
CD47-SIRPα Checkpoint Disruption in Metastases Requires Tumor-Targeting Antibody for Molecular and Engineered Macrophage Therapies

 , , , , , , and
, , , , , , and 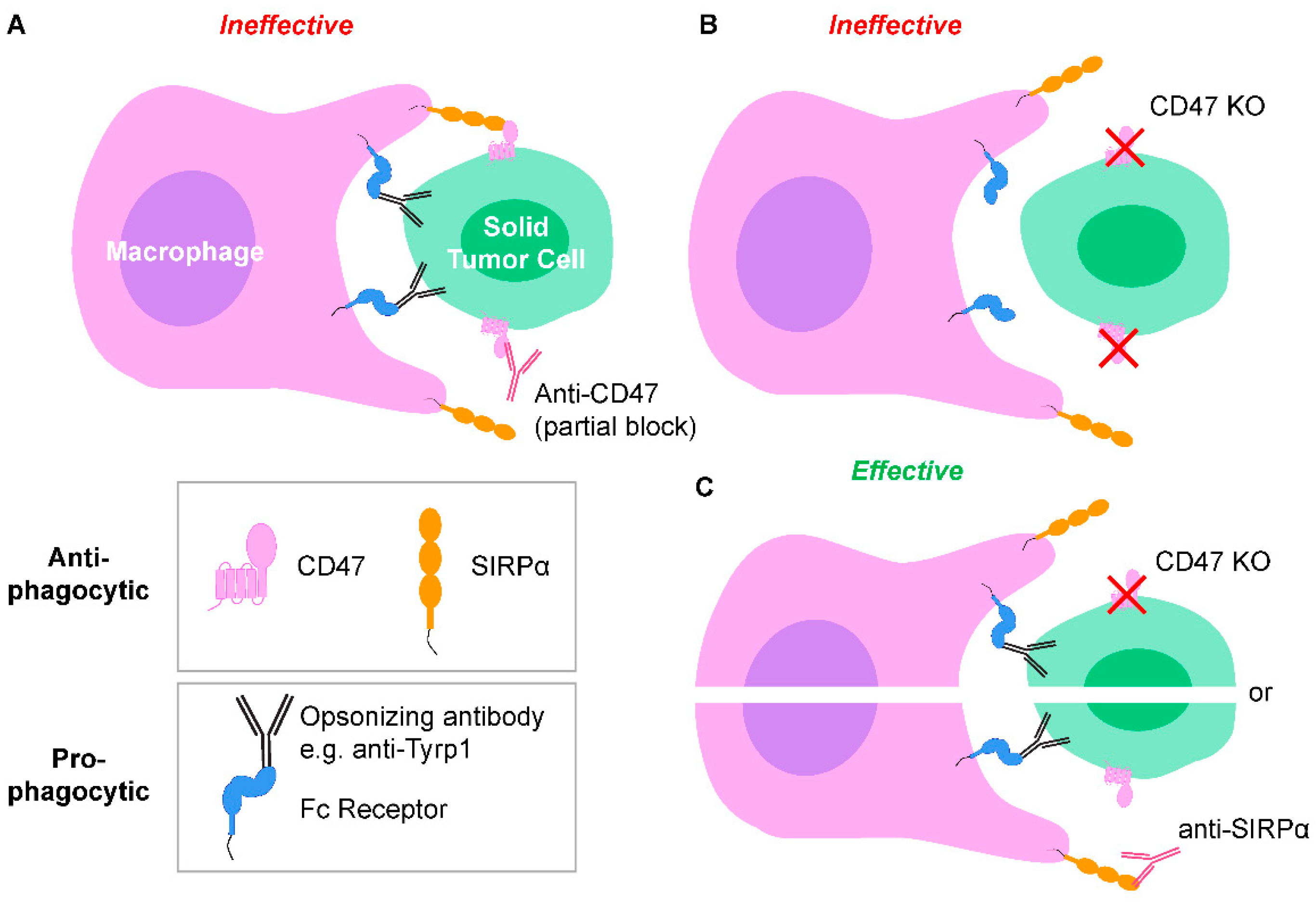{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
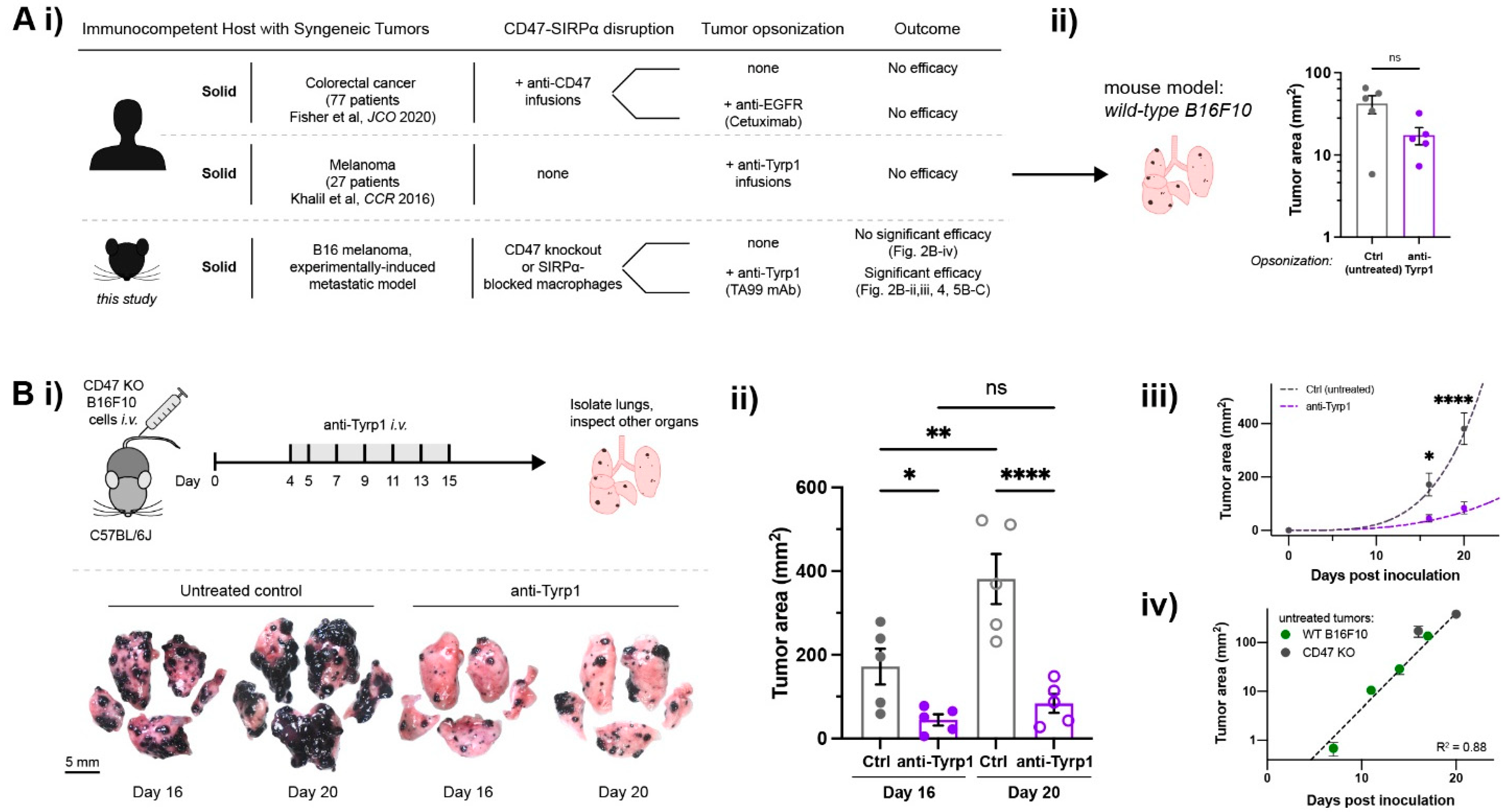
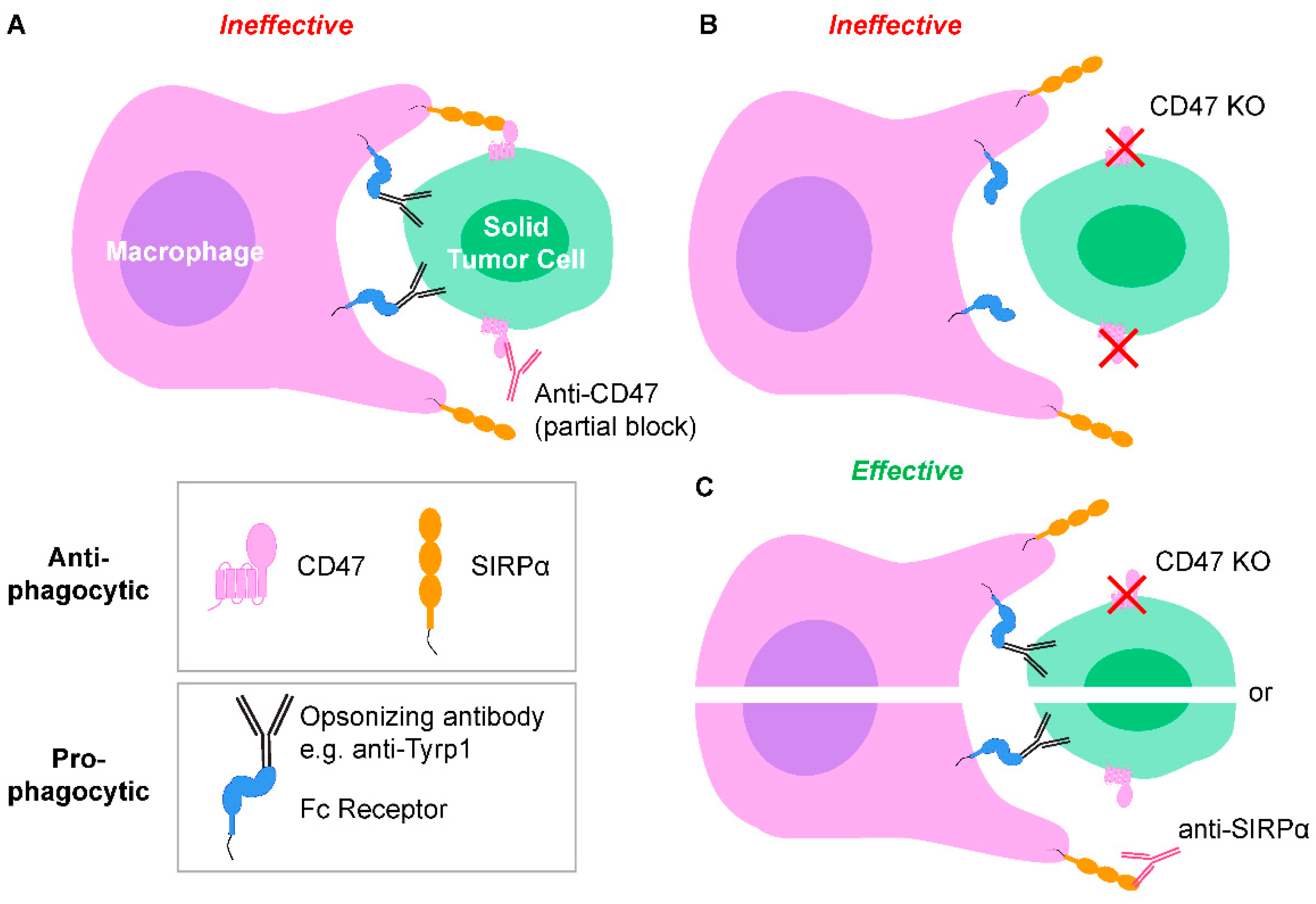
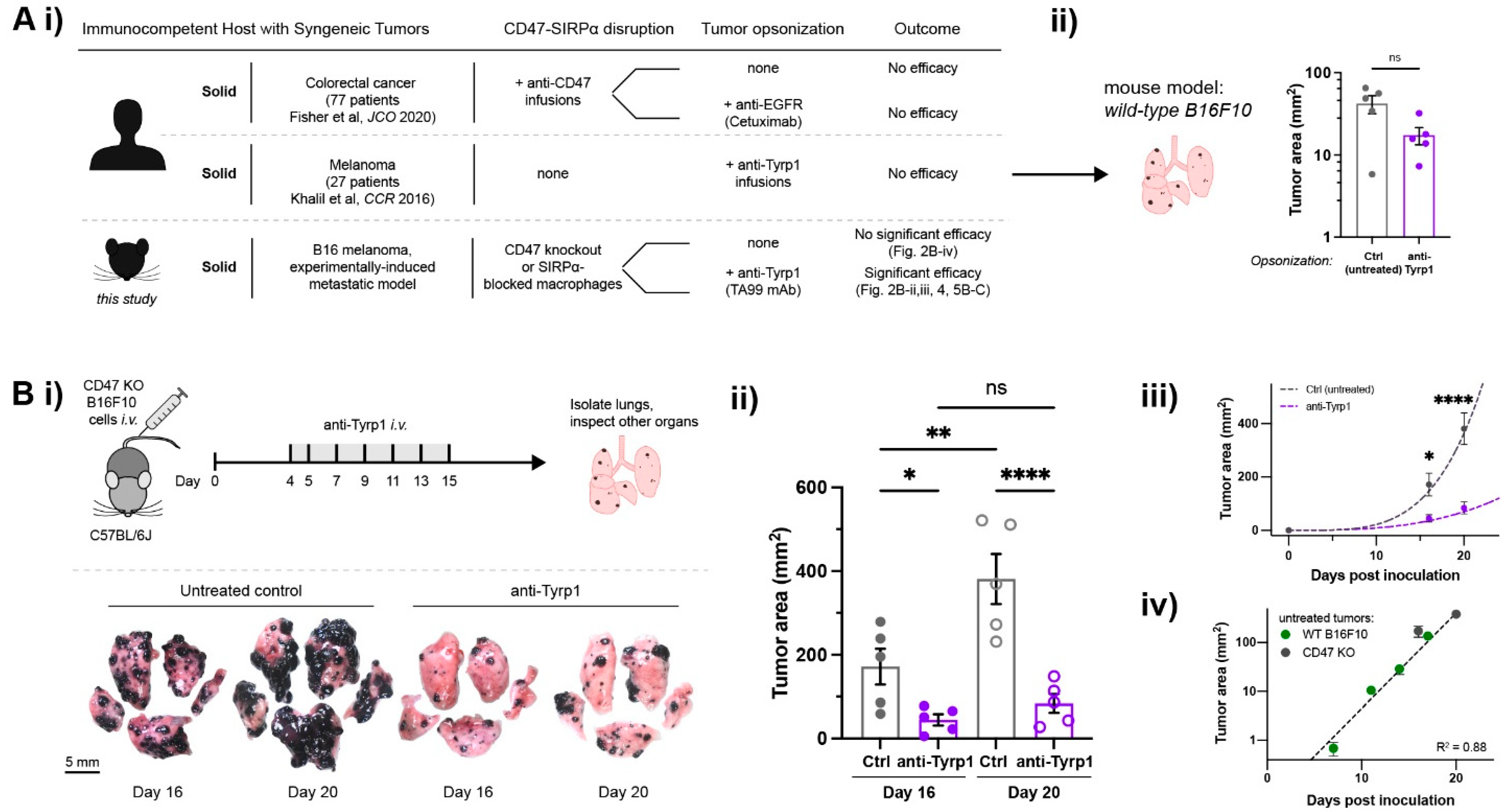
2.1. Tumor-Opsonizing Antibodies Drive Regression of Metastases Only When CD47 Is Disrupted
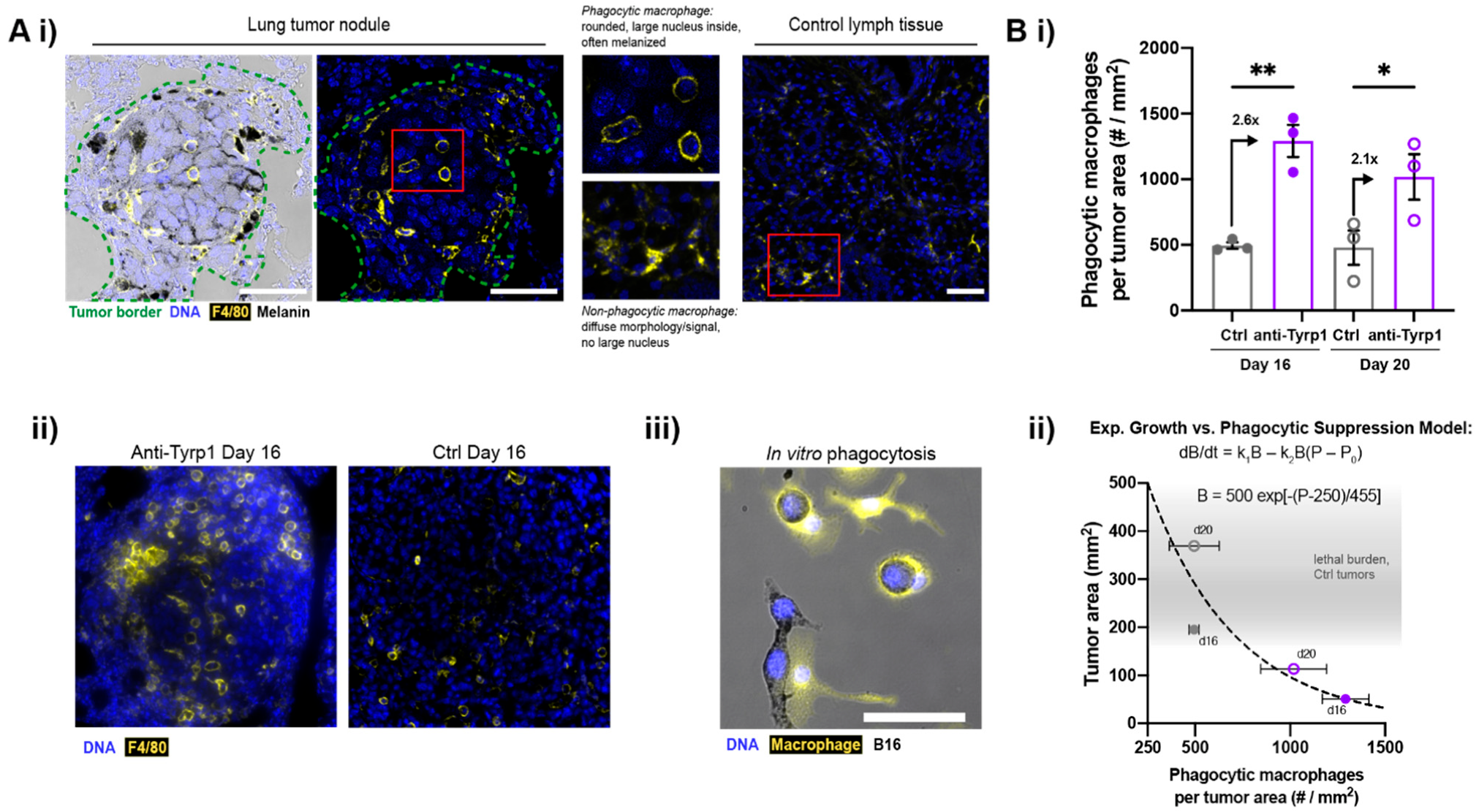
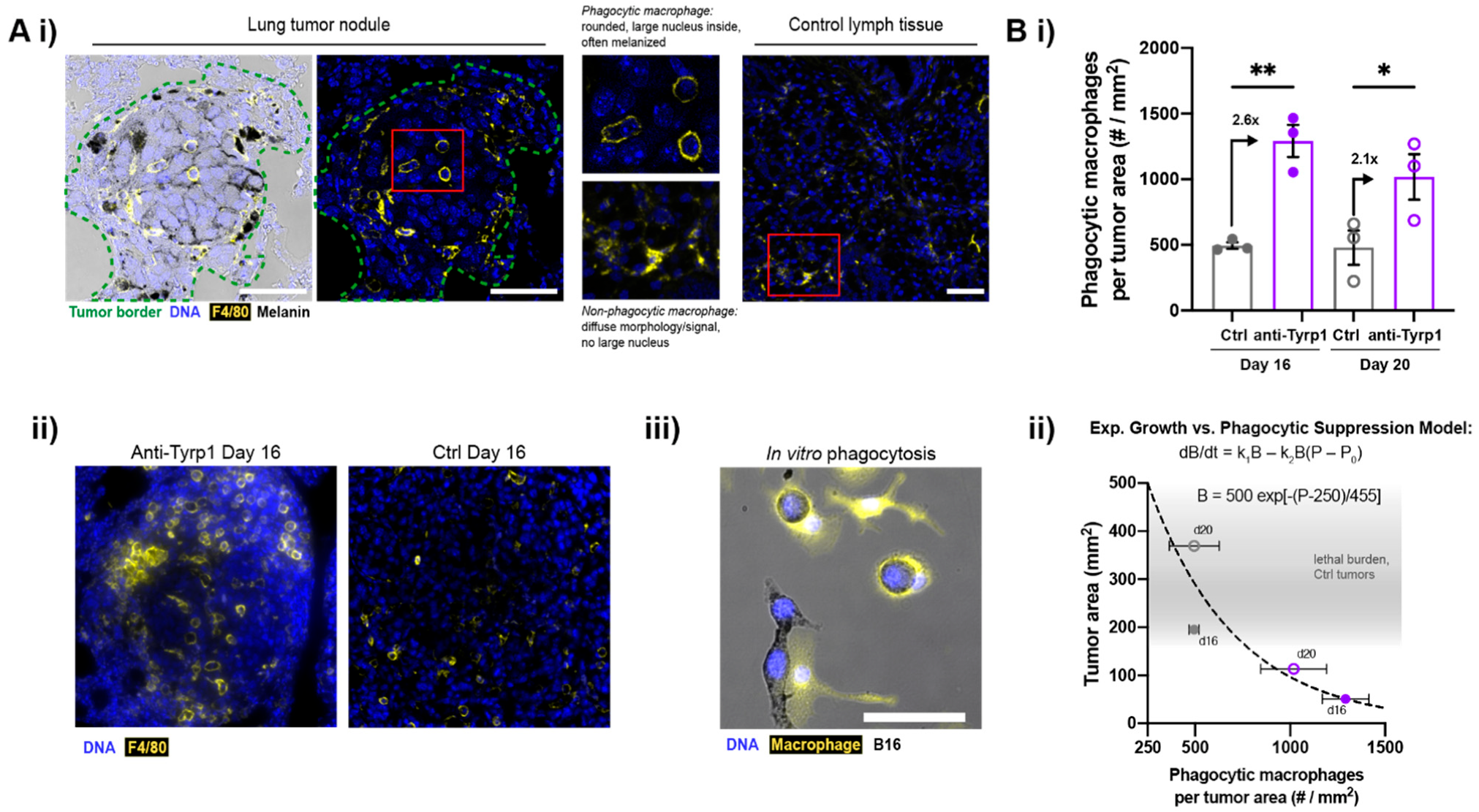
2.2. Macrophages Infiltrate and Phagocytose CD47-Depleted, IgG-Opsonized B16F10 Nodules
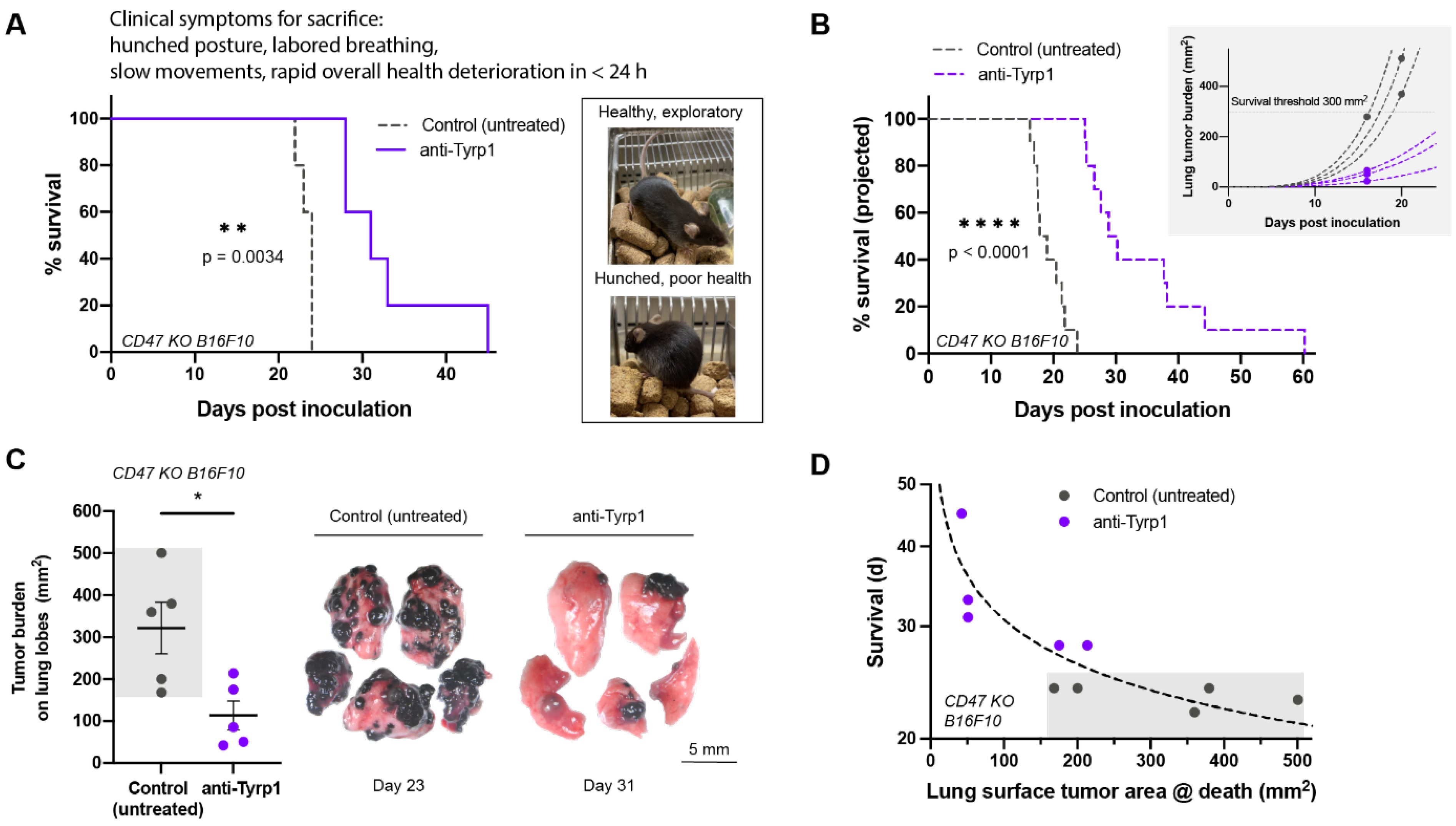
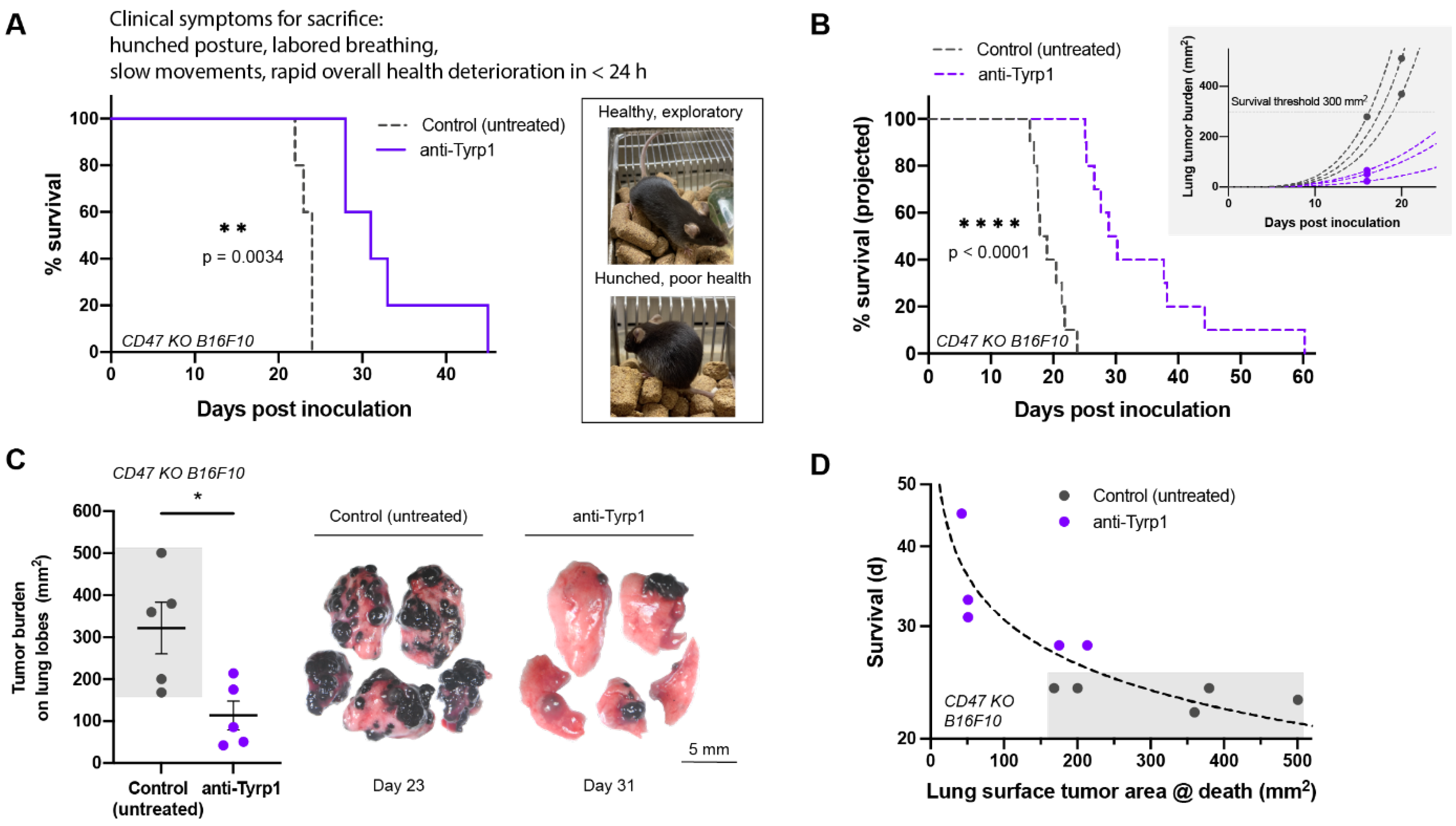
2.3. Tumor Clearance Induced by Opsonization and CD47 Deletion Prolongs Survival, but Some Tumors Escape
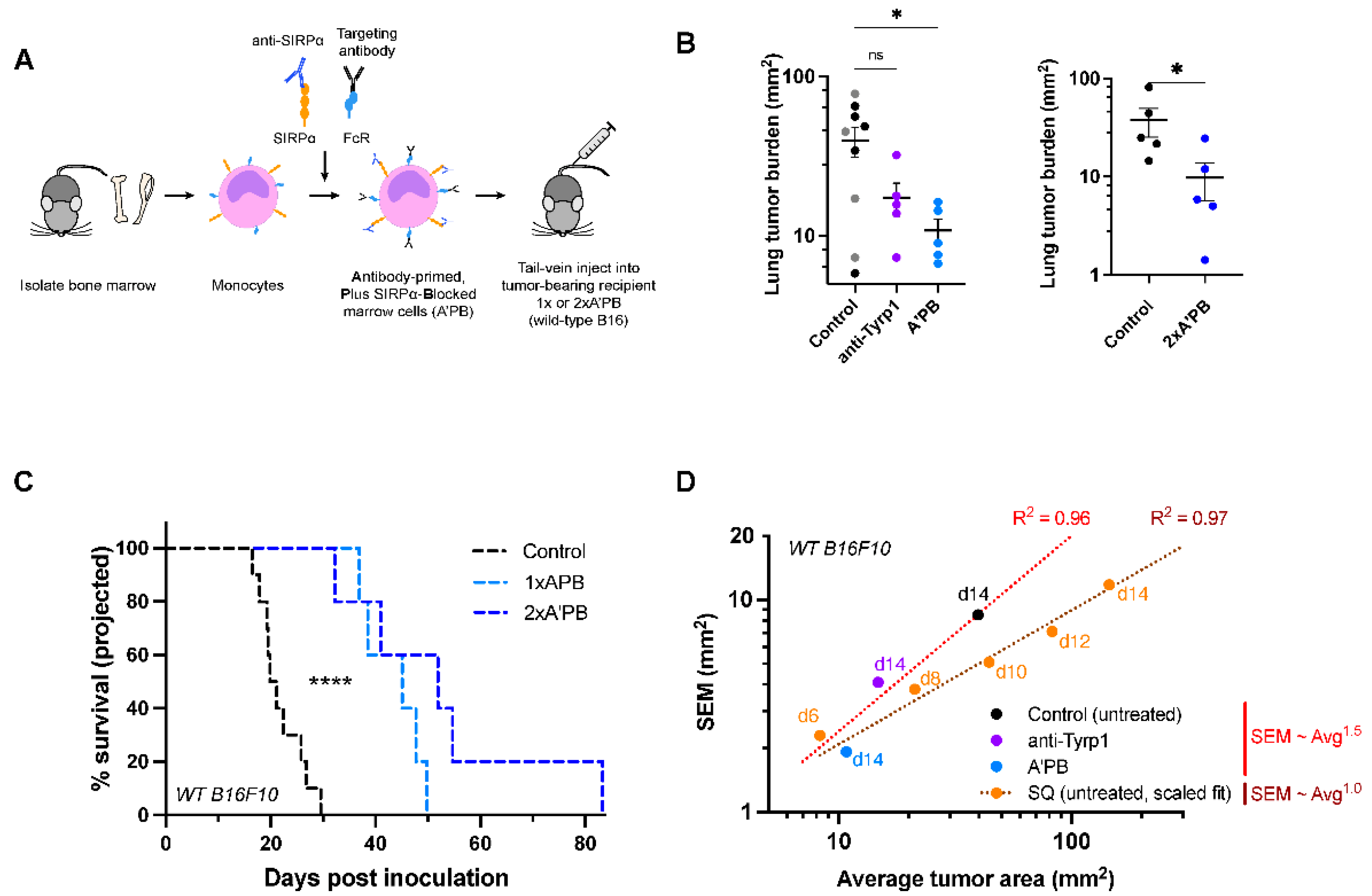
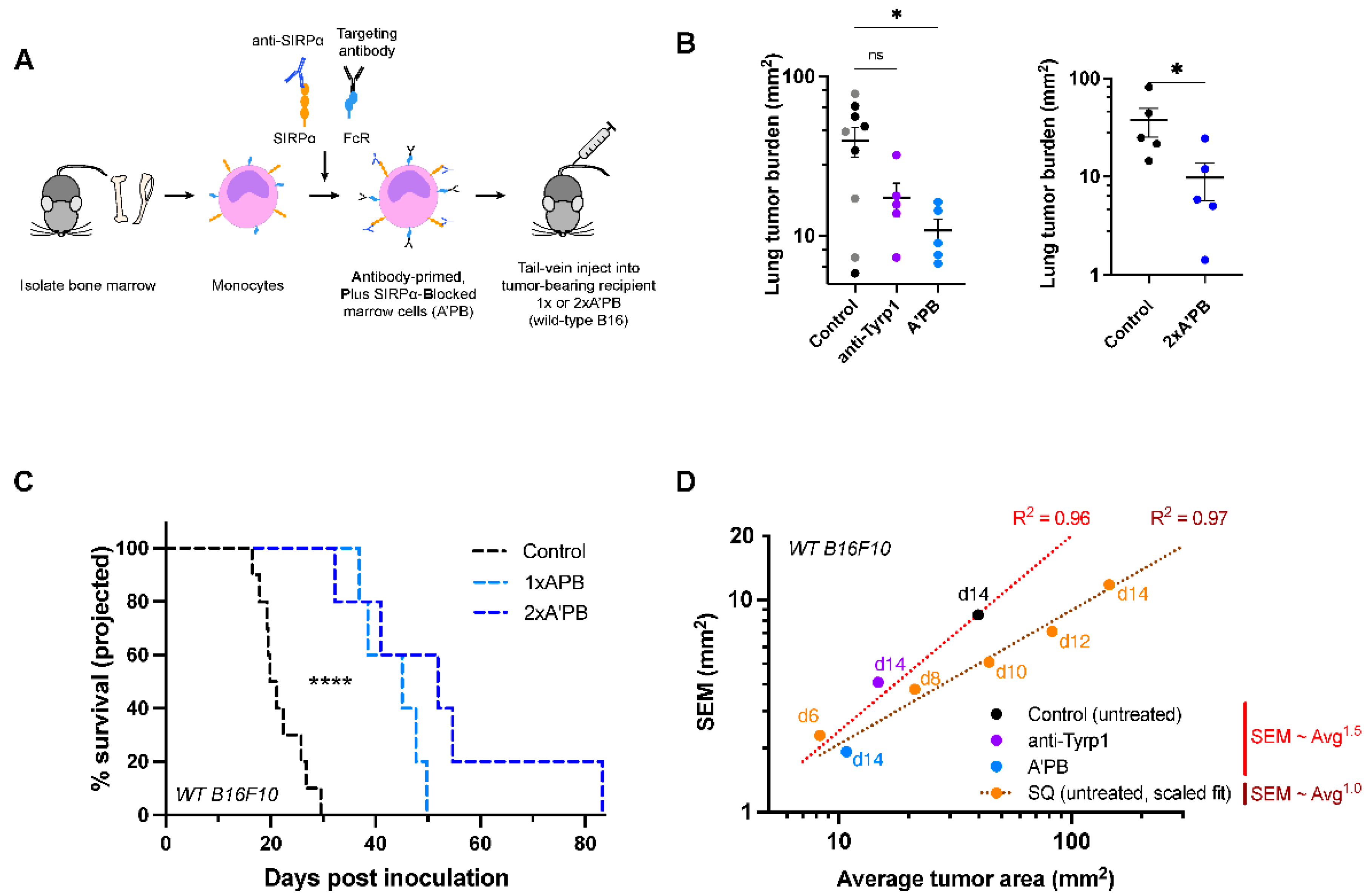
2.4. Antibody-Engineered Marrow Macrophages/Monocytes Eliminate Lung Tumor Nodules in WT Metastases, While Anti-Tyrp1 Treatment Alone Does Not
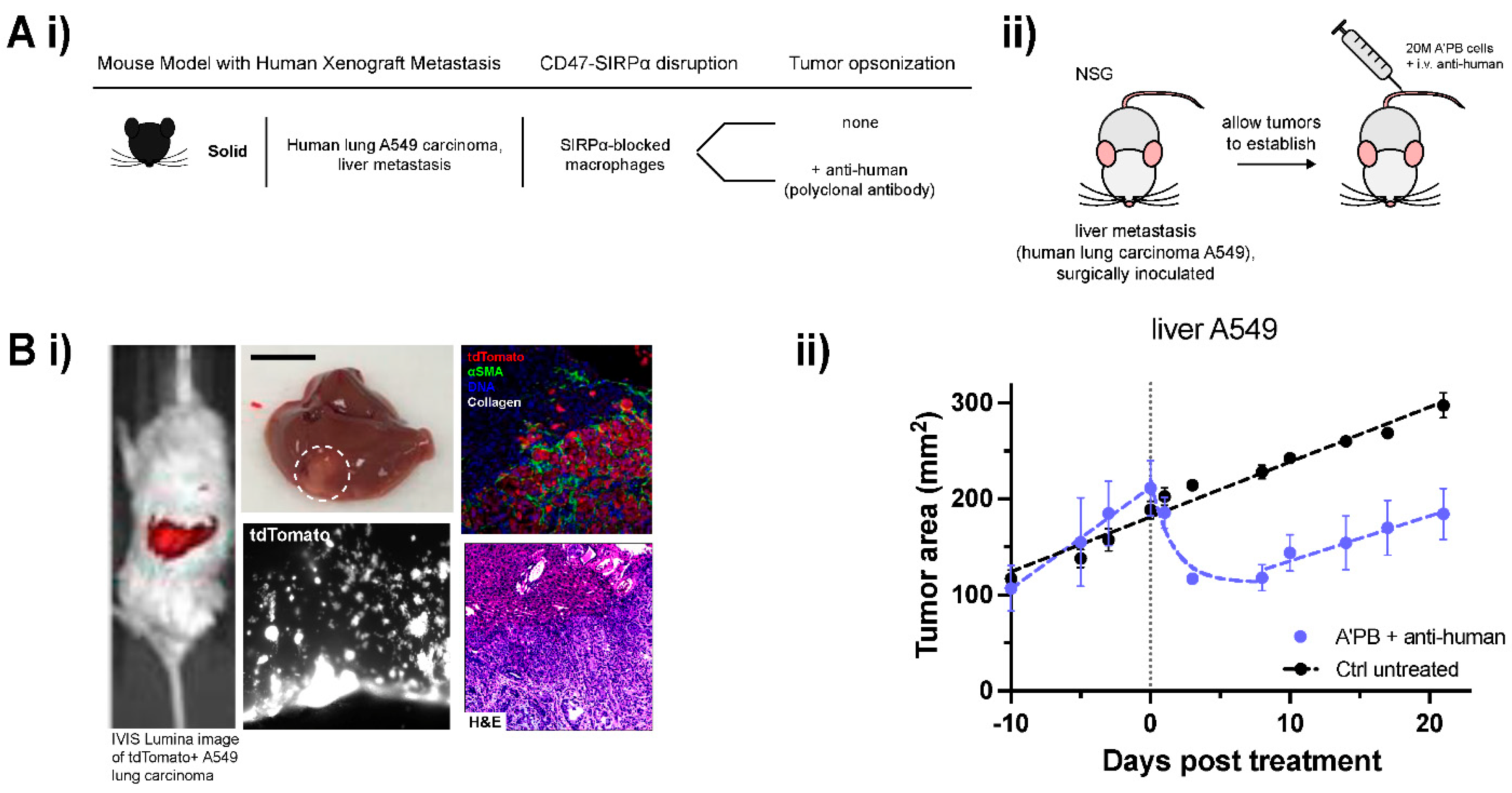
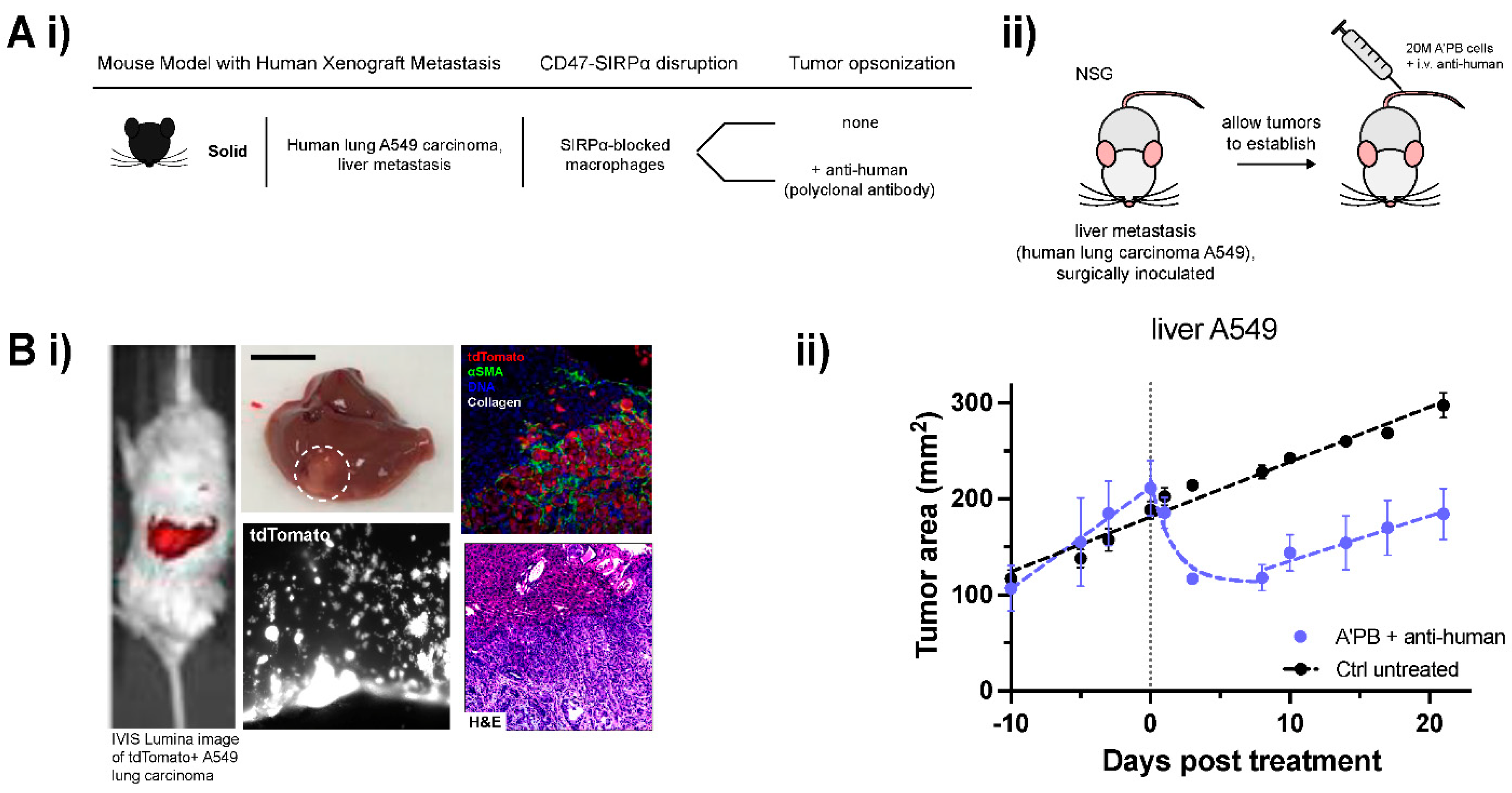
2.5. WT Human Lung Cancer Metastases in Liver Suppressed by Antibody-Engineered Marrow Macrophages/Monocytes
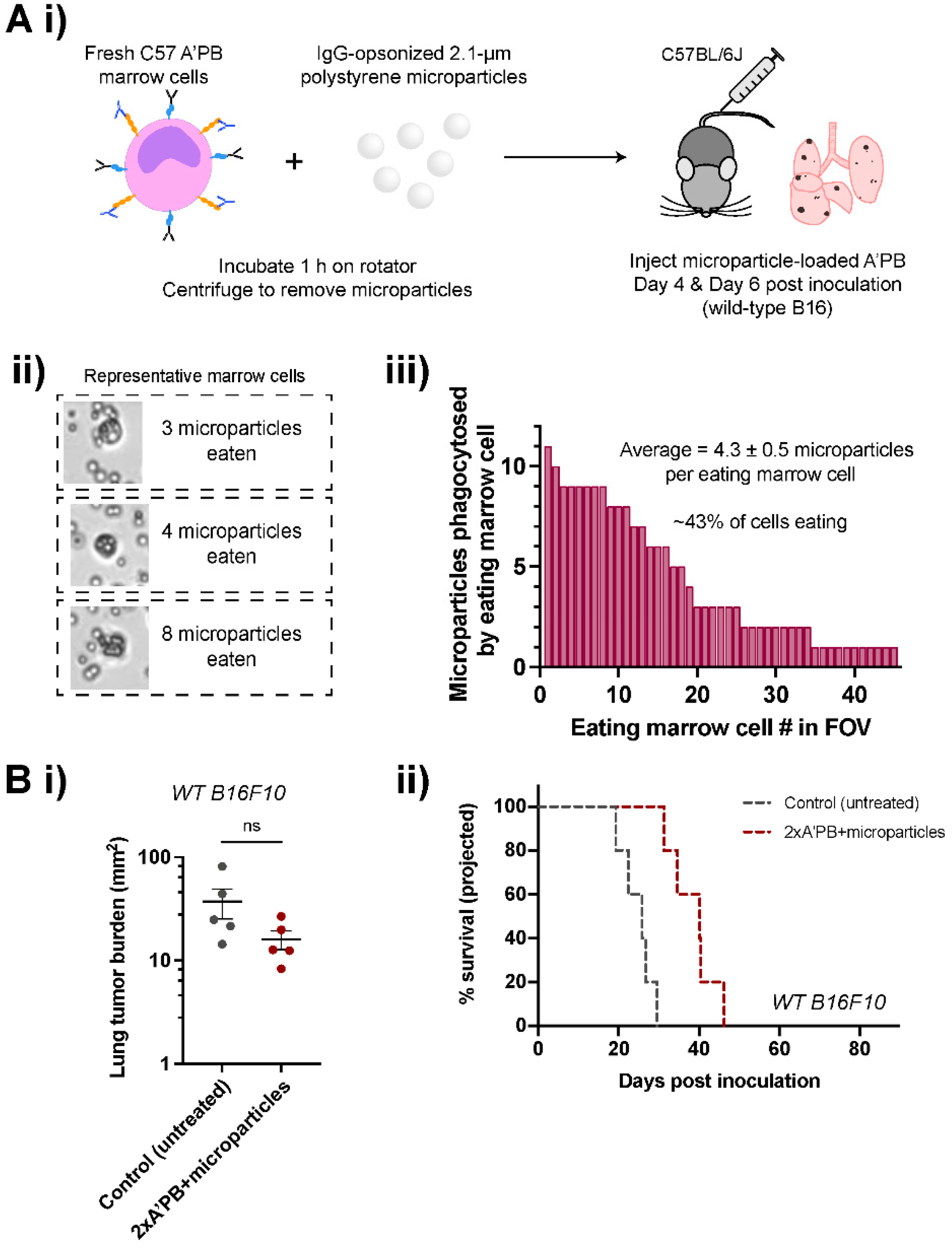
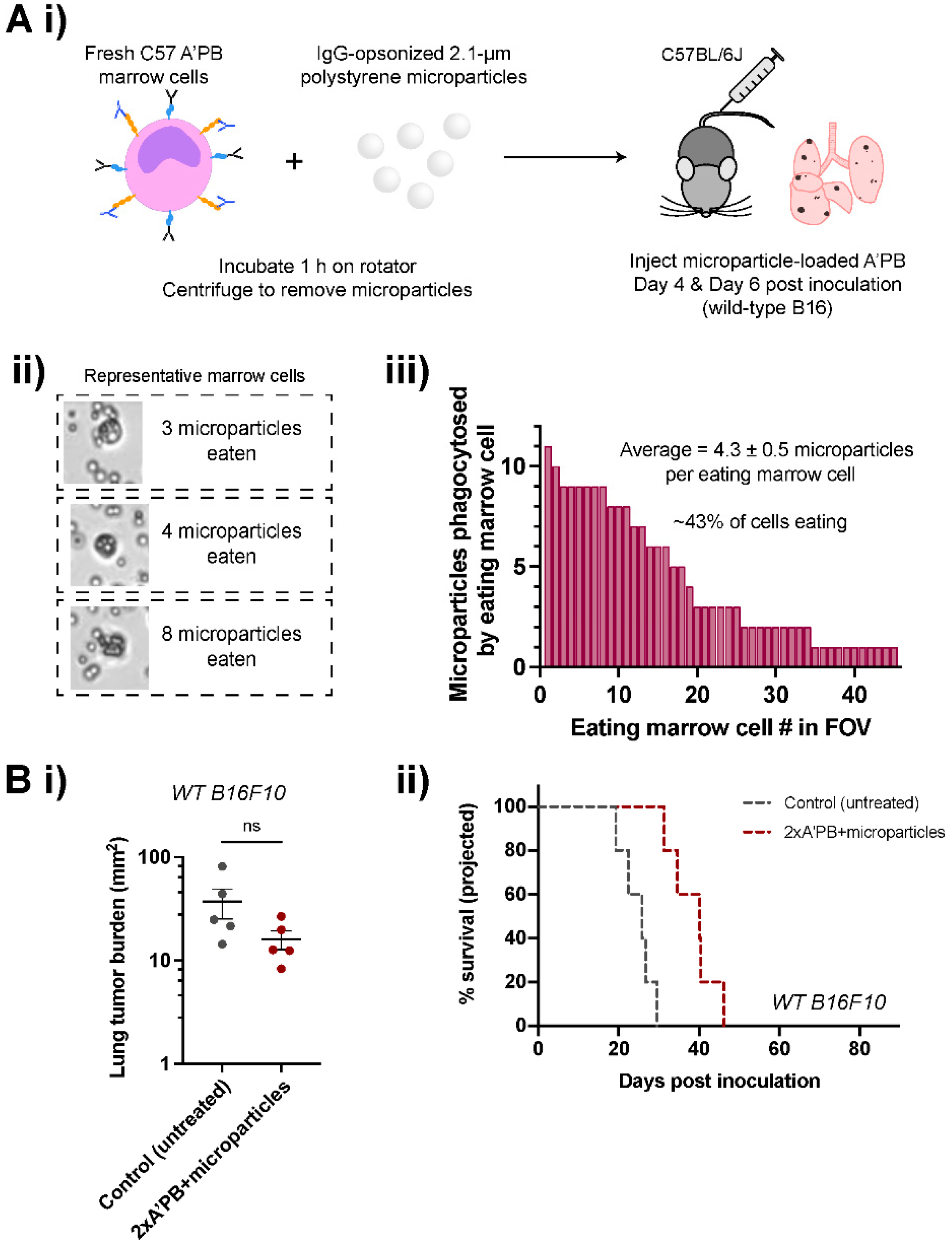
2.6. Antibody-Engineered Marrow Macrophages/Monocytes Are Less Effective When Pre-Loaded with Microparticles
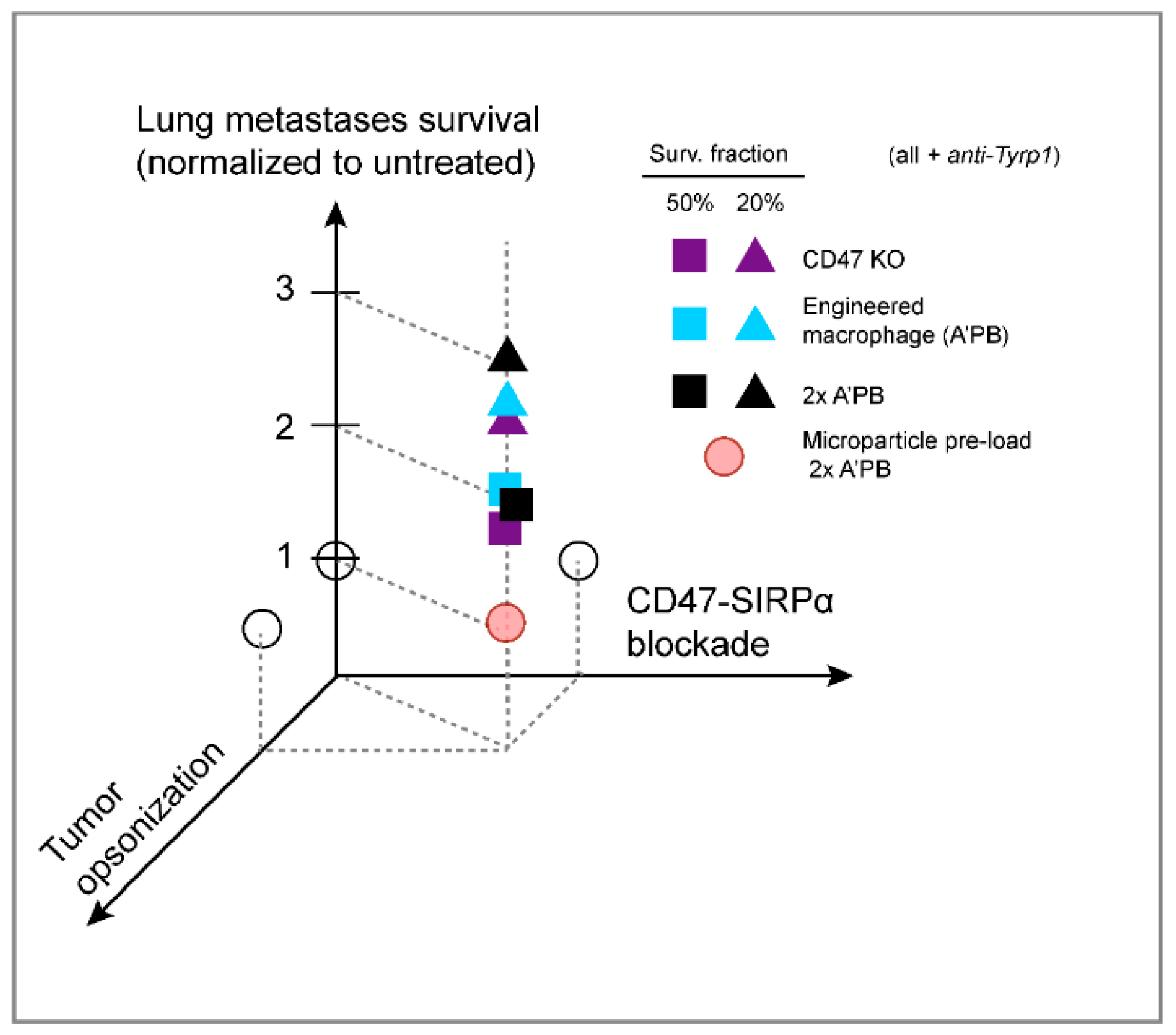
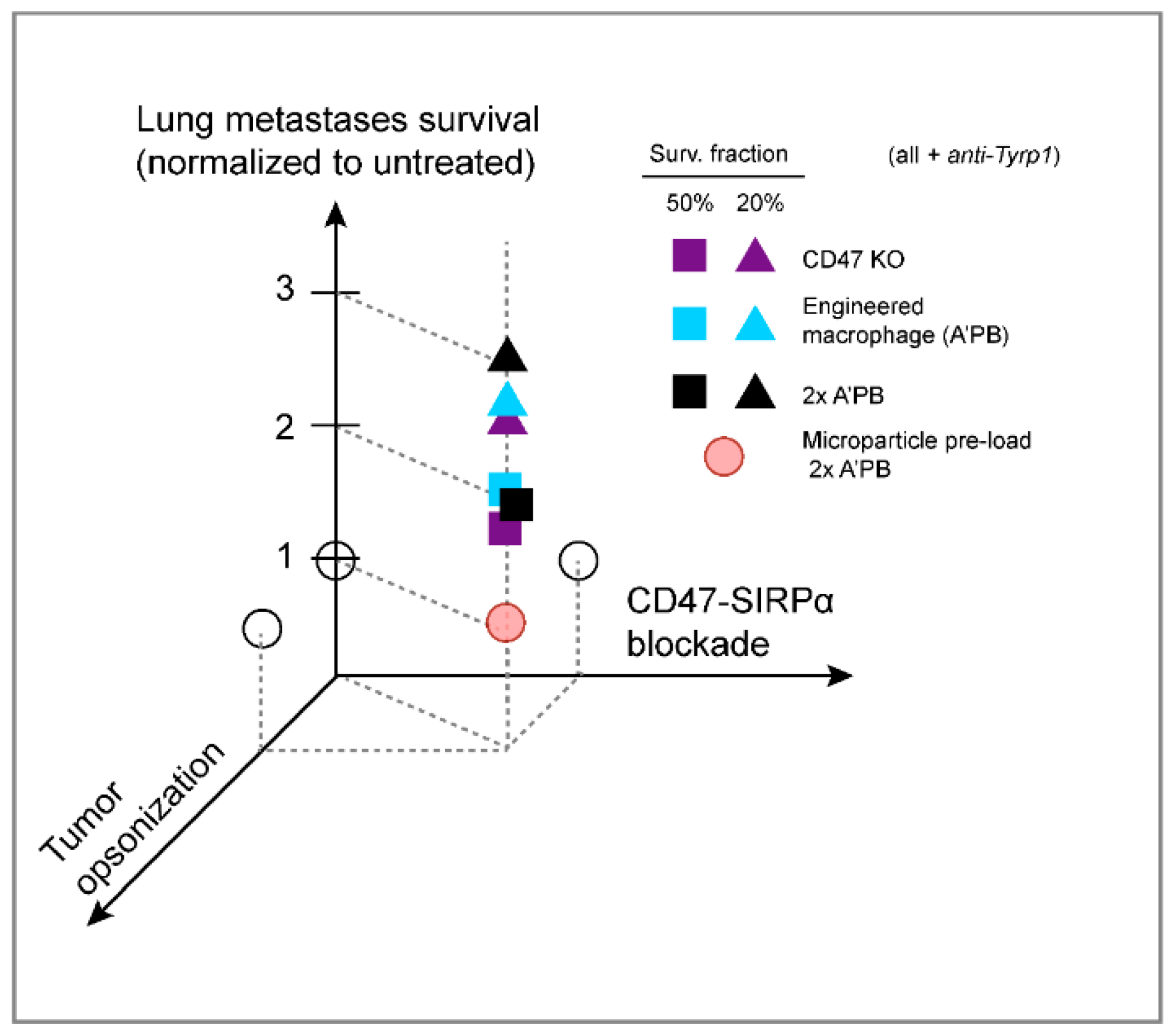
