
Metabolic Vulnerabilities in Multiple Myeloma


Abstract
Simple Summary
Abstract
1. Introduction
Metabolic Deