
Acquired Mechanisms of Resistance to Osimertinib—The Next Challenge


Abstract
Simple Summary
Abstract
1. Introduction
2. EGFR-Dependent Mechanisms of Resistance
2.1. C797X Mutations
2.2. Less Common EGFR-Dependent Alterations
2.3. Fourth-Generation EGFR TKIs as a Strategy to Overcome Resistance to Osimertinib
2.3.1. EAI045
2.3.2. JBJ-04-125-02
2.3.3. CH7233163
2.3.4. BLU-945
2.3.5. Other Drugs
3. Histologic and Phenotypic Transformation
3.1. Small Cell Transformation
3.2. Squamous Cell Transformation
4. MET Amplification
5. HER2 Alterations
6. RET Alterations
7. BRAF Alterations
8. KRAS Mutations
9. PI3K Alterations
10. Cell Cycle Gene Alterations
11. AXL Overexpression
12. Insulin-like Growth Factor (IGF)-1 Receptor Activation
13. Epithelial-Mesenchymal Transition (EMT)
14. Other Rare Acquired Resistance Mechanisms
15. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.; Faivre-Finn, C.; Mok, T.; Reck, M.; Van Schil, P.; Hellmann, M.; et al. Correction to: “Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up”. Ann. Oncol. 2019, 30, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Andrini, E.; Sisi, M.; Rizzo, A.; Parisi, C.; Di Federico, A.; Gelsomino, F.; Ardizzoni, A. Beyond EGFR, ALK and ROS1: Current evidence and future perspectives on newly targetable oncogenic drivers in lung adenocarcinoma. Crit. Rev. Oncol. 2020, 156, 103119. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-H.; Yang, J.C.-H.; Mok, T.; Loong, H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 2018, 29, i3–i9. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Picone, A.; Ferraro, G.; Zanghì, M.; Toscano, G.; Giordano, A.; Adamo, V. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget 2015, 6, 26814–26825. [Google Scholar] [CrossRef]
- Midha, A.; Dearden, S.; McCormack, R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am. J. Cancer Res. 2015, 5, 2892–2911. [Google Scholar]
- Cote, M.L.; Haddad, R.; Edwards, D.J.; Atikukke, G.; Gadgeel, S.; Soubani, A.O.; Lonardo, F.; Bepler, G.; Schwartz, A.G.; Ethier, S.P. Frequency and Type of Epidermal Growth Factor Receptor Mutations in African Americans with Non-small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 627–630. [Google Scholar] [CrossRef]
- Karachaliou, N.; Fernandez-Bruno, M.; Bracht, J.W.P.; Rosell, R. EGFR first- and second-generation TKIs—there is still place for them in EGFR-mutant NSCLC patients. Transl. Cancer Res. 2018, 8, S23–S47. [Google Scholar] [CrossRef]
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef]
- Remon, J.; Steuer, C.; Ramalingam, S.; Felip, E. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann. Oncol. 2018, 29, i20–i27. [Google Scholar] [CrossRef]
- Lee, K.-O.; Cha, M.Y.; Kim, M.; Song, J.Y.; Lee, J.-H.; Kim, Y.H.; Lee, Y.-M.; Suh, K.H.; Son, J. Abstract LB-100: Discovery of HM61713 as an orally available and mutant EGFR selective inhibitor. In Proceedings of the Experimental and Molecular Therapeutics; American Association for Cancer Research: Philadelphia, PA, USA, 2014. [Google Scholar]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.-W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Yang, J.C.-H.; Yu, H.; Kim, S.-W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Jänne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhao, Y.; Shen, S.; Gu, L.; Niu, X.; Xu, Y.; Zhang, T.; Xiang, J.; Mao, X.; Lu, S. Durable Clinical Response of Lung Adenocarcinoma Harboring EGFR 19Del/T790M/in trans-C797S to Combination Therapy of First- and Third-Generation EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2019, 14, e157–e159. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, N.; Zhang, Y.; Li, L.; Han, R.; Zhu, M.; Feng, M.; Chen, H.; Lizaso, A.; Qin, T.; et al. Effective Treatment of Lung Adenocarcinoma Harboring EGFR-Activating Mutation, T790M, and cis-C797S Triple Mutations by Brigatinib and Cetuximab Combination Therapy. J. Thorac. Oncol. 2020, 15, 1369–1375. [Google Scholar] [CrossRef]
- Jänne, P.A.; Baik, C.; Su, W.-C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor–Resistant, EGFR-Mutated Non–Small Cell Lung Cancer. Cancer Discov. 2021, 12, 74–89. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2016, 385, 51–54. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Venkatachalam, G.; Lim, H.H.; Surana, U.; Verma, C. Conformational landscape of the epidermal growth factor receptor kinase reveals a mutant specific allosteric pocket. Chem. Sci. 2018, 9, 5212–5222. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020, 19, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.; Dineen, T.; Lim, S.; Park, C.; Hsieh, J.; Woessner, R.; Zhang, Z.; Wilson, K.; Eno, M.; Wilson, D.; et al. 384P BLU-945, a highly potent and selective 4th generation EGFR TKI for the treatment of EGFR T790M/C797S resistant NSCLC. Ann. Oncol. 2020, 31, S1391. [Google Scholar] [CrossRef]
- Lim, S.M.; Park, C.W.; Zhang, Z.; Woessner, R.; Dineen, T.; Stevison, F.; Hsieh, J.; Eno, M.; Wilson, D.; Campbell, J.; et al. Abstract 1467: BLU-945, a fourth-generation, potent and highly selective epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) with intracranial activity, demonstrates robust in vivo antitumor activity in models of osimertinib-resistant non-small cell lung cancer (NSCLC). Cancer Res. 2021, 81, 1467. [Google Scholar] [CrossRef]
- Marcoux, N.; Gettinger, S.N.; O’kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Bonomi, P.H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2018, 37, 278–285. [Google Scholar] [CrossRef]
- A Study of the Combination of Osimertinib, Platinum and Etoposide for Patients with Metastatic EGFR Mutant Lung Cancers. NCT03567642. Available online: https://clinicaltrials.gov/ct2/show/NCT03567642 (accessed on 31 March 2022).