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Quantification of Tumor Burden
4.3. Quantitative Analyses for Estimation of Tumor Growth and Projected Survival
4.4. Marrow Harvest and Engineering
4.5. Cell Culture
4.6. CRISPR-Cas9 Deletion of CD47
4.7. Phagocytosis Assays
4.8. Immunohistochemistry
4.9. Statistical Analyses and Equation Fitting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oldenborg, P.-A. Role of CD47 as a Marker of Self on Red Blood Cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrechak, J.C.; Dooling, L.J.; Discher, D.E. The macrophage checkpoint CD47: SIRPα for recognition of ‘self’ cells: From clinical trials of blocking antibodies to mechanobiological fundamentals. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180217. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Horrigan, S.K.; Iorns, E.; Williams, S.R.; Perfito, N.; Errington, T.M. Replication study: The CD47-signal regulatory protein alpha (SIRPA) interaction is a therapeutic target for human solid tumors. Elife 2017, 6, e18173. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.K.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.M.; Volkmer, J.-P.; Levin, A.M.; Volkmer, A.K.; Özkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPα Variants as Immunotherapeutic Adjuvants to Anticancer Antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Abrisqueta, P.; Sancho, J.-M.; Cordoba, R.; Persky, D.O.; Andreadis, C.; Huntington, S.F.; Carpio, C.; Morillo Giles, D.; Wei, X.; Li, Y.F.; et al. Anti-CD47 Antibody, CC-90002, in Combination with Rituximab in Subjects with Relapsed and/or Refractory Non-Hodgkin Lymphoma (R/R NHL). Blood 2019, 134, 4089. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalil, A.R.; Andrechak, J.C.; Discher, D.E. Macrophage checkpoint blockade: Results from initial clinical trials, binding analyses, and CD47-SIRPα structure–function. Antib. Ther. 2020, 3, 80–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, G.A.; Lakhani, N.J.; Eng, C.; Hecht, J.R.; Bendell, J.C.; Philip, P.A.; O’Dwyer, P.J.; Johnson, B.; Kardosh, A.; Ippolito, T.M.; et al. A phase Ib/II study of the anti-CD47 antibody magrolimab with cetuximab in solid tumor and colorectal cancer patients. J. Clin. Oncol. 2020, 38, 114. [Google Scholar] [CrossRef]
- Stanchina, M.; Soong, D.; Zheng-Lin, B.; Watts, J.M.; Taylor, J. Advances in acute myeloid leukemia: Recently approved therapies and drugs in development. Cancers 2020, 12, 3225. [Google Scholar] [CrossRef]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.-P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [Green Version]
- Andrejeva, G.; Capoccia, B.J.; Hiebsch, R.R.; Donio, M.J.; Darwech, I.M.; Puro, R.J.; Pereira, D.S. Novel SIRPα Antibodies That Induce Single-Agent Phagocytosis of Tumor Cells while Preserving T Cells. J. Immunol. 