EpCAM). Med. Sci. Monit. 2018, 24, 412–420. [Google Scholar] [CrossRef]
- Kazachkov, M.; Chen, K.; Babiy, S.; Yu, P.H. Evidence for in Vivo Scavenging by Aminoguanidine of Formaldehyde Produced via Semicarbazide-Sensitive Amine Oxidase-Mediated Deamination. J. Pharmacol. Exp. Ther. 2007, 322, 1201–1207. [Google Scholar] [CrossRef]
- Zhang, Q.-S.; Tang, W.; Deater, M.; Phan, N.; Marcogliese, A.N.; Li, H.; Al-Dhalimy, M.; Major, A.; Olson, S.; Monnat, R.J.; et al. Metformin improves defective hematopoiesis and delays tumor formation in Fanconi anemia mice. Blood 2016, 128, 2774–2784. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Marquez-Loza, L.; Eaton, L.; Duncan, A.W.; Goldman, D.C.; Anur, P.; Watanabe-Smith, K.; Keaney Rathbun, R.; Fleming, W.H.; Bagby, G.C.; et al. Fancd2−/− mice have hematopoietic defects that can be partially corrected by resveratrol. Blood 2010, 116, 5140–5148. [Google Scholar] [CrossRef]
- Zhang, Q.-S.; Deater, M.; Schubert, K.; Marquez-Loza, L.; Pelz, C.; Sinclair, D.A.; Grompe, M. The Sirt1 activator SRT3025 expands hematopoietic stem and progenitor cells and improves hematopoiesis in Fanconi anemia mice. Stem Cell Res. 2015, 15, 130–140. [Google Scholar] [CrossRef]
- Zhang, Q.-S.; Loza, L.M.; Sheehan, A.M.; Watanabe-Smith, K.; Eaton, L.; Bs, E.B.; Major, A.; Bs, K.S.; Bs, M.D.; Joseph, E.; et al. Evaluation of resveratrol and N -acetylcysteine for cancer chemoprevention in a Fanconi anemia murine model. Pediatr. Blood Cancer 2013, 61, 740–742. [Google Scholar] [CrossRef][Green Version]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef]
- Yu, H.; Zhong, X.; Gao, P.; Shi, J.; Wu, Z.; Guo, Z.; Wang, Z.; Song, Y. The Potential Effect of Metformin on Cancer: An Umbrella Review. Front. Endocrinol. 2019, 10, 617. [Google Scholar] [CrossRef] [PubMed]
- Clifford, G.M.; Smith, J.S.; Plummer, M.; Muñoz, N.; Franceschi, S. Human papillomavirus types in invasive cervical cancer worldwide: A meta-analysis. Br. J. Cancer 2003, 88, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Broker, T.R.; Steinberg, B. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 2010, 118, 422–449. [Google Scholar] [CrossRef] [PubMed]
- Hasche, D.; Vinzón, S.E.; Rösl, F. Cutaneous Papillomaviruses and Non-melanoma Skin Cancer: Causal Agents or Innocent Bystanders? Front. Microbiol. 2018, 9, 874. [Google Scholar] [CrossRef]
- Burchell, A.N.; Winer, R.L.; de Sanjosé, S.; Franco, E. Chapter 6: Epidemiology and transmission dynamics of genital HPV infection. Vaccine 2006, 24, S52–S61. [Google Scholar] [CrossRef]
- Althoff, K.N.; Paul, P.; Burke, A.E.; Viscidi, R.; Sangaramoorthy, M.; Gravitt, P.E. Correlates of Cervicovaginal Human Papillomavirus Detection in Perimenopausal Women. J. Women’s Health 2009, 18, 1341–1346. [Google Scholar] [CrossRef]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef]
- Bosch, F.X.; Lorincz, A.; Munoz, N.; Meijer, C.J.L.M.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32 (Suppl. S1), 7–15. [Google Scholar] [CrossRef]
- Hagensee, M.E.; Yaegashi, N.; Galloway, D.A. Self-assembly of human papillomavirus type 1 capsids by expression of the L1 protein alone or by coexpression of the L1 and L2 capsid proteins. J. Virol. 1993, 67, 315–322. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A.; Khurana, S. Multiple Roles of Brd4 in the Infectious Cycle of Human Papillomaviruses. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P. The Human Papillomavirus 16 E6 Protein Binds to Fas-associated Death Domain and Protects Cells from Fas-triggered Apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Yim, E.-K.; Park, J.-S. The Role of HPV E6 and E7 Oncoproteins in HPV-associated Cervical Carcinogenesis. Cancer Res. Treat. 2005, 37, 319–324. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef]
- Rashid, N.N.; Rothan, H.A.; Yusoff, M.S.M. The association of mammalian DREAM complex and HPV16 E7 proteins. Am. J. Cancer Res. 2015, 5, 3525–3533. [Google Scholar]
- Seavey, S.E.; Holubar, M.; Saucedo, L.J.; Perry, M.E. The E7 Oncoprotein of Human Papillomavirus Type 16 Stabilizes p53 through a Mechanism Independent of p19 ARF. J. Virol. 1999, 73, 7590–7598. [Google Scholar] [CrossRef]
- Okun, M.M.; Day, P.M.; Greenstone, H.L.; Booy, F.P.; Lowy, D.R.; Schiller, J.T.; Roden, R.B.S. L1 Interaction Domains of Papillomavirus L2 Necessary for Viral Genome Encapsidation. J. Virol. 2001, 75, 4332–4342. [Google Scholar] [CrossRef]
- Sauter, S.; Zhang, X.; Romick-Rosendale, L.; Wells, S.; Myers, K.; Brusadelli, M.; Poff, C.; Brown, D.; Panicker, G.; Unger, E.; et al. Human Papillomavirus Oral- and Sero- Positivity in Fanconi Anemia. Cancers 2021, 13, 1368. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Laimins, L.A. FANCD2 Binds Human Papillomavirus Genomes and Associates with a Distinct Set of DNA Repair Proteins to Regulate Viral Replication. mBio 2017, 8, e02340-16. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi’s anemia and malignancies. Am. J. Hematol. 1996, 53, 99–110. [Google Scholar] [CrossRef]
- Chlon, T.; Hoskins, E.E.; Mayhew, C.; Wikenheiser-Brokamp, K.A.; Davies, S.M.; Mehta, P.; Myers, K.C.; Wells, J.M.; Wells, S.I. High-Risk Human Papillomavirus E6 Protein Promotes Reprogramming of Fanconi Anemia Patient Cells through Repression of p53 but Does Not Allow for Sustained Growth of Induced Pluripotent Stem Cells. J. Virol. 2014, 88, 11315–11326. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morris, T.A.; Higginbotham, J.M.; Spardy, N.; Cha, E.; Kelly, P.; Williams, D.A.; Wikenheiser-Brokamp, K.A.; Duensing, S.; Wells, S.I. Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene 2009, 28, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Shin, M.-K.; Lambert, P.F. High incidence of female reproductive tract cancers in FA-deficient HPV16-transgenic mice correlates with E7’s induction of DNA damage response, an activity mediated by E7’s inactivation of pocket proteins. Oncogene 2014, 33, 3383–3391. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Pan, Y.; Savage, S.A.; Pinto, L.A. Antibody response to human papillomavirus vaccine in subjects with inherited bone marrow failure syndromes. Vaccine 2013, 32, 1169–1173. [Google Scholar] [CrossRef]
- De Araujo, M.R.; Rubira-Bullen, I.R.F.; Santos, C.F.; Dionísio, T.J.; Bonfim, C.M.S.; De Marco, L.; Gillio-Tos, A.; Merletti, F. High prevalence of oral human papillomavirus infection in Fanconi’s anemia patients. Oral Dis. 2011, 17, 572–576. [Google Scholar] [CrossRef]
- Han, T.-J.; Lee, C.-H.; Yoo, C.-W.; Shin, H.-J.; Park, H.-J.; Cho, K.H.; Park, J.-Y.; Choi, S.-W.; Kim, J.-Y. Synchronous multifocal HPV-related neoplasm involving both the genital tract and the head-and-neck area: A case report of Fanconi anemia. Radiother. Oncol. 2009, 92, 138–141. [Google Scholar] [CrossRef]
- Katzenellenbogen, R.A.; Carter, J.J.; Stern, J.E.; Kovacic, M.S.B.; Mehta, P.A.; Sauter, S.L.; Galloway, D.A.; Winer, R.L. Skin and Mucosal Human Papillomavirus Seroprevalence in Persons with Fanconi Anemia. Clin. Vaccine Immunol. 2015, 22, 413–420. [Google Scholar] [CrossRef]
- van Zeeburg, H.J.T.; Snijders, P.J.F.; Wu, T.; Gluckman, E.; Soulier, J.; Surralles, J.; Castella, M.; van der Wal, J.E.; Wennerberg, J.; Califano, J.; et al. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J. Natl. Cancer Inst. 2008, 100, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Winer, R.L.; Huang, C.E.; Cherne, S.; Stern, J.E.; Kovacic, M.S.B.; Mehta, P.A.; Sauter, S.L.; Galloway, D.A.; Katzenellenbogen, R.A. Detection of human papillomavirus in the oral cavities of persons with Fanconi anemia. Oral Dis. 2014, 21, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Park, J.W.; Pitot, H.C.; Lambert, P.F. Loss of Dependence on Continued Expression of the Human Papillomavirus 16 E7 Oncogene in Cervical Cancers and Precancerous Lesions Arising in Fanconi Anemia Pathway-Deficient Mice. mBio 2016, 7, e00628-16. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morreale, R.J.; Werner, S.P.; Higginbotham, J.M.; Laimins, L.A.; Lambert, P.F.; Brown, D.R.; Gillison, M.L.; Nuovo, G.J.; Witte, D.P.; et al. The Fanconi Anemia Pathway Limits Human Papillomavirus Replication. J. Virol. 2012, 86, 8131–8138. [Google Scholar] [CrossRef] [PubMed]
- Banks, L.; Spence, P.; Androphy, E.; Hubbert, N.; Matlashewski, G.; Murray, A.; Crawford, L. Identification of Human Papillomavirus Type 18 E6 Polypeptide in Cells Derived from Human Cervical Carcinomas. J. Gen. Virol. 1987, 68, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; DiMaio, D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 12513–12518. [Google Scholar] [CrossRef]
- Martínez-Ramírez, I.; Carrillo-García, A.; Contreras-Paredes, A.; Ortiz-Sánchez, E.; Cruz-Gregorio, A.; Lizano, M. Regulation of Cellular Metabolism by High-Risk Human Papillomaviruses. Int. J. Mol. Sci. 2018, 19, 1839. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.-Z.; Liu, Y.; Wang, K.; Ding, W.; Wang, H.; Liu, X.; Zhou, S.; Lu, X.-C.; Yang, H.-B.; et al. Nuclear lactate dehydrogenase A senses ROS to produce α-hydroxybutyrate for HPV-induced cervical tumor growth. Nat. Commun. 2018, 9, 4429. [Google Scholar] [CrossRef]
- Lee, S.-A.; Ho, C.; Troxler, M.; Lin, C.-Y.; Chung, S.-H. Non-Metabolic Functions of PKM2 Contribute to Cervical Cancer Cell Proliferation Induced by the HPV16 E7 Oncoprotein. Viruses 2021, 13, 433. [Google Scholar] [CrossRef]
- Fleming, J.; Woo, J.; Moutasim, K.; Mellone, M.; Frampton, S.J.; Mead, A.; Ahmed, W.; Wood, O.; Robinson, H.; Ward, M.; et al. HPV, tumour metabolism and novel target identification in head and neck squamous cell carcinoma. Br. J. Cancer 2019, 120, 356–367. [Google Scholar] [CrossRef]
- Krupar, R.; Robold, K.; Gaag, D.; Spanier, G.; Kreutz, M.; Renner, K.; Hellerbrand, C.; Hofstaedter, F.; Bosserhoff, A.K. Immunologic and metabolic characteristics of HPV-negative and HPV-positive head and neck squamous cell carcinomas are strikingly different. Virchows Arch. 2014, 465, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Meng, X.; Ma, J.; Zheng, Y.; Wang, Q.; Wang, Y.; Shang, H. Human Papillomavirus 16 E6 Contributes HIF-1α Induced Warburg Effect by Attenuating the VHL-HIF-1α Interaction. Int. J. Mol. Sci. 2014, 15, 7974–7986. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H. Cellular life span and the Warburg effect. Exp. Cell Res. 2008, 314, 1923–1928. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Complex | Gene | Alternative Name | Function |
|---|---|---|---|
| Core complex | FANCA | FA core complex assembly required to mono-ubiquitinate FANCD2 and FANCI | |
| FANCB | FA core complex assembly required to mono-ubiquitinate FANCD2 and FANCI | ||
| FANCC | FA core complex assembly required to mono-ubiquitinate FANCD2 and FANCI | ||
| FANCE | FA core complex assembly required to mono-ubiquitinate FANCD2 and FANCI | ||
| FANCF | FA core complex assembly required to mono-ubiquitinate FANCD2 and FANCI | ||
| FANCG | XRCC9 | FA core complex assembly required to mono-ubiquitinate FANCD2 and FANCI | |
| FANCL | POG | E3 ubiquitin ligase for FANCD2 and FANCI mono-ubiquitination | |
| FANCM | Recognizes ICL lesions; Recruits the FA core complex and BLM helicase; Activates ATR-Chk1 signaling; 5′-3′ DNA helicase involved in the repair of Holliday junctions and replication forks | ||
| FANCT | UBE2T | E2 ubiquitin ligase for FANCD2 and FANCI mono-ubiquitination | |
| ID Complex | FANCD2 | Binds to FANCI; Recruits nucleases and TLS polymerases for DNA damage repair; Histone chaperone, fork protection | |
| FANCI | Binds FANCD2; Recruits DNA repair proteins | ||
| Downstream Effectors and DNA Repair Proteins | FANCD1 | BRCA2 | Controls DNA repair via HR and effector recruitment; Required for RAD51 loading and replication fork stabilization |
| FANCJ | BRIP1 | 3′-5′ DNA helicase; Essential for DNA repair via HR and TLS | |
| FANCN | PALB2 | Regulates BRCA2 localization to DNA damage sites; Required for DNA repair via HR | |
| FANCO | RAD51C | Required for DNA repair via HR | |
| FANCP | SLX4 | Endonuclease required for the resolution of Holliday junctions; Interacts with several nucleases, including FANCQ | |
| FANCQ | ERCC4/XPF | DNA repair endonuclease; Functions in DNA nucleotide excision repair | |
| FANCR | RAD51 | Recombinase required for DNA repair via HR by promoting homology search and strand invasion; Required for fork stabilization | |
| FANCS | BRCA1 | Required for DNA repair via HR and fork stabilization; Promotes end-resection; Ubiquitin ligase activity towards histone H2A and CtIP | |
| FANCU | XRCC2 | Required for DNA repair via HR; Stabilizes the levels of RAD51C and other RAD51 paralogs | |
| FANCV | REV7/MAD2L2 | Required for TLS repair | |
| FANCW | RFWD3 | E3 ligase required for DNA repair via HR; Facilitates removal of RPA and RAD51 from DNA damage sites |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chihanga, T.; Vicente-Muñoz, S.; Ruiz-Torres, S.; Pal, B.; Sertorio, M.; Andreassen, P.R.; Khoury, R.; Mehta, P.; Davies, S.M.; Lane, A.N.; et al. Head and Neck Cancer Susceptibility and Metabolism in Fanconi Anemia. Cancers 2022, 14, 2040. https://doi.org/10.3390/cancers14082040
Chihanga T, Vicente-Muñoz S, Ruiz-Torres S, Pal B, Sertorio M, Andreassen PR, Khoury R, Mehta P, Davies SM, Lane AN, et al. Head and Neck Cancer Susceptibility and Metabolism in Fanconi Anemia. Cancers. 2022; 14(8):2040. https://doi.org/10.3390/cancers14082040
Chicago/Turabian StyleChihanga, Tafadzwa, Sara Vicente-Muñoz, Sonya Ruiz-Torres, Bidisha Pal, Mathieu Sertorio, Paul R. Andreassen, Ruby Khoury, Parinda Mehta, Stella M. Davies, Andrew N. Lane, and et al. 2022. "Head and Neck Cancer Susceptibility and Metabolism in Fanconi Anemia" Cancers 14, no. 8: 2040. https://doi.org/10.3390/cancers14082040
APA StyleChihanga, T., Vicente-Muñoz, S., Ruiz-Torres, S., Pal, B., Sertorio, M., Andreassen, P. R., Khoury, R., Mehta, P., Davies, S. M., Lane, A. N., Romick-Rosendale, L. E., & Wells, S. I. (2022). Head and Neck Cancer Susceptibility and Metabolism in Fanconi Anemia. Cancers, 14(8), 2040. https://doi.org/10.3390/cancers14082040