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020, 38, 2926–2936. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.-Y.; Ahn, M.-J.; Cho, B.C.; Yu, H.; Kim, S.-W.; Yang, J.C.-H.; Lee, J.S.; Su, W.-C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Bauml, J.; Cho, B.C.; Park, K.; Lee, K.H.; Cho, E.K.; Kim, D.-W.; Kim, S.-W.; Haura, E.B.; Sabari, J.K.; Sanborn, R.E.; et al. Amivantamab in combination with lazertinib for the treatment of osimertinib-relapsed, chemotherapy-naïve EGFR mutant (EGFRm) non-small cell lung cancer (NSCLC) and potential biomarkers for response. J. Clin. Oncol. 2021, 39, 9006. [Google Scholar] [CrossRef]
- A Study of Tepotinib Plus Osimertinib in Osimertinib Relapsed MET Amplified NSCLC (INSIGHT 2). NCT03940703. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03940703 (accessed on 31 March 2022).
- Giroux-Leprieur, E.; Dumenil, C.; Chinet, T. Combination of Crizotinib and Osimertinib or Erlotinib Might Overcome MET-Mediated Resistance to EGFR Tyrosine Kinase Inhibitor in EGFR-Mutated Adenocarcinoma. J. Thorac. Oncol. 2018, 13, e232–e234. [Google Scholar] [CrossRef] [PubMed]
- Zhu, V.W.; Schrock, A.B.; Ali, S.M.; Ou, S.-H.I. Differential response to a combination of full-dose osimertinib and crizotinib in a patient with EGFR-mutant non-small cell lung cancer and emergent MET amplification. Lung Cancer Targets Ther. 2019, 10, 21–26. [Google Scholar] [CrossRef]
- Ross, H.J.; Blumenschein, G.R.; Aisner, J.; Damjanov, N.; Dowlati, A.; Garst, J.; Rigas, J.R.; Smylie, M.; Hassani, H.; Allen, K.E.; et al. Randomized Phase II Multicenter Trial of Two Schedules of Lapatinib as First- or Second-Line Monotherapy in Patients with Advanced or Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2010, 16, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2-Mutant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Kim, C.; Reuss, J.E.; Kiedrowski, L.A.; Garg, R.J.; Liu, S.V. Divergent RET- and BRAF-Mediated Resistance to Osimertinib in EGFR-Mutant NSCLC: A Case Report. JCO Precis. Oncol. 2021, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.F.S.A.; Knebel, F.H.; Bettoni, F.; Saddi, R.; Sacardo, K.P.; Canedo, F.S.N.A.; Alessi, J.V.M.; Shimada, A.K.; Marin, J.F.G.; Camargo, A.A.; et al. Impressive response to dabrafenib, trametinib, and osimertinib in a metastatic EGFR-mutant/BRAF V600E lung adenocarcinoma patient. NPJ Precis. Oncol. 2021, 5, 1–7. [Google Scholar] [CrossRef]
- Xie, Z.; Gu, Y.; Xie, X.; Lin, X.; Ouyang, M.; Qin, Y.; Zhang, J.; Lizaso, A.; Chen, S.; Zhou, C. Lung Adenocarcinoma Harboring Concomitant EGFR Mutations and BRAF V600E Responds to a Combination of Osimertinib and Vemurafenib to Overcome Osimertinib Resistance. Clin. Lung Cancer 2020, 22, e390–e394. [Google Scholar] [CrossRef]
- La Monica, S.; Minari, R.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Galetti, M.; Digiacomo, G.; Riccardi, F.; Petronini, P.G.; et al. Acquired BRAF G469A Mutation as a Resistance Mechanism to First-Line Osimertinib Treatment in NSCLC Cell Lines Harboring an EGFR Exon 19 Deletion. Target. Oncol. 2019, 14, 619–626. [Google Scholar] [CrossRef]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.A.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients with Advanced KRASG12C Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Huang, Y.; Gu, W.; Gan, J.; Wang, W.; Zhang, S.; Wang, K.; Zhan, J.; Yang, Y.; Huang, Y.; et al. PI3K-AKT-mTOR pathway alterations in advanced NSCLC patients after progression on EGFR-TKI and clinical response to EGFR-TKI plus everolimus combination therapy. Transl. Lung Cancer Res. 2020, 9, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Huang, Y.; Gan, J.; Yang, Y.; Wu, Y.; Huang, J.; Xu, Z.; Wang, W.; Zhang, L. Abstract 327: The impact of PIK3CA/PTEN/AKT1 genes in advanced NSCLC patients with acquired EGFR-TKI resistance and clinical response to EGFR-TKI plus everolimus combination therapy. In Proceedings of the Experimental and Molecular Therapeutics; American Association for Cancer Research: Philadelphia, PA, USA, 2019; p. 327. [Google Scholar]
- Vaclova, T.; Grazini, U.; Ward, L.; O’Neill, D.; Markovets, A.; Huang, X.; Chmielecki, J.; Hartmaier, R.; Thress, K.S.; Smith, P.D.; et al. Clinical impact of subclonal EGFR T790M mutations in advanced-stage EGFR-mutant non-small-cell lung cancers. Nat. Commun. 2021, 12, 1780. [Google Scholar] [CrossRef]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef]