2021, 206, 712–721. [Google Scholar] [CrossRef]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 2013, 36, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.H.; Tsai, R.K.; Dooling, L.J.; Kadu, S.; Lee, J.Y.; Pantano, D.; Rodriguez, P.L.; Subramanian, S.; Shin, J.W.; Discher, D.E. Macrophages show higher levels of engulfment after disruption of cis interactions between CD47 and the checkpoint receptor SIRPα. J. Cell Sci. 2020, 133, jcs237800. [Google Scholar] [CrossRef] [Green Version]
- Clynes, R.; Takechi, Y.; Moroi, Y.; Houghton, A.; Ravetch, J.V. Fc receptors are required in passive and active immunity to melanoma. Proc. Natl. Acad. Sci. USA 1998, 95, 652–656. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, M.; Mancardi, D.A.; Macdonald, L.E.; Iannascoli, B.; Zitvogel, L.; Murphy, A.J.; Leusen, J.H.; Bruhns, P. Cutting Edge: FcγRIII (CD16) and FcγRI (CD64) Are Responsible for Anti-Glycoprotein 75 Monoclonal Antibody TA99 Therapy for Experimental Metastatic B16 Melanoma. J. Immunol. 2012, 189, 5513–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, I.; Takechi, Y.; Houghton, A.N. Implicating a role for immune recognition of self in tumor rejection: Passive immunization against the brown locus protein. J. Exp. Med. 1995, 182, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.N.; Postow, M.A.; Ibrahim, N.; Ludwig, D.L.; Cosaert, J.; Kambhampati, S.R.P.; Tang, S.; Grebennik, D.; Kauh, J.S.W.; Lenz, H.; et al. An Open-Label, Dose–Escalation Phase I Study of Anti-TYRP1 Monoclonal Antibody IMC-20D7S for Patients with Relapsed or Refractory Melanoma. Clin. Cancer Res. 2016, 22, 5204–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sockolosky, J.T.; Dougan, M.; Ingram, J.R.; Ho, C.C.M.; Kauke, M.J.; Almo, S.C.; Ploegh, H.L.; Garcia, K.C. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2646–E2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, J.R.; Blomberg, O.S.; Sockolosky, J.T.; Ali, L.; Schmidt, F.I.; Pishesha, N.; Espinosa, C.; Dougan, S.K.; Garcia, K.C.; Ploegh, H.L.; et al. Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc. Natl. Acad. Sci. USA 2017, 114, 10184–10189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvey, C.; Discher, D.E. Engineering macrophages to eat cancer: From “marker of self” CD47 and phagocytosis to differentiation. J. Leukoc. Biol. 2017, 102, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmann, C.T.; Figueroa, S.D.; Rold, T.L.; Volkert, W.A.; Hoffman, T.J. Microimaging Characterization of a B16-F10 Melanoma Metastasis Mouse Model. Mol. Imaging 2006, 5, 105–114. [Google Scholar] [CrossRef]
- Stephenson, E.M. Karyotype Analysis of the B16 Mouse Melanoma With Reassessment of the Normal Mouse Idiogram. JNCI J. Natl. Cancer Inst. 1970, 45, 789–800. [Google Scholar] [CrossRef]
- Alvey, C.M.; Spinler, K.R.; Irianto, J.; Pfeifer, C.R.; Hayes, B.; Xia, Y.; Cho, S.; Dingal, P.C.P.D.; Hsu, J.; Smith, L.; et al. SIRPA-Inhibited, Marrow-Derived Macrophages Engorge, Accumulate, and Differentiate in Antibody-Targeted Regression of Solid Tumors. Curr. Biol. 2017, 27, 2065–2077. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.L.; Harada, T.; Christian, D.A.; Pantano, D.A.; Tsai, R.K.; Discher, D.E. Minimal “self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Nair, P.R.; Alvey, C.; Jin, X.; Irianto, J.; Ivanovska, I.; Discher, D.E. Filomicelles Deliver a Chemo-Differentiation Combination of Paclitaxel and Retinoic Acid That Durably Represses Carcinomas in Liver to Prolong Survival. Bioconjug. Chem. 2018, 29, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.S.; Brown, M.H.; Barclay, A.N.; Hatherley, D. Signal-regulatory protein α from the NOD mouse binds human CD47 with an exceptionally high affinity—Implications for engraftment of human cells. Immunology 2014, 143, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Prasolava, T.K.; Wang, J.C.Y.; Mortin-Toth, S.M.; Khalouei, S.; Gan, O.I.; Dick, J.E.; Danska, J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007, 8, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dowling, J.P.; Murray, W.K.; McArthur, G.A.; Thompson, J.F.; Wolfe, R.; Kelly, J.W. Rate of Growth in Melanomas. Arch. Dermatol. 2006, 142, 1551–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Vijaykumar, A.; Raghavan, J.V.; Rananaware, S.R.; Alakesh, A.; Bodele, J.; Rehman, J.U.; Shukla, S.; Wagde, V.; Nadig, S.; et al. Particle uptake driven phagocytosis in macrophages and neutrophils enhances bacterial clearance. J. Control. Release 2022, 343, 131–141. [Google Scholar] [CrossRef]
