
The Role of TP53 Mutations in EGFR-Mutated Non-Small-Cell Lung Cancer: Clinical Significance and Implications for Therapy

 ,
,  , and
, and 
Abstract
Simple Summary
Abstract
1. Introduction
2. EGFR-Mutated NSCLC
2.1. EGFR+ Clinical Management: Clinical Trials Demonstrating First-, Second- and Third-Generation TKIs Efficacy
2.1.1. First-Generation of EGFR TKIs
2.1.2. First-Generation EGFR TKIs Combined with Other Agents
2.1.3. Second-Generation of EGFR TKIs
2.1.4. Third-Generation of EGFR TKIs
2.2. EGFR TKIs for the Treatment of Uncommon EGFR Mutations
3. Common Resistance Mechanisms to EGFR-TKIs
3.1. On-Target Mechanisms of Resistance
3.2. Off-Target Mechanisms of Resistance
3.2.1. MET Amplification
3.2.2. HER2
3.2.3. KRAS, BRAF and PI3K
4. TP53: The Master Regulator of the Genome
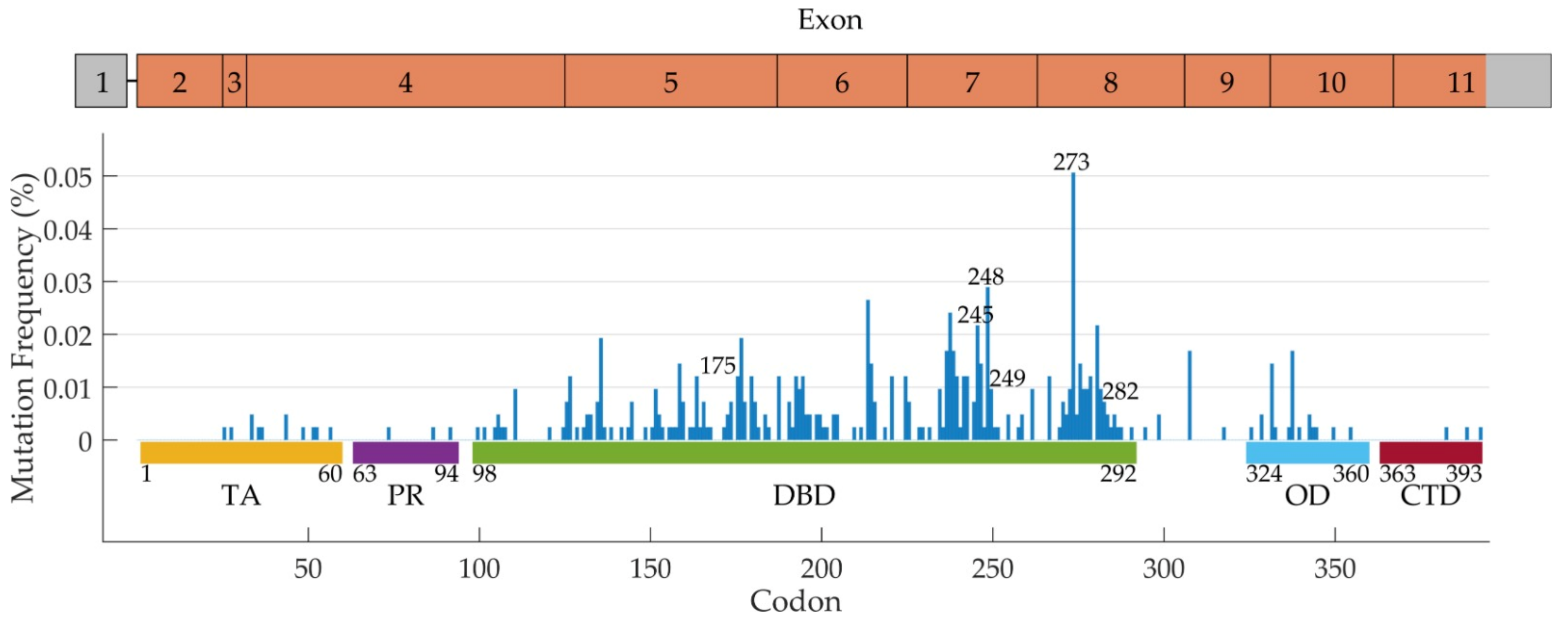
TP53 Mutations in Human Cancer
5. TP53 Mutations in NSCLC
5.1. TP53 Mutations in EGFR-Positive NSCLC: Clinical Significance
5.2. TP53 Mutations in EGFR-Positive NSCLC: Phenotypic Changes
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ulivi, P.; Petracci, E.; Canale, M.; Priano, I.; Capelli, L.; Calistri, D.; Chiadini, E.; Cravero, P.; Rossi, A.; Delmonte, A.; et al. Liquid Biopsy for EGFR Mutation Analysis in Advanced Non-Small-Cell Lung Cancer Patients: Thoughts Drawn from a Real-Life Experience. Biomedicines 2021, 9, 1299. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming Therapy Resistance in EGFR-Mutant Lung Cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D. Therapeutic Targeting of p53: All Mutants Are Equal, but Some Mutants Are More Equal than Others. Nat. Rev. Clin. Oncol. 2017, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.-D.; Qin, B.-D.; You, P.; Cai, J.; Zang, Y.-S. The Prognostic Value of TP53 and Its Correlation with EGFR Mutation in Advanced Non-Small Cell Lung Cancer, an Analysis Based on cBioPortal Data Base. Lung Cancer 2018, 123, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare Epidermal Growth Factor Receptor (EGFR) Mutations in Non-Small Cell Lung Cancer. Semin. Cancer Biol. 2019, 61, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR Mutations and Lung Cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR -Mutated Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-Targeted Therapies in the Post-Genomic Era. Cancer Metastasis Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef]
- Talukdar, S.; Emdad, L.; Das, S.K.; Fisher, P.B. EGFR: An Essential Receptor Tyrosine Kinase-Regulator of Cancer Stem Cells. Adv. Cancer Res. 2020, 147, 161–188. [Google Scholar] [CrossRef]
- Gelatti, A.C.; Drilon, A.; Santini, F.C. Optimizing the Sequencing of Tyrosine Kinase Inhibitors (TKIs) in Epidermal Growth Factor Receptor (EGFR) Mutation-Positive Non-Small Cell Lung Cancer (NSCLC). Lung Cancer 2019, 137, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal Growth Factor Receptor Mutations in Lung Cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, E.; Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Saijo, Y.; Yoshizawa, H.; et al. Efficacy of Chemotherapy after First-Line Gefitinib Therapy in EGFR Mutation-Positive Advanced Non-Small Cell Lung Cancer-Data from a Randomized Phase III Study Comparing Gefitinib with Carboplatin Plus Paclitaxel (NEJ002). Jpn. J. Clin. Oncol. 2015, 45, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-K.; Hahn, S.; Kim, D.-W.; Suh, K.J.; Keam, B.; Kim, T.M.; Lee, S.-H.; Heo, D.S. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors vs. Conventional Chemotherapy in Non–Small Cell Lung Cancer Harboring Wild-Type Epidermal Growth Factor Receptor. JAMA 2014, 311, 1430–1437. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib Versus Cisplatin Plus Docetaxel in Patients with Non-Small-Cell Lung Cancer Harbouring Mutations of the Epidermal Growth Factor Receptor (WJTOG3405): An Open Label, Randomised Phase 3 Trial. Lancet Oncol. 2009, 11, 121–128. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib Versus Chemotherapy as First-Line Treatment for Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib Versus Standard Chemotherapy as First-Line Treatment for European Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Liam, C.-K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-Line Erlotinib Versus Gemcitabine/Cisplatin in Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer: Analyses from the Phase III, Randomized, Open-Label, ENSURE Study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef]
- Lee, C.K.; Davies, L.; Wu, Y.-L.; Mitsudomi, T.; Inoue, A.; Rosell, R.; Zhou, C.; Nakagawa, K.; Thongprasert, S.; Fukuoka, M.; et al. Gefitinib or Erlotinib vs. Chemotherapy for EGFR Mutation-Positive Lung Cancer: Individual Patient Data Meta-Analysis of Overall Survival. JNCI J. Natl. Cancer Inst. 2017, 109, djw279. [Google Scholar] [CrossRef]