- La Monica, S.; Fumarola, C.; Cretella, D.; Bonelli, M.; Minari, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Volta, F.; Mancini, M.; et al. Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib. Cancers 2020, 13, 6. [Google Scholar] [CrossRef]
- Koopman, L.A.; Terp, M.G.; Zom, G.G.; Janmaat, M.L.; Jacobsen, K.; Heuvel, E.G.-V.D.; Brandhorst, M.; Forssmann, U.; De Bree, F.; Pencheva, N.; et al. Enapotamab vedotin, an AXL-specific antibody-drug conjugate, shows preclinical antitumor activity in non-small cell lung cancer. JCI Insight 2019, 4, e128199. [Google Scholar] [CrossRef]
- Enapotamab Vedotin (HuMax-AXL-ADC) Safety Study in Patients with Solid Tumors. NCT02988817. Available online: https://clinicaltrials.gov/ct2/show/NCT02988817 (accessed on 31 March 2022).
- Hayakawa, D.; Takahashi, F.; Mitsuishi, Y.; Tajima, K.; Hidayat, M.; Winardi, W.; Ihara, H.; Kanamori, K.; Matsumoto, N.; Asao, T.; et al. Activation of insulin-like growth factor-1 receptor confers acquired resistance to osimertinib in non-small cell lung cancer withEGFRT790M mutation. Thorac. Cancer 2019, 11, 140–149. [Google Scholar] [CrossRef]
- Garon, E.; Johnson, M.; Lisberg, A.; Spira, A.; Yamamoto, N.; Heist, R.; Sands, J.; Yoh, K.; Meric-Bernstam, F.; Kitazono, S.; et al. LBA49 Efficacy of datopotamab deruxtecan (Dato-DXd) in patients (pts) with advanced/metastatic (adv/met) non-small cell lung cancer (NSCLC) and actionable genomic alterations (AGAs): Preliminary results from the phase I TROPION-PanTumor01 study. Ann. Oncol. 2021, 32, S1326–S1327. [Google Scholar] [CrossRef]
- Ercan, D.; Choi, H.G.; Yun, C.-H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef]
- Shi, P.; Oh, Y.-T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-C.; Liao, B.-C.; Liao, W.-Y.; Markovets, A.; Stetson, D.; Thress, K.; Yang, J.C.-H. Exon 16–Skipping HER2 as a Novel Mechanism of Osimertinib Resistance in EGFR L858R/T790M–Positive Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 15, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-S.; Lim, S.-N.; An, J.Y.; Lee, K.M.; Choe, K.H.; Kim, S.T.; Son, S.-M.; Choi, S.-Y.; Lee, H.-C.; Lee, O.-J. Detection of EGFR Mutation Status in Lung Adenocarcinoma Specimens with Different Proportions of Tumor Cells Using Two Methods of Differential Sensitivity. J. Thorac. Oncol. 2012, 7, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Abudula, M.; Li, C.; Chen, Z.; Zhang, Y.; Chen, Y. Icotinib-resistant HCC827 cells produce exosomes with mRNA MET oncogenes and mediate the migration and invasion of NSCLC. Respir. Res. 2019, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Truini, A.; Starrett, J.H.; Stewart, T.F.; Ashtekar, K.D.; Walther, Z.; Wurtz, A.; Lu, D.; Park, J.H.; Deveaux, M.; Song, X.; et al. The EGFR Exon 19 Mutant L747-A750>P Exhibits Distinct Sensitivity to Tyrosine Kinase Inhibitors in Lung Adenocarcinoma. Clin. Cancer Res. 2019, 25, 6382–6391. [Google Scholar] [CrossRef]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2018, 38, 455–468. [Google Scholar] [CrossRef]
- Nukaga, S.; Yasuda, H.; Tsuchihara, K.; Hamamoto, J.; Masuzawa, K.; Kawada, I.; Naoki, K.; Matsumoto, S.; Mimaki, S.; Ikemura, S.; et al. Amplification of EGFR Wild-Type Alleles in Non–Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2017, 77, 2078–2089. [Google Scholar] [CrossRef]
- Yu, Z.; Boggon, T.J.; Kobayashi, S.; Jin, C.; Ma, P.C.; Dowlati, A.; Kern, J.; Tenen, D.; Halmos, B. Resistance to an Irreversible Epidermal Growth Factor Receptor (EGFR) Inhibitor in EGFR-Mutant Lung Cancer Reveals Novel Treatment Strategies. Cancer Res. 2007, 67, 10417–10427. [Google Scholar] [CrossRef]
- Warmuth, M.; Kim, S.; Gu, X.-J.; Xia, G.; Adrián, F. Ba/F3 cells and their use in kinase drug discovery. Curr. Opin. Oncol. 2007, 19, 55–60. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.H.; Bulusu, K.C.; Laus, G.J.A.O.O.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, E.; Mileham, K.F.; Husain, H.; Costa, D.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFRT790M–Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, J.-J.; Huang, J.; Ye, J.-Y.; Zhang, X.-C.; Tu, H.-Y.; Han-Zhang, H.; Wu, Y.-L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef]
- HERTHENA-Lung01: Patritumab Deruxtecan in Subjects with Metastatic or Locally Advanced EGFR-Mutated Non-Small Cell Lung Cancer. NCT04619004. Available online: https://clinicaltrials.gov/ct2/show/NCT04619004 (accessed on 31 March 2022).