- Lehmann, B.; Biburger, M.; Brückner, C.; Ipsen-escobedo, A.; Gordan, S.; Lehmann, C.; Voehringer, D.; Winkler, T.; Schaft, N.; Dudziak, D.; et al. Tumor location determines tissue-specific recruitment of tumor-associated macrophages and antibody-dependent immunotherapy response. Sci. Immunol. 2017, 2, 6413. [Google Scholar] [CrossRef]
- Querfeld, C.; Thompson, J.; Taylor, M.H.; Pillai, R.; Johnson, L.D.; Catalano, T.; Petrova, P.S.; Thompson, T.; Uger, R.A.; Shou, Y.; et al. Intralesional Administration of the CD47 Antagonist TTI-621 (SIRPαFc) Induces Responses in Both Injected and Non-Injected Lesions in Patients with Relapsed/Refractory Mycosis Fungoides and Sézary Syndrome: Interim Results of a Multicenter Phase I Trial. Blood 2018, 132, 1653. [Google Scholar] [CrossRef]
- Querfeld, C.; Thompson, J.; Taylor, M.; Pillai, R.; Johnson, L.D.S.; Catalano, T.; Petrova, P.S.; Uger, R.A.; Irwin, M.; Sievers, E.L.; et al. A single direct intratumoral injection of TTI-621 (SIRPαFc) induces antitumor activity in patients with relapsed/refractory mycosis fungoides and Sézary syndrome: Preliminary findings employing an immune checkpoint inhibitor blocking the CD47 “do not eat”. Blood 2017, 130, 4076. [Google Scholar]
- Tsai, R.K.; Rodriguez, P.L.; Discher, D.E. Self inhibition of phagocytosis: The affinity of “marker of self” CD47 for SIRPα dictates potency of inhibition but only at low expression levels. Blood Cells Mol. Dis. 2010, 45, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Andrechak, J.C.; Dooling, L.J.; Hayes, B.H.; Kadu, S.; Zhang, W.; Pan, R.; Vashisth, M.; Irianto, J.; Alvey, C.M.; Discher, D.E. Cooperative phagocytosis underlies macrophage immunotherapy of solid tumours and initiates a broad anti-tumour IgG response. bioRxiv 2022. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrechak, J.C.; Dooling, L.J.; Tobin, M.P.; Zhang, W.; Hayes, B.H.; Lee, J.Y.; Jin, X.; Irianto, J.; Discher, D.E. CD47-SIRPα Checkpoint Disruption in Metastases Requires Tumor-Targeting Antibody for Molecular and Engineered Macrophage Therapies. Cancers 2022, 14, 1930. https://doi.org/10.3390/cancers14081930
Andrechak JC, Dooling LJ, Tobin MP, Zhang W, Hayes BH, Lee JY, Jin X, Irianto J, Discher DE. CD47-SIRPα Checkpoint Disruption in Metastases Requires Tumor-Targeting Antibody for Molecular and Engineered Macrophage Therapies. Cancers. 2022; 14(8):1930. https://doi.org/10.3390/cancers14081930
Chicago/Turabian StyleAndrechak, Jason C., Lawrence J. Dooling, Michael P. Tobin, William Zhang, Brandon H. Hayes, Justine Y. Lee, Xiaoling Jin, Jerome Irianto, and Dennis E. Discher. 2022. "CD47-SIRPα Checkpoint Disruption in Metastases Requires Tumor-Targeting Antibody for Molecular and Engineered Macrophage Therapies" Cancers 14, no. 8: 1930. https://doi.org/10.3390/cancers14081930
APA StyleAndrechak, J. C., Dooling, L. J., Tobin, M. P., Zhang, W., Hayes, B. H., Lee, J. Y., Jin, X., Irianto, J., & Discher, D. E. (2022). CD47-SIRPα Checkpoint Disruption in Metastases Requires Tumor-Targeting Antibody for Molecular and Engineered Macrophage Therapies. Cancers, 14(8), 1930. https://doi.org/10.3390/cancers14081930