- Shi, Y.K.; Wang, L.; Han, B.H.; Li, W.; Yu, P.; Liu, Y.P.; Ding, C.M.; Song, X.; Ma, Z.Y.; Ren, X.L.; et al. First-Line Icotinib Versus Cisplatin/Pemetrexed Plus Pemetrexed Maintenance Therapy for Patients with Advanced EGFR Mutation-Positive Lung Adenocarcinoma (CONVINCE): A Phase 3, Open-Label, Randomized Study. Ann. Oncol. 2017, 28, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Zhou, Q.; Yan, H.H.; Zhang, X.C.; Chen, H.J.; Tu, H.Y.; Wang, Z.; Xu, C.R.; Su, J.; Wang, B.C.; et al. A Phase III Randomised Controlled Trial of Erlotinib vs. Gefitinib in Advanced Non-Small Cell Lung Cancer with EGFR Mutations. Br. J. Cancer 2017, 116, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Liu, X.; Zhou, C.; Zhang, S.; Wang, D.; Li, Q.; Qin, S.; Hu, C.; Zhang, Y.; et al. Icotinib Versus Gefitinib in Previously Treated Advanced Non-Small-Cell Lung Cancer (ICOGEN): A Randomised, Double-Blind Phase 3 Non-Inferiority Trial. Lancet Oncol. 2013, 14, 953–961. [Google Scholar] [CrossRef]
- Yamamoto, N.; Seto, T.; Nishio, M.; Goto, K.; Okamoto, I.; Yamanaka, T.; Tanaka, M.; Takahashi, K.; Fukuoka, M. Erlotinib Plus Bevacizumab vs. Erlotinib Monotherapy as First-Line Treatment for Advanced EGFR Mutation-Positive Non-Squamous Non-Small-Cell Lung Cancer: Survival Follow-Up Results of the Randomized JO25567 Study. Lung Cancer 2020, 151, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Aix, S.P.; Paz-Ares, L.; Chiu, C.-H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab Plus Erlotinib in Patients with Untreated, EGFR-Mutated, Advanced Non-Small-Cell Lung Cancer (RELAY): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef]
- Nakagawa, K.; Nadal, E.; Garon, E.B.; Nishio, M.; Seto, T.; Yamamoto, N.; Park, K.; Shih, J.-Y.; Paz-Ares, L.; Frimodt-Moller, B.; et al. RELAY Subgroup Analyses by EGFR Ex19del and Ex21L858R Mutations for Ramucirumab Plus Erlotinib in Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 5258–5271. [Google Scholar] [CrossRef]
- Wu, Q.; Luo, W.; Li, W.; Wang, T.; Huang, L.; Xu, F. First-Generation EGFR-TKI Plus Chemotherapy Versus EGFR-TKI Alone as First-Line Treatment in Advanced NSCLC with EGFR Activating Mutation: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Oncol. 2021, 11, 883. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an Irreversible EGFR/HER2 Inhibitor Highly Effective in Preclinical Lung Cancer Models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.-M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations that Are Resistant to Gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.-H.; Hirsh, V.; Schuler, M.; Yamamoto, N.; O’Byrne, K.J.; Mok, T.; Zazulina, V.; Shahidi, M.; Lungershausen, J.; Massey, D.; et al. Symptom Control and Quality of Life in LUX-Lung 3: A Phase III Study of Afatinib or Cisplatin/Pemetrexed in Patients with Advanced Lung Adenocarcinoma with EGFR Mutations. J. Clin. Oncol. 2013, 31, 3342–3350. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Hirsh, V.; Boyer, M.; Yang, J.C.-H.; Mok, T.; Lee, K.H.; Lu, S.; et al. Afatinib Versus Gefitinib in Patients with EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer: Overall Survival Data from the Phase IIb LUX-Lung 7 Trial. Ann. Oncol. 2017, 28, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib Versus Gefitinib as First-Line Treatment for Patients with EGFR-Mutation-Positive Non-Small-Cell Lung Cancer (ARCHER 1050): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Chawla, A.; Rosell, R.; Corral, J.; Migliorino, M.R.; et al. Updated Overall Survival in a Randomized Study Comparing Dacomitinib with Gefitinib as First-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. Drugs 2020, 81, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib Versus Cisplatin-Based Chemotherapy for EGFR Mutation-Positive Lung Adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of Overall Survival Data from Two Randomised, Phase 3 Trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Sequist, L.V.; Geater, S.L.; Tsai, C.-M.; Mok, T.; Schuler, M.; Yamamoto, N.; Yu, C.-J.; Ou, S.-H.I.; Zhou, C.; et al. Clinical Activity of Afatinib in Patients with Advanced Non-Small-Cell Lung Cancer Harbouring Uncommon EGFR Mutations: A Combined Post-Hoc Analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Chiu, C.-H.; Yang, C.-T.; Shih, J.-Y.; Huang, M.-S.; Su, W.-C.; Lai, R.-S.; Wang, C.-C.; Hsiao, S.-H.; Lin, Y.-C.; Ho, C.-L.; et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment Response in Advanced Lung Adenocarcinomas with G719X/L861Q/S768I Mutations. J. Thorac. Oncol. 2015, 10, 793–799. [Google Scholar] [CrossRef]
- Haura, E.B.; Cho, B.C.; Lee, J.S.; Han, J.-Y.; Lee, K.H.; Sanborn, R.E.; Govindan, R.; Cho, E.K.; Kim, S.-W.; Reckamp, K.L.; et al. JNJ-61186372 (JNJ-372), an EGFR-cMet Bispecific Antibody, in EGFR-Driven Advanced Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2019, 37, 9009. [Google Scholar] [CrossRef]
- Cho, J.H.; Lim, S.H.; An, H.J.; Kim, K.H.; Park, K.U.; Kang, E.J.; Choi, Y.H.; Ahn, M.S.; Lee, M.H.; Sun, J.-M.; et al. Osimertinib for Patients with Non–Small-Cell Lung Cancer Harboring Uncommon EGFR Mutations: A Multicenter, Open-Label, Phase II Trial (KCSG-LU15-09). J. Clin. Oncol. 2020, 38, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Reita, D.; Pabst, L.; Pencreach, E.; Guérin, E.; Dano, L.; Rimelen, V.; Voegeli, A.-C.; Vallat, L.; Mascaux, C.; Beau-Faller, M. Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers 2021, 13, 4926. [Google Scholar] [CrossRef]
- Pao, W.; Girard, N. New Driver Mutations in Non-Small-Cell Lung Cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of Dosing for EGFR-Mutant Non–Small Cell Lung Cancer with Evolutionary Cancer Modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired Resistance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer: Mechanisms and Therapeutic Strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of Acquired Resistance to First-Line Osimertinib: Preliminary Data from the Phase III FLAURA Study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of Novel and Dominant Acquired EGFR Solvent-Front Mutations at Gly796 (G796S/R) Together with C797S/G and L792F/H Mutations in One EGFR (L858R/T790M) NSCLC Patient Who Progressed on Osimertinib. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Gan, J.; Huang, Y.; Zhou, H.; Zhang, L. Acquired EGFR L718V Mutation and Loss of T790M-Mediated Resistance to Osimertinib in a Patient with NSCLC Who Responded to Afatinib. J. Thorac. Oncol. 2019, 14, e274–e275. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Chan, J.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations as Early Resistance Mechanisms to First-Line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, X.-C.; Yang, J.-J.; Yang, Z.-F.; Bai, Y.; Su, J.; Wang, Z.; Zhang, Z.; Shao, Y.; Zhou, Q.; et al. EGFR L792H and G796R: Two Novel Mutations Mediating Resistance to the Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib. J. Thorac. Oncol. 2018, 13, 1415–1421. [Google Scholar] [CrossRef]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D Mutation Mediates Resistance to Osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef]
- Zhang, Y.; He, B.; Zhou, D.; Li, M.; Hu, C. Newly Emergent Acquired EGFR Exon 18 G724S Mutation after Resistance of a T790M Specific EGFR Inhibitor Osimertinib in Non-Small-Cell Lung Cancer: A Case Report. OncoTargets Ther. 2018, 12, 51–56. [Google Scholar] [CrossRef]
- Oztan, A.; Fischer, S.; Schrock, A.; Erlich, R.; Lovly, C.; Stephens, P.; Ross, J.; Miller, V.; Ali, S.; Ou, S.-H.I.; et al. Emergence of EGFR G724S Mutation in EGFR-Mutant Lung Adenocarcinoma Post Progression on Osimertinib. Lung Cancer 2017, 111, 84–87. [Google Scholar] [CrossRef]
- Nukaga, S.; Yasuda, H.; Tsuchihara, K.; Hamamoto, J.; Masuzawa, K.; Kawada, I.; Naoki, K.; Matsumoto, S.; Mimaki, S.; Ikemura, S.; et al. Amplification of EGFR Wild-Type Alleles in Non–Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2017, 77, 2078–2089. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, P.; Han, X.; Wang, S.; Liu, Y.; Liu, P.; Li, J.; Chang, L.; Guan, Y.; Zhang, Z.; et al. P1.13-18 Exploring the Resistance Mechanism of Osimertinib and Monitoring the Treatment Response Using Plasma ctDNA in Chinese NSCLC Patients. J. Thorac. Oncol. 2018, 13, S589. [Google Scholar] [CrossRef]
- Graham, T.A.; McDonald, S.A.; Wright, N.A. Field Cancerization in the GI Tract. Futur. Oncol. 2011, 7, 981–993. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Jänne, P.A. Mechanisms of Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small Cell Lung Cancer: Figure 1. Clin. Cancer Res. 2008, 14, 2895–2899. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Mok, T.S.; Han, J.Y.; Ahn, M.J.; Delmonte, A.; Ramalingam, S.S.; Kim, S.W.; Shepherd, F.A.; Laskin, J.; He, Y.; et al. Osimertinib Versus Platinum–Pemetrexed for Patients with EGFR T790M Advanced NSCLC and Progression on a Prior EGFR-Tyrosine Kinase Inhibitor: AURA3 Overall Survival Analysis. Ann. Oncol. 2020, 31, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; De Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 Amplification: A Potential Mechanism of Acquired Resistance to EGFR Inhibition in EGFR-Mutant Lung Cancers That Lack the Second-Site EGFRT790M Mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-Independent Mechanisms of Acquired Resistance to AZD9291 in EGFR T790M-Positive NSCLC Patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical Definition of Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, E.; Mileham, K.F.; Husain, H.; Costa, D.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients WithEGFRT790M–Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, X.B.; Qiu, X.P.; Shuai, Z.; Wang, C.; Zheng, F. CDKN2A Promoter Methylation and Hepatocellular Carcinoma Risk: A Meta-Analysis. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 529–541. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.S.; Lee, B.; Kim, H.K.; Sun, J.; Ahn, J.S.; Ahn, M.; Park, K.; Lee, S. Genomic Landscape of Acquired Resistance to Third-Generation EGFR Tyrosine Kinase Inhibitors in EGFR T790M-Mutant Non–Small Cell Lung Cancer. Cancer 2020, 126, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The Multiple Mechanisms That Regulate P53 Activity and Cell Fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in Survival, Death and Metabolic Health: A Lifeguard with a Licence to Kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of P53 Protein in the Cellular Response to DNA Damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M. Genome Stability Requires P53. Cold Spring Harb. Perspect. Med. 2016, 6, a026096. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jaeger, N.; Remke, M.; Shih, D.J.H.; Northcott, P.A.; et al. Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements with TP53 Mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Levine, A.J.; Ting, D.; Greenbaum, B.D. P53 and the Defenses against Genome Instability Caused by Transposons and Repetitive Elements. BioEssays 2016, 38, 508–513. [Google Scholar] [CrossRef]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.-J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. P53 Genes Function to Restrain Mobile Elements. Genes Dev. 2015, 30, 64–77. [Google Scholar] [CrossRef]
- Schlereth, K.; Heyl, C.; Krampitz, A.-M.; Mernberger, M.; Finkernagel, F.; Scharfe, M.; Jarek, M.; Leich, E.; Rosenwald, A.; Stiewe, T. Characterization of the P53 Cistrome–DNA Binding Cooperativity Dissects P53′s Tumor Suppressor Functions. PLoS Genet. 2013, 9, e1003726. [Google Scholar] [CrossRef] [PubMed]
- Nikulenkov, F.; Spinnler, C.; Li, H.; Tonelli, C.; Shi, Y.; Turunen, M.; Kivioja, T.; Ignatiev, I.; Kel, A.; Taipale, J.; et al. Insights into P53 Transcriptional Function via Genome-Wide Chromatin Occupancy and Gene Expression Analysis. Cell Death Differ. 2012, 19, 1992–2002. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A Global Map of P53 Transcription-Factor Binding Sites in the Human Genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef]
- Laptenko, O.; Beckerman, R.; Freulich, E.; Prives, C. P53 Binding to Nucleosomes within the P21 Promoter in Vivo Leads to Nucleosome Loss and Transcriptional Activation. Proc. Natl. Acad. Sci. USA 2011, 108, 10385–10390. [Google Scholar] [CrossRef] [PubMed]
- Botcheva, K.; McCorkle, S.; McCombie, W.R.; Dunn, J.J.; Anderson, C.W. Distinct P53 Genomic Binding Patterns in Normal and Cancer-Derived Human Cells. Cell Cycle 2011, 10, 4237–4249. [Google Scholar] [CrossRef][Green Version]
- Min, S.; Kim, K.; Kim, S.-G.; Cho, H.; Lee, Y. Chromatin-Remodeling Factor, RSF1, Controls P53-Mediated Transcription in Apoptosis Upon DNA Strand Breaks. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef]
- Dearth, L.R.; Qian, H.; Wang, T.; Baroni, T.E.; Zeng, J.; Chen, S.W.; Yi, S.Y.; Brachmann, R.K. Inactive Full-Length P53 Mutants Lacking Dominant Wild-Type P53 Inhibition Highlight Loss of Heterozygosity as an Important Aspect of P53 Status in Human Cancers. Carcinogenesis 2007, 28, 289–298. [Google Scholar] [CrossRef]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of Function Mutations in P53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Noll, J.E.; Jeffery, J.; Al-Ejeh, F.; Kumar, R.; Khanna, K.K.; Callen, D.; Neilsen, P. Mutant P53 Drives Multinucleation and Invasion through a Process That Is Suppressed by ANKRD11. Oncogene 2011, 31, 2836–2848. [Google Scholar] [CrossRef] [PubMed]
- El-Hizawi, S.; Lagowski, J.P.; Kulesz-Martin, M.; Albor, A. Induction of Gene Amplification as a Gain-of-Function Phenotype of Mutant P53 Proteins. Cancer Res. 2002, 62, 3264–3270. [Google Scholar] [PubMed]