- Patritumab Deruxtecan in Combination with Osimertinib in Subjects with Locally Advanced or Metastatic EGFR-Mutated Non-Small Cell Lung Cancer. NCT04676477. Available online: https://clinicaltrials.gov/ct2/show/NCT04676477 (accessed on 31 March 2022).
- Rotow, J.K.; Costa, D.B.; Paweletz, C.P.; Awad, M.M.; Marcoux, P.; Rangachari, D.; Barbie, D.A.; Sands, J.; Cheng, M.L.; Johnson, B.E.; et al. Concurrent osimertinib plus gefitinib for first-line treatment of EGFR-mutated non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9507. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef]
- Fairclough, S.R.; Kiedrowski, L.A.; Lin, J.J.; Zelichov, O.; Tarcic, G.; Stinchcombe, T.E.; Odegaard, J.I.; Lanman, R.B.; Shaw, A.T.; Nagy, R.J. Identification of osimertinib-resistant EGFR L792 mutations by cfDNA sequencing: Oncogenic activity assessment and prevalence in large cfDNA cohort. Exp. Hematol. Oncol. 2019, 8, 1–6. [Google Scholar] [CrossRef]
- Bersanelli, M.; Minari, R.; Bordi, P.; Gnetti, L.; Bozzetti, C.; Squadrilli, A.; Lagrasta, C.A.M.; Bottarelli, L.; Osipova, G.; Capelletto, E.; et al. L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR-Mutated NSCLC. J. Thorac. Oncol. 2016, 11, e121–e123. [Google Scholar] [CrossRef]
- Yang, X.; Huang, C.; Chen, R.; Zhao, J. Resolving Resistance to Osimertinib Therapy with Afatinib in an NSCLC Patient with EGFR L718Q Mutation. Clin. Lung Cancer 2020, 21, e258–e260. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Chan, J.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Phase 2 Platform Study in Patients with Advanced Non-Small Lung Cancer Who Progressed on First-Line Osimertinib Therapy (ORCHARD). NCT03944772. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03944772 (accessed on 31 March 2022).
- (SYMPHONY) Phase 1/2 Study Targeting EGFR Resistance Mechanisms in NSCLC. NCT04862780. Available online: https://clinicaltrials.gov/ct2/show/NCT04862780 (accessed on 31 March 2022).
- Liu, X.; Zhang, X.; Yang, L.; Tian, X.; Dong, T.; Ding, C.Z.; Hu, L.; Wu, L.; Zhao, L.; Mao, J.; et al. Abstract 1320: Preclinical evaluation of TQB3804, a potent EGFR C797S inhibitor. In Proceedings of the Experimental and Molecular Therapeutics, Atlanta, GA, USA, 29 March–3 April 2019; American Association for Cancer Research: Atlanta, GA, USA, 2019; p. 1320. [Google Scholar]
- Choe, H.; Jeon, B.U.; Jung, M.E.; Jeon, M.-K.; Shin, I.; Cho, B.C.; Choi, G.; Chae, C.H.; Lee, K. Structure-Activity Relationship Study of 2,4-Dianilinopyrimidine Containing Methanesulfonamide (TRE-069) as Potent and Selective Epidermal Growth Factor Receptor T790M/C797S Mutant Inhibitor for Anticancer Treatment. Bull. Korean Chem. Soc. 2017, 38, 1353–1357. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, Á.; Chan, J.M.; Yu, H.A.; Pe’Er, D.; Sawyers, C.L.; Sen, T.; Rudin, C.M. Lineage plasticity in cancer: A shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 2020, 17, 360–371. [Google Scholar] [CrossRef]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.S.; Lee, B.; Kim, H.K.; Sun, J.; Ahn, J.S.; Ahn, M.; Park, K.; Lee, S. Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non–small cell lung cancer. Cancer 2020, 126, 2704–2712. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.-Y.; Tang, Y.; Xu, Y.; Guo, W.-H.; Li, Y.-C.; Liu, X.-K.; Huang, C.-Y.; Wang, Y.-S.; Wei, Y.-Q. Rapid increase of serum neuron specific enolase level and tachyphylaxis of EGFR-tyrosine kinase inhibitor indicate small cell lung cancer transformation from EGFR positive lung adenocarcinoma? Lung Cancer 2013, 81, 302–305. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Zhao, J.; Wang, M.-Z.; Huo, Z.; Zhang, J.; Zhong, W.; Xu, Y. Small-Cell Lung Cancer Transformation in Patients with Pulmonary Adenocarcinoma. Medicine 2016, 95, e2752. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef]
- Vokes, N.I.; Chambers, E.; Nguyen, T.; Coolidge, A.; Lydon, C.A.; Le, X.; Sholl, L.; Heymach, J.V.; Nishino, M.; Van Allen, E.M.; et al. Concurrent TP53 mutations facilitate resistance evolution in EGFR mutant lung adenocarcinoma. J. Thorac. Oncol. 2022, S1556-0864, 135907. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Cappuzzo, F.; Ou, S.-H.I.; Camidge, D.R. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J. Thorac. Oncol. 2017, 12, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Raghav, K.P.S.; Gonzalez-Augulo, A.M.; Blumenschein, G.R. Role of HGF/MET axis in resistance of lung cancer to contemporary management. Transl. Lung Cancer Res. 2012, 1, 179–193. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Osimertinib Plus Savolitinib in EGFRm+/MET+ NSCLC Following Prior Osimertinib (SAVANNAH). NCT03778229. Available online: https://clinicaltrials.gov/ct2/show/NCT03778229 (accessed on 31 March 2022).
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.-Y.; Kim, S.-W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion–Mutated Non–Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- York, E.R.; Varella-Garcia, M.; Bang, T.J.; Aisner, D.L.; Camidge, D.R. Tolerable and Effective Combination of Full-Dose Crizotinib and Osimertinib Targeting MET Amplification Sequentially Emerging after T790M Positivity in EGFR- Mutant Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, e85–e88. [Google Scholar] [CrossRef]
- Janne, P.A.; Baik, C.S.; Su, W.-C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and safety of patritumab deruxtecan (HER3-DXd) in EGFR inhibitor-resistant, EGFR-mutated (EGFRm) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2021, 39, 9007. [Google Scholar] [CrossRef]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 expression status in diverse cancers: Review of results from 37,992 patients. Cancer Metastasis Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Boone, M.; Meuris, L.; Lemmens, I.; Van Roy, N.; Soete, A.; Reumers, J.; Moisse, M.; Plaisance, S.; Drmanac, R.T.; et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014, 5, 4767. [Google Scholar] [CrossRef]
- Wu, L.; Ke, L.; Zhang, Z.; Yu, J.; Meng, X. Development of EGFR TKIs and Options to Manage Resistance of Third-Generation EGFR TKI Osimertinib: Conventional Ways and Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 2778. [Google Scholar] [CrossRef]
- Du, X.; Yang, B.; An, Q.; Assaraf, Y.G.; Cao, X.; Xia, J. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovation 2021, 2, 100103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xia, Y. Targeting HER2 Alterations in Non–Small-Cell Lung Cancer: A Comprehensive Review. JCO Precis. Oncol. 2020, 4, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.A.; Reckamp, K.L.; Chae, Y.K.; Doebele, R.C.; Iams, W.T.; Oh, M.; Raymond, V.M.; Lanman, R.B.; Riess, J.W.; Stinchcombe, T.E.; et al. Analysis of Cell-Free DNA from 32,989 Advanced Cancers Reveals Novel Co-occurring Activating RET Alterations and Oncogenic Signaling Pathway Aberrations. Clin. Cancer Res. 2019, 25, 5832–5842. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, T.; Shiraishi, K.; Shimada, Y.; Ogiwara, H.; Tsuta, K.; Ichikawa, H.; Sakamoto, H.; Kato, M.; Shibata, T.; Nakano, T.; et al. Molecular Mechanisms Underlying Oncogenic RET Fusion in Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Iams, W.; Chae, Y. P3.02-034 Acquired Resistance to Osimertinib by CCDC6-RET Fusion in a Patient with EGFR T790M Mutant Metastatic Lung Adenocarcinoma. J. Thorac. Oncol. 2017, 12, S2249–S2250. [Google Scholar] [CrossRef]