- Sarig, R.; Rivlin, N.; Brosh, R.; Bornstein, C.; Kamer, I.; Ezra, O.; Molchadsky, A.; Goldfinger, N.; Brenner, O.; Rotter, V. Mutant P53 Facilitates Somatic Cell Reprogramming and Augments the Malignant Potential of Reprogrammed Cells. J. Exp. Med. 2010, 207, 2127–2140. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant P53 as a Guardian of the Cancer Cell. Cell Death Differ. 2018, 26, 199–212. [Google Scholar] [CrossRef]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. P53 Dynamics Control Cell Fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-Cell Variation in P53 Dynamics Leads to Fractional Killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer Therapeutic Approach Based on Conformational Stabilization of Mutant P53 Protein by Small Peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.N.; Wiman, K.G. Mutant P53 Reactivation by Small Molecules Makes Its Way to the Clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Volckmar, A.-L.; Leichsenring, J.; Kirchner, M.; Christopoulos, P.; Neumann, O.; Budczies, J.; Morais de Oliveira, C.M.; Rempel, E.; Buchhalter, I.; Brandt, R.; et al. Combined Targeted DNA and RNA Sequencing of Advanced NSCLC in Routine Molecular Diagnostics: Analysis of the First 3000 Heidelberg Cases. Int. J. Cancer 2019, 145, 649–661. [Google Scholar] [CrossRef]
- Never-smoker, N.E.S. The Cancer Genome Atlas Research Network Erratum: Corrigendum: Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 514, 262. [Google Scholar] [CrossRef]
- Shi, J.; Hua, X.; Zhu, B.; Ravichandran, S.; Wang, M.; Nguyen, C.; Brodie, S.A.; Palleschi, A.; Alloisio, M.; Pariscenti, G.; et al. Somatic Genomics and Clinical Features of Lung Adenocarcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002162. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Yatabe, Y.; Onozato, R.; Kuwano, H.; Mitsudomi, T. Prognostic Implication of EGFR, KRAS, and TP53 Gene Mutations in a Large Cohort of Japanese Patients with Surgically Treated Lung Adenocarcinoma. J. Thorac. Oncol. 2009, 4, 22–29. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation Patterns in a Population-Based Non-Small Cell Lung Cancer Cohort and Prognostic Impact of Concomitant Mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Scoccianti, C.; Vésin, A.; Martel, G.; Olivier, M.; Brambilla, E.; Timsit, J.-F.; Tavecchio, L.D.; Brambilla, C.; Field, J.K.; Hainaut, P.; et al. Prognostic Value ofTP53, KRAS and EGFR Mutations in Non-Small Cell Lung Cancer: The EUELC Cohort. Eur. Respir. J. 2012, 40, 177–184. [Google Scholar] [CrossRef]
- Tomasini, P.; Mascaux, C.; Jao, K.; Labbe, C.; Kamel-Reid, S.; Stockley, T.; Hwang, D.M.; Leighl, N.B.; Liu, G.; Bradbury, P.A.; et al. Effect of Coexisting KRAS and TP53 Mutations in Patients Treated with Chemotherapy for Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2018, 20, e338–e345. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, C.Y.; Duan, F.G.; Fan, X.-X.; Yao, X.-J.; Parks, R.J.; Tang, Y.-J.; Wang, M.-F.; Liu, L.; Tsang, B.K.; et al. P53 Sensitizes Chemoresistant Non-Small Cell Lung Cancer via Elevation of Reactive Oxygen Species and Suppression of EGFR/PI3K/AKT Signaling. Cancer Cell Int. 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Ma, X.; Le Teuff, G.; Lacas, B.; Tsao, M.S.; Graziano, S.; Pignon, J.-P.; Douillard, J.-Y.; Le Chevalier, T.; Seymour, L.; Filipits, M.; et al. Prognostic and Predictive Effect of TP53 Mutations in Patients with Non–Small Cell Lung Cancer from Adjuvant Cisplatin–Based Therapy Randomized Trials: A LACE-Bio Pooled Analysis. J. Thorac. Oncol. 2016, 11, 850–861. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Bao, H.; Wang, Q.; Mao, W.-M.; Chen, Y.; Tong, X.; Xu, S.-T.; Wu, L.; Wei, Y.-C.; Liu, Y.-Y.; et al. Genomic Signatures Define Three Subtypes of EGFR-Mutant Stage II–III Non-Small-Cell Lung Cancer with Distinct Adjuvant Therapy Outcomes. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Ma, X.; Rousseau, V.; Sun, H.; Lantuejoul, S.; Filipits, M.; Pirker, R.; Popper, H.; Mendiboure, J.; Vataire, A.-L.; Le Chevalier, T.; et al. Significance ofTP53 Mutations as Predictive Markers of Adjuvant Cisplatin-Based Chemotherapy in Completely Resected Non-Small-Cell Lung Cancer. Mol. Oncol. 2014, 8, 555–564. [Google Scholar] [CrossRef]
- Molinavila, M.A.; Bertran-Alamillo, J.; Gascó, A.; Mayo-De-Las-Casas, C.; Sánchez-Ronco, M.; Pujantell-Pastor, L.; Bonanno, L.; Favaretto, A.; Cardona, A.F.; Vergnenègre, A.; et al. Nondisruptive P53 Mutations Are Associated with Shorter Survival in Patients with Advanced Non–Small Cell Lung Cancer. Clin. Cancer Res. 2014, 20, 4647–4659. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Davis, C.W.; Mick, R.; Thompson, J.C.; Ahmed, S.; Jeffries, S.; Bagley, S.; Gabriel, P.; Evans, T.L.; Bauml, J.M.; et al. Influence of TP53 Mutation on Survival in Patients with Advanced EGFR-Mutant Non–Small-Cell Lung Cancer. JCO Precis. Oncol. 2018, 2018, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Aisner, D.L.; Sholl, L.M.; Berry, L.D.; Rossi, M.R.; Chen, H.; Fujimoto, J.; Moreira, A.L.; Ramalingam, S.S.; Villaruz, L.C.; Otterson, G.A.; et al. The Impact of Smoking and TP53 Mutations in Lung Adenocarcinoma Patients with Targetable Mutations—The Lung Cancer Mutation Consortium (LCMC2). Clin. Cancer Res. 2017, 24, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2016, 23, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Petracci, E.; Delmonte, A.; Bronte, G.; Chiadini, E.; Ludovini, V.; Dubini, A.; Papi, M.; Baglivo, S.; De Luigi, N.; et al. Concomitant TP53 Mutation Confers Worse Prognosis in EGFR-Mutated Non-Small Cell Lung Cancer Patients Treated with TKIs. J. Clin. Med. 2020, 9, 1047. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, F.; Wang, Y.; Wu, Q.; Wang, B.; Yao, Y.; Zhang, Y.; Han-Zhang, H.; Ye, J.; Zhang, L.; et al. Mutations in Exon 8 of TP53 Are Associated with Shorter Survival in Patients with Advanced Lung Cancer. Oncol. Lett. 2019, 18, 3159–3169. [Google Scholar] [CrossRef]
- Labbé, C.; Cabanero, M.; Korpanty, G.J.; Tomasini, P.; Doherty, M.K.; Mascaux, C.; Jao, K.; Pitcher, B.; Wang, R.; Pintilie, M.; et al. Prognostic and Predictive Effects of TP53 Co-Mutation in Patients with EGFR-Mutated Non-Small Cell Lung Cancer (NSCLC). Lung Cancer 2017, 111, 23–29. [Google Scholar] [CrossRef]
- Ko, J.-L.; Cheng, Y.-W.; Chang, S.-L.; Su, J.-M.; Chen, C.-Y.; Lee, H. MDM2 mRNA Expression Is a Favorable Prognostic Factor in Non-Small-Cell Lung Cancer. Int. J. Cancer 2000, 89, 265–270. [Google Scholar] [CrossRef]
- Rachiglio, A.M.; Fenizia, F.; Piccirillo, M.C.; Galetta, D.; Crinò, L.; Vincenzi, B.; Barletta, E.; Pinto, C.; Ferraù, F.; Lambiase, M.; et al. The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers 2019, 11, 341. [Google Scholar] [CrossRef]
- Michels, S.; Heydt, C.; Van Veggel, B.; Deschler-Baier, B.; Pardo, N.; Monkhorst, K.; Rüsseler, V.; Stratmann, J.; Griesinger, F.; Steinhauser, S.; et al. Genomic Profiling Identifies Outcome-Relevant Mechanisms of Innate and Acquired Resistance to Third-Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy in Lung Cancer. JCO Precis. Oncol. 2019, 3, 1–14. [Google Scholar] [CrossRef]
- Jin, Y.; Shi, X.; Zhao, J.; He, Q.; Chen, M.; Yan, J.; Ou, Q.; Wu, X.; Shao, Y.W.; Yu, X. Mechanisms of Primary Resistance to EGFR Targeted Therapy in Advanced Lung Adenocarcinomas. Lung Cancer 2018, 124, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, B.; Shim, J.H.; Lee, S.-H.; Park, W.-Y.; Choi, Y.-L.; Sun, J.-M.; Ahn, J.S.; Ahn, M.-J.; Park, K. Concurrent Genetic Alterations Predict the Progression to Target Therapy in EGFR-Mutated Advanced NSCLC. J. Thorac. Oncol. 2019, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Bria, E.; Pilotto, S.; Amato, E.; Fassan, M.; Novello, S.; Peretti, U.; Vavalà, T.; Kinspergher, S.; Righi, L.; Santo, A.; et al. Molecular Heterogeneity Assessment by Next-Generation Sequencing and Response to Gefitinib of EGFR Mutant Advanced Lung Adenocarcinoma. Oncotarget 2015, 6, 12783–12795. [Google Scholar] [CrossRef] [PubMed]
- Helena, A.Y.; Suzawa, K.; Jordan, E.J.; Zehir, A.; Ni, A.; Kim, H.R.; Kris, M.G.; Hellmann, M.D.; Li, B.T.; Somwar, R.; et al. Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clin. Cancer Res. 2018, 24, 3108–3118. [Google Scholar] [CrossRef]
- Roeper, J.; Falk, M.; Chalaris-Rißmann, A.; Lueers, A.C.; Ramdani, H.; Wedeken, K.; Stropiep, U.; Diehl, L.; Tiemann, M.; Heukamp, L.C.; et al. TP53 Co-Mutations in EGFR Mutated Patients in NSCLC Stage IV: A Strong Predictive Factor of ORR, PFS and OS in EGFR mt+ NSCLC. Oncotarget 2020, 11, 250–264. [Google Scholar] [CrossRef]
- Xu, Y.; Tong, X.; Yan, J.; Wu, X.; Shao, Y.W.; Fan, Y. Short-Term Responders of Non–Small Cell Lung Cancer Patients to EGFR Tyrosine Kinase Inhibitors Display High Prevalence of TP53 Mutations and Primary Resistance Mechanisms. Transl. Oncol. 2018, 11, 1364–1369. [Google Scholar] [CrossRef]
- Hou, H.; Qin, K.; Liang, Y.; Zhang, C.; Liu, D.; Jiang, H.; Liu, K.; Zhu, J.; Lv, H.; Li, T.; et al. Concurrent TP53 Mutations Predict Poor Outcomes of EGFR-TKI Treatments in Chinese Patients with Advanced NSCLC. Cancer Manag. Res. 2019, 11, 5665–5675. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, H.R.; Cho, E.K.; Min, Y.J.; Ahn, J.S.; Ahn, M.-J.; Park, K.; Cho, B.C.; Lee, J.-H.; Jeong, H.C.; et al. Targeted Sequencing Identifies Genetic Alterations That Confer Primary Resistance to EGFR Tyrosine Kinase Inhibitor (Korean Lung Cancer Consortium). Oncotarget 2016, 7, 36311–36320. [Google Scholar] [CrossRef]
- Tsui, D.W.Y.; Murtaza, M.; Wong, A.S.C.; Rueda, O.M.; Smith, C.G.; Chandrananda, D.; Soo, R.A.; Lim, H.L.; Goh, B.C.; Caldas, C.; et al. Dynamics of Multiple Resistance Mechanisms in Plasma DNA during EGFR-Targeted Therapies in Non-Small Cell Lung Cancer. EMBO Mol. Med. 2018, 10, e7945. [Google Scholar] [CrossRef]