- Sun, Y.; Pei, L.; Luo, N.; Chen, D.; Meng, L. A Novel MYH9-RET Fusion Occurrence and EGFR T790M Loss as an Acquired Resistance Mechanism to Osimertinib in a Patient with Lung Adenocarcinoma: A Case Report. OncoTargets Ther. 2020, 13, 11177–11181. [Google Scholar] [CrossRef]
- Klempner, S.; Bazhenova, L.A.; Braiteh, F.S.; Nikolinakos, P.G.; Gowen, K.; Cervantes, C.M.; Chmielecki, J.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.J.; et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer 2015, 89, 357–359. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Ho, C.-C.; Liao, W.-Y.; Lin, C.-A.; Shih, J.-Y.; Yu, C.-J.; Yang, J.C.-H. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 567–572. [Google Scholar] [CrossRef]
- Huang, Y.; Gan, J.; Guo, K.; Deng, Y.; Fang, W. Acquired BRAF V600E Mutation Mediated Resistance to Osimertinib and Responded to Osimertinib, Dabrafenib, and Trametinib Combination Therapy. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, e236–e237. [Google Scholar] [CrossRef]
- Roper, N.; Brown, A.-L.; Wei, J.S.; Pack, S.; Trindade, C.; Kim, C.; Restifo, O.; Gao, S.; Sindiri, S.; Mehrabadi, F.; et al. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep. Med. 2020, 1, 100007. [Google Scholar] [CrossRef] [PubMed]
- Vojnic, M.; Kubota, D.; Kurzatkowski, C.; Offin, M.; Suzawa, K.; Benayed, R.; Schoenfeld, A.J.; Plodkowski, A.J.; Poirier, J.; Rudin, C.M.; et al. Acquired BRAF Rearrangements Induce Secondary Resistance to EGFR therapy in EGFR-Mutated Lung Cancers. J. Thorac. Oncol. 2019, 14, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Piotrowska, Z.; Cobb, R.; Banwait, M.; Lennerz, J.K.; Hata, A.N.; Digumarthy, S.R.; Sequist, L.V. Response to the Combination of Osimertinib and Trametinib in a Patient with EGFR-Mutant NSCLC Harboring an Acquired BRAF Fusion. J. Thorac. Oncol. 2019, 14, e226–e228. [Google Scholar] [CrossRef] [PubMed]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, P.; Han, X.; Wang, S.; Liu, Y.; Liu, P.; Li, J.; Chang, L.; Guan, Y.; Zhang, Z.; et al. P1.13-18 Exploring the Resistance Mechanism of Osimertinib and Monitoring the Treatment Response Using Plasma ctDNA in Chinese NSCLC Patients. J. Thorac. Oncol. 2018, 13, S589. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef]
- Hong, M.; Kim, M.; Kim, S.-Y.; Heo, S.; Kang, H.-N.; Park, C.-W.; Barrett, J.; Stetson, D.; Chmielecki, J.; Markovets, A.; et al. Molecular landscape of osimertinib resistance revealed by targeted panel sequencing and patient-derived cancer models in non-small cell lung cancer patients. Ann. Oncol. 2018, 29, viii516. [Google Scholar] [CrossRef]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef]
- Fay, A.P.; Moreira, R.B.; Filho, P.R.S.N.; Albuquerque, C.; Barrios, C.H. The management of immune-related adverse events associated with immune checkpoint blockade. Expert Rev. Qual. Life Cancer Care 2016, 1, 89–97. [Google Scholar] [CrossRef][Green Version]
- Choi, Y.J.; Anders, L. Signaling through cyclin D-dependent kinases. Oncogene 2014, 33, 1890–1903. [Google Scholar] [CrossRef]
- Osoegawa, A.; Yamaguchi, M.; Nakamura, T.; Morinaga, R.; Tanaka, K.; Kashiwabara, K.; Miura, T.; Suetsugu, T.; Harada, T.; Asoh, T.; et al. High Incidence of C797S Mutation in Patients with Long Treatment History of EGFR Tyrosine Kinase Inhibitors Including Osimertinib. JTO Clin. Res. Rep. 2021, 2, 100191. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- G1T38, a CDK 4/6 Inhibitor, in Combination with Osimertinib in EGFR-Mutant Non-Small Cell Lung Cancer. NCT03455829. Available online: https://clinicaltrials.gov/ct2/show/NCT03455829 (accessed on 31 March 2022).
- Antony, J.; Huang, R.Y.-J. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [PubMed]
- Nonagase, Y.; Takeda, M.; Azuma, K.; Hayashi, H.; Haratani, K.; Tanaka, K.; Yonesaka, K.; Ishii, H.; Hoshino, T.; Nakagawa, K. Tumor tissue and plasma levels of AXL and GAS6 before and after tyrosine kinase inhibitor treatment in EGFR-mutated non–small cell lung cancer. Thorac. Cancer 2019, 10, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Jang, Y.; Lee, S.H.; Kang, B.; Lim, S.M. AXL/MET Dual Inhibitor, CB469, Has Activity in Non-Small Cell Lung Cancer with Acquired Resistance to EGFR TKI with AXL or MET Activation; Elsevier Ireland Ltd.: Shannon, Ireland, 2020; Volume 146, ISBN 8231780343. [Google Scholar]
- Namba, K.; Shien, K.; Takahashi, Y.; Torigoe, H.; Sato, H.; Yoshioka, T.; Takeda, T.; Kurihara, E.; Ogoshi, Y.; Yamamoto, H.; et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non–Small Cell Lung Cancer Cells. Mol. Cancer Res. 2018, 17, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Kim, T.M.; Song, A.; Kim, D.-W.; Kim, S.; Ahn, Y.-O.; Keam, B.; Jeon, Y.K.; Lee, S.-H.; Chung, D.H.; Heo, D.S. Mechanisms of Acquired Resistance to AZD9291: A Mutation-Selective, Irreversible EGFR Inhibitor. J. Thorac. Oncol. 2015, 10, 1736–1744. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Yamada, T.; Yoshimura, A.; Kaira, K.; Atagi, S.; Yano, S.; Takayama, K. 411P Impact of pre-treatment AXL expression on osimertinib efficacy in patients with non-small cell lung cancer with EGFR mutation. Ann. Oncol. 2020, 31, S1402–S1403. [Google Scholar] [CrossRef]
- Li, J.; Choi, E.; Yu, H.; Bai, X.-C. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat. Commun. 2019, 10, 4567. [Google Scholar] [CrossRef]
- Manabe, T.; Yasuda, H.; Terai, H.; Kagiwada, H.; Hamamoto, J.; Ebisudani, T.; Kobayashi, K.; Masuzawa, K.; Ikemura, S.; Kawada, I.; et al. IGF2 Autocrine-Mediated IGF1R Activation Is a Clinically Relevant Mechanism of Osimertinib Resistance in Lung Cancer. Mol. Cancer Res. 2020, 18, 549–559. [Google Scholar] [CrossRef]
- Ohara, S.; Suda, K.; Mitsudomi, T. Cell Line Models for Acquired Resistance to First-Line Osimertinib in Lung Cancers—Applications and Limitations. Cells 2021, 10, 354. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-M.; Xu, Y.-L.; Yuan, L.-W.; Zhang, L.-L.; Huang, M.-Y.; Ye, Z.-H.; Su, M.-X.; Chen, X.-P.; Zhu, H.; Ye, R.D.; et al. TGFβ2-mediated epithelial–mesenchymal transition and NF-κB pathway activation contribute to osimertinib resistance. Acta Pharmacol. Sin. 2020, 42, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. Targeting the EMT transcription factor Snail overcomes resistance to osimertinib in EGFR-mutant non-small cell lung cancer. Thorac. Cancer 2021, 12, 1708–1715. [Google Scholar] [CrossRef]
- Zeng, L.; Yang, N.; Zhang, Y. GOPC-ROS1 Rearrangement as an Acquired Resistance Mechanism to Osimertinib and Responding to Crizotinib Combined Treatments in Lung Adenocarcinoma. J. Thorac. Oncol. 2018, 13, e114–e116. [Google Scholar] [CrossRef]
- Schrock, A.B.; Zhu, V.W.; Hsieh, W.-S.; Madison, R.; Creelan, B.; Silberberg, J.; Costin, D.; Bharne, A.; Bonta, I.; Bosemani, T.; et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 1312–1323. [Google Scholar] [CrossRef]


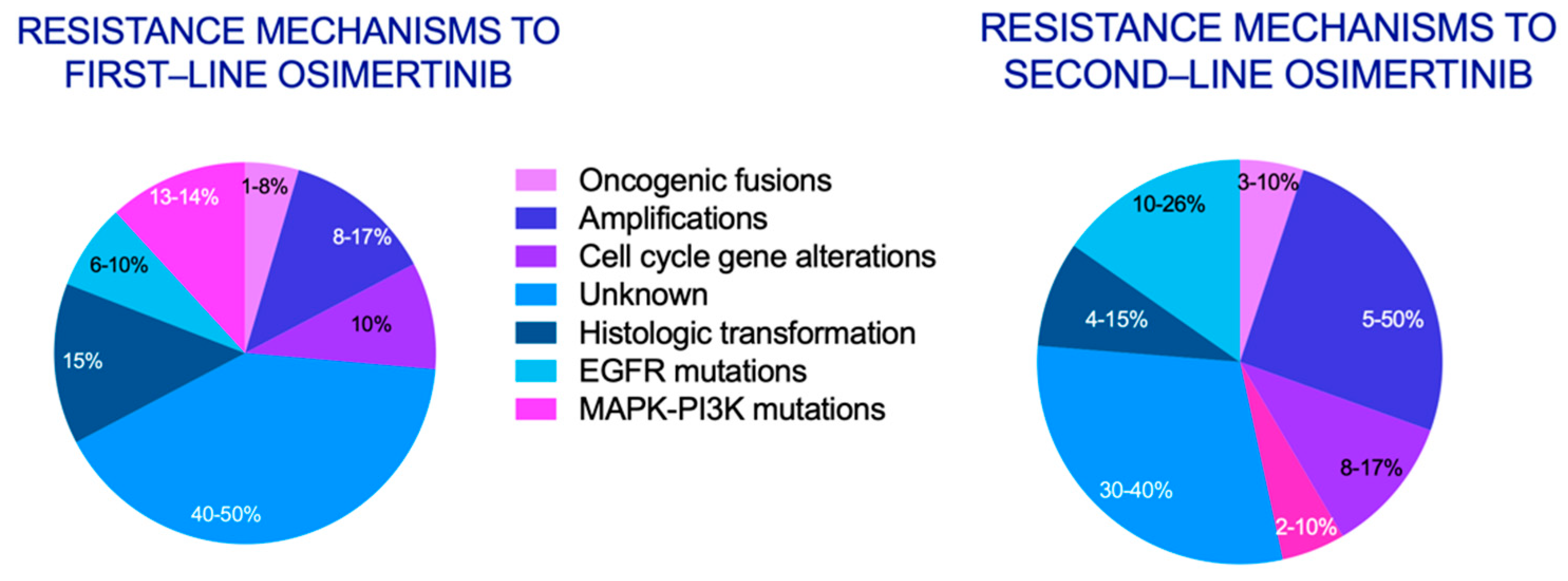{kind=link}
| Mechanism of Resistance | Therapeutic Strategies | References |
|---|---|---|
| C797X | Gefitinib, erlotinib | [17] |
| Osimertinib + erlotinib | [18] | |
| Brigatinib + cetuximab | [19] | |
| Patritumab deruxtecan | [20] | |
| EAI045 | [21,22,23] | |
| JBJ-04-125-02 | [24] | |
| CH7233163 | [25] | |
| BLU-945 | [26,27] | |
| Small cell transformation | Platinum-etoposide | [28,29] |
| Squamous cell transformation | Histology based approach | [30] |
| MET amplification | Osimertinib + savolitinib | [31] |
| Lazertinib + amivantamab | [32] | |
| Tepotinib + osimertinib | [33] | |
| Osimertinib + crizotinib | [34,35] | |
| Patritumab deruxtecan | [20] | |
| HER2 alterations | Patritumab deruxtecan | [20] |
| Osimertinib + lapatinib * | [36] | |
| Osimertinib + neratinib * | [37] | |
| Osimertinib + T-DXd * | [38] | |
| RET alterations | Osimertinib + selpercatinib | [39] |
| BRAF alterations | Osimertinib + dabrafenib + trametinib | [40] |
| Osimertinib + vemurafenib | [41] | |
| Osimertinib + selumetinib or trametinib | [42] | |
| RAS | Osimertinib + selumetinib or Aurora kinase b inhibitor | [43] |
| Osimertinib + sotorasib * | [44] | |
| Osimertinib + adagrasib * | [45] | |
| PIK3 | EGFR TKIs and everolimus | [46,47] |
| Osimertinib + alpelisib | [48] | |
| Cell cycle gene alterations | Osimertinib + palbociclib | [49] |
| Osimertinib + abemaciclib | [50] | |
| AXL overexpression | Enapotamab vedotin | [51,52] |
| IGF-1 receptor activation | Osimertinib + linsitinib | [53] |
| Non-specific alterations | Datopotamab deruxtecan | [54] |
| Induced Resistance Mechanism | Cell Lines | Mechanism of Induction | References |
|---|---|---|---|
| del19/T790M/C797S and L858R/T790M/C797S | NIH3T3 cells (immortalized mouse embryonic fibroblast cell line) | Transduction with lentiviruses | [25] |
| del19/L858R +/− T790M | Ba/F3 cells (a murine, IL-3 dependent, hematopoietic cell line) | Transduction with retroviral JP1540 or lentiviral JP1698 vectors | [55] |
| MET amplification | HCC827 cells (EGFR del19) | Exposure to osimertinib through a stepwise escalation process | [56] |
| HER2 exon 16 skipping | HEK293 cells (human embryonic kidney cell line and H1975 (T790M/L858R) | Plasmid transfection | [57] |
| BRAF G469A | PC9 cells (EGFR del 19) | Exposure to osimertinib through a stepwise escalation process | [42] |
| RAS alterations | PC9 cells | Exposure to osimertinib through a stepwise escalation process and a single concentration of osimertinib | [43] |
| Cell cycle gene alterations | H1975 cells (EGFR L858R/T790M) | Exposure to osimertinib through a stepwise escalation process | [49] |
| AXL overexpression | HCC827 cells (EGFR del19), PC9, H1975, and HCC4006 cells (EGFR del19) | Exposure to osimertinib through a stepwise escalation process a single concentration of osimertinib | [58,59,60] |
| Activation of IGF-1 receptor | PC9 cells | Exposure to gefitinib, developing resistance through the T790M, subsequently culture with stepwise escalation with osimertinib | [53] |
| H1975 cells | Exposure to osimertinib using a high-concentration method | [53] | |
| EMT | H1975/AR cells (gefitinib resistant) | Exposure to osimertinib through a stepwise escalation process | [61] |
| Other mechanisms: Src-AKT pathway and EGFR wild-type amplification | PC9 and H1975 cells | Exposure to osimertinib through a stepwise escalation process | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ríos-Hoyo, A.; Moliner, L.; Arriola, E. Acquired Mechanisms of Resistance to Osimertinib—The Next Challenge. Cancers 2022, 14, 1931. https://doi.org/10.3390/cancers14081931
Ríos-Hoyo A, Moliner L, Arriola E. Acquired Mechanisms of Resistance to Osimertinib—The Next Challenge. Cancers. 2022; 14(8):1931. https://doi.org/10.3390/cancers14081931
Chicago/Turabian StyleRíos-Hoyo, Alejandro, Laura Moliner, and Edurne Arriola. 2022. "Acquired Mechanisms of Resistance to Osimertinib—The Next Challenge" Cancers 14, no. 8: 1931. https://doi.org/10.3390/cancers14081931
APA StyleRíos-Hoyo, A., Moliner, L., & Arriola, E. (2022). Acquired Mechanisms of Resistance to Osimertinib—The Next Challenge. Cancers, 14(8), 1931. https://doi.org/10.3390/cancers14081931