- Qin, K.; Hou, H.; Liang, Y.; Zhang, X. Prognostic Value of TP53 Concurrent Mutations for EGFR-TKIs and ALK-TKIs Based Targeted Therapy in Advanced Non-Small Cell Lung Cancer: A Meta-Analysis. BMC Cancer 2020, 20, 1–16. [Google Scholar] [CrossRef]
- Zhang, R.; Tian, P.; Chen, B.; Wang, T.; Li, W. The Prognostic Impact of TP53 Comutation in EGFR Mutant Lung Cancer Patients: A Systematic Review and Meta-Analysis. Postgrad. Med. 2019, 131, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, N.; Gao, G.; Deng, H.-B.; Wang, Z.-H.; Deng, L.-L.; Yang, Y.; Lu, C. Prognostic Value of TP53 Co-Mutation Status Combined with EGFR Mutation in Patients with Lung Adenocarcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Jeon, S.Y.; Jeong, G.S.; Lee, H.W.; Jeong, S.H.; Kang, S.Y.; Park, J.S.; Choi, J.-H.; Koh, Y.W.; Han, J.H.; et al. EGFR Exon 19 Deletion Is Associated with Favorable Overall Survival after First-Line Gefitinib Therapy in Advanced Non–Small Cell Lung Cancer Patients. Am. J. Clin. Oncol. 2018, 41, 385–390. [Google Scholar] [CrossRef]
- Hong, W.; Wu, Q.; Zhang, J.; Zhou, Y. Prognostic Value of EGFR 19-del and 21-L858R Mutations in Patients with Non-Small Cell Lung Cancer. Oncol. Lett. 2019, 18, 3887–3895. [Google Scholar] [CrossRef]
- VanderLaan, P.; Rangachari, D.; Mockus, S.M.; Spotlow, V.; Reddi, H.V.; Malcolm, J.; Huberman, M.S.; Joseph, L.J.; Kobayashi, S.S.; Costa, D.B. Mutations in TP53, PIK3CA, PTEN and Other Genes in EGFR Mutated Lung Cancers: Correlation with Clinical Outcomes. Lung Cancer 2017, 106, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Bai, H.; Li, T.; Gao, B.; Han, J.; Chang, G.; Zhang, P.; Fei, K.; He, X.; Wang, J. TP53 Mutations in Circulating Tumor DNA in Advanced Epidermal Growth Factor Receptor-Mutant Lung Adenocarcinoma Patients Treated with Gefitinib. Transl. Oncol. 2021, 14, 101163. [Google Scholar] [CrossRef]
- Li, X.-M.; Li, W.-F.; Lin, J.-T.; Yan, H.-H.; Tu, H.-Y.; Chen, H.-J.; Wang, B.-C.; Wang, Z.; Zhou, Q.; Zhang, X.-C.; et al. Predictive and Prognostic Potential of TP53 in Patients with Advanced Non–Small-Cell Lung Cancer Treated with EGFR-TKI: Analysis of a Phase III Randomized Clinical Trial (CTONG 0901). Clin. Lung Cancer 2020, 22, 100–109. [Google Scholar] [CrossRef]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at Risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef]
- Lee, J.-K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.-W.; Heo, D.S.; et al. Clonal History and Genetic Predictors of Transformation into Small-Cell Carcinomas from Lung Adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB Loss in Resistant EGFR Mutant Lung Adenocarcinomas That Transform to Small-Cell Lung Cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020, 38, 2926–2936. [Google Scholar] [CrossRef] [PubMed]
- Clinical Lung Cancer Genome Project. The Clinical Lung Cancer Genome Project (CLCGP) and Network Genomic Medicine (NGM) A Genomics-Based Classification of Human Lung Tumors. Sci. Transl. Med. 2013, 5, 209ra153. [Google Scholar] [CrossRef]
- Jung, S.; Kim, D.H.; Choi, Y.J.; Kim, S.Y.; Park, H.; Lee, H.; Choi, C.-M.; Sung, Y.H.; Lee, J.C.; Rho, J.K. Contribution of P53 in Sensitivity to EGFR Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Pailler, E.; Oulhen, M.; Borget, I.; Remon, J.; Ross, K.; Auger, N.; Billiot, F.; Camus, M.N.; Commo, F.; Lindsay, C.R.; et al. Circulating Tumor Cells with Aberrant ALK Copy Number Predict Progression-Free Survival during Crizotinib Treatment in ALK-Rearranged Non–Small Cell Lung Cancer Patients. Cancer Res. 2017, 77, 2222–2230. [Google Scholar] [CrossRef]
- Wei, Y.; Shen, K.; Lv, T.; Wang, X.; Li, C.; Fan, H.; Lv, Y.; Liu, H.; Song, Y. Three New Disease-Progression Modes in NSCLC Patients after EGFR-TKI Treatment by Next-Generation Sequencing Analysis. Lung Cancer 2018, 125, 43–50. [Google Scholar] [CrossRef]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.; Olivas, V.R.; et al. Evolution and Clinical Impact of Co-Occurring Genetic Alterations in Advanced-Stage EGFR-Mutant Lung Cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]


{kind=link}
| Study | Treatment Arms | mPFS (Months) | mOS (Months) | ORR (%) | Ref. |
|---|---|---|---|---|---|
| IPASS | Gefitinib vs. carboplatin/paclitaxel | 9.8 vs. 6.4 p < 0.001 | 21.6 vs. 21.9 p = 0.99 | 71.2 vs. 47.3 | [15] |
| NEJ002 | Gefitinib vs. carboplatin/paclitaxel | 10.8 vs. 5.4 p < 0.001 | 27.7 vs. 26.6 p = 0.48 | 73.7 vs. 30.7 | [13] |
| WJT0G3405 | Gefitinib vs. cisplatin/docetaxel | 9.2 vs. 6.3 p < 0.0001 | 36.0 vs. 39.0 | 62.1 vs. 32.2 | [16] |
| OPTIMAL | Erlotinib vs. carboplatin/gemcitabine | 13.1 vs. 4.6 p < 0.0001 | 22.8 vs. 27.2 p = 0.27 | 83.0 vs. 36.0 | [17] |
| EURTAC | Erlotinib vs. cisplatin/docetaxel | 9.7 vs. 5.2 p < 0.0001 | 19.3 vs. 19.5 p = 0.87 | 64.0 vs. 18.0 | [18] |
| ENSURE | Erlotinib vs. cisplatin/gemcitabine | 11 vs. 5.6 | 26.3 vs. 25.5 | 62.7 vs. 33.6 | [20] |
| CONVINCE | Icotinib vs. cisplatin/pemetrexed | 11.2 vs. 7.9 p = 0.006 | 30.5 vs. 32.1 p = 0.89 | NR | [21] |
| LUX-Lung 3 | Afatinib vs. cisplatin/Pemetrexed | 11.1 vs. 6.9 p = 0.001 | 28.2 vs. 28.2 p = 0.39 | 56.1 vs. 22.6 | [22] |
| LUX-Lung 6 | Afatinib vs. cisplatin/gemcitabine | 11.0 vs. 5.6 p < 0.0001 | 23.1 vs. 23.5 p = 0.61 | 66.9 vs. 23.0 | [23] |
| AURA 3 | Osimertinib vs. platinum/pemetrexed | 10.1 vs. 4.4 p < 0.001 | NA | 26.8 vs. 22.5 | [24] |
| CTONG | Erlotinib vs. gefitinib | 13.2 vs. 11.1 | 22.4 vs. 20.7 | NR | [25] |
| LUX-Lung 7 | Afatinib vs. gefitinib | 13.7 vs. 11.5 p = 0.007 | 27.9 vs. 24.5 | 70 vs. 56 | [26] |
| FLAURA | Osimertinib vs. erlotinib or gefitinib | 18.9 vs. 10.2 p < 0.001 | NR | 80.0 vs. 76 | [27] |
| Clinical Trial Number | Phase | Treatment Arms | Primary Endpoint | Link to the Clinical Trial |
|---|---|---|---|---|
| NCT03515837 | III | Pembrolizumab + pemetrexed + chemo vs. placebo + pemetrexed + chemo | PFS, OS | https://clinicaltrials.gov/ct2/show/NCT03515837 (accessed on 28 January 2022) |
| NCT03778229 | II | Osimertinib + savolitinib | ORR | https://www.clinicaltrials.gov/ct2/show/NCT03778229 (accessed on 28 January 2022) |
| NCT03944772 | II | Osimertinib + savolitinib vs. osimertinib + gefitinib vs. osimertinib + necitumumab vs. durvalumab + carboplatin + pemetrexed | ORR | https://www.clinicaltrials.gov/ct2/show/NCT03944772 (accessed on 28 January 2022) |
| NCT03940703 | II | Tepotinib + osimertinib | Safety, ORR | https://clinicaltrials.gov/ct2/show/NCT03940703 (accessed on 28 January 2022) |
| NCT04136535 | II | Pemetrexed and carboplatin with or without anlotinib | PFS | https://www.clinicaltrials.gov/ct2/show/NCT4136535 (accessed on 28 January 2022) |
| NCT03532698 | II | Osimertinib + aspirin | ORR | https://www.clinicaltrials.gov/ct2/show/NCT03532698 (accessed on 28 January 2022) |
| NCT04316351 | II | Toripalimab + pemetrexed + anlotinib | ORR | https://www.clinicaltrials.gov/ct2/show/NCT04316351 (accessed on 28 January 2022) |
| NCT03784599 | I/II | Trastuzumab emtansine and osimertinib | Safety, ORR | https://www.clinicaltrials.gov/ct2/show/NCT03784599 (accessed on 28 January 2022) |
| NCT03891615 | I | Osimertinib + Niraparib | MTD | https://www.clinicaltrials.gov/ct2/show/NCT03891615 (accessed on 28 January 2022) |
| NCT03516214 | I | Nazartinib and trametinib | MTD; RP2D | https://www.clinicaltrials.gov/ct2/show/NCT03516214 (accessed on 28 January 2022) |
| TP53 Status | Number of Patients (Generation of TKI) | Result | Ref. |
|---|---|---|---|
| Non-disruptive mutations | 193 (I) | OS | [105] |
| Any mutation | 131 (I–II) | OS | [106] |
| Any mutation | 116 (I–II) | OS | [107] |
| Exon 8 mutations | 123 (I–II) | DCR, PFS, OS | [108] |
| Exon 8 mutations | 136 (I–II) | OS | [109] |
| Exon 8 mutations | 379 (I–II) | OS | [110] |
| Missense mutations | 60 (I–II) | PFS | [111] |
| Any mutation | 75; 82 (I–II; III) | PFS, PFS | [116] |
| Any mutation | 18 (I) | TTP | [117] |
| Any mutation | 374 (I–II) | TTP | [118] |
| Any mutation | 28 (I) | TTP | [119] |
| Any mutation | 132 (I) | PFS, OS | [121] |
| Any mutation | 71 (I) | TTP, OS | [120] |
| Any mutation | 50 (I) | OS | [122] |
| Any mutationExon 6, 7 mutations | 368 (I) | PFS, OS | [129] |
| Exon 4, 7 mutations | 256 (I) | PFS, OS | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canale, M.; Andrikou, K.; Priano, I.; Cravero, P.; Pasini, L.; Urbini, M.; Delmonte, A.; Crinò, L.; Bronte, G.; Ulivi, P. The Role of TP53 Mutations in EGFR-Mutated Non-Small-Cell Lung Cancer: Clinical Significance and Implications for Therapy. Cancers 2022, 14, 1143. https://doi.org/10.3390/cancers14051143
Canale M, Andrikou K, Priano I, Cravero P, Pasini L, Urbini M, Delmonte A, Crinò L, Bronte G, Ulivi P. The Role of TP53 Mutations in EGFR-Mutated Non-Small-Cell Lung Cancer: Clinical Significance and Implications for Therapy. Cancers. 2022; 14(5):1143. https://doi.org/10.3390/cancers14051143
Chicago/Turabian StyleCanale, Matteo, Kalliopi Andrikou, Ilaria Priano, Paola Cravero, Luigi Pasini, Milena Urbini, Angelo Delmonte, Lucio Crinò, Giuseppe Bronte, and Paola Ulivi. 2022. "The Role of TP53 Mutations in EGFR-Mutated Non-Small-Cell Lung Cancer: Clinical Significance and Implications for Therapy" Cancers 14, no. 5: 1143. https://doi.org/10.3390/cancers14051143
APA StyleCanale, M., Andrikou, K., Priano, I., Cravero, P., Pasini, L., Urbini, M., Delmonte, A., Crinò, L., Bronte, G., & Ulivi, P. (2022). The Role of TP53 Mutations in EGFR-Mutated Non-Small-Cell Lung Cancer: Clinical Significance and Implications for Therapy. Cancers, 14(5), 1143. https://doi.org/10.3390/cancers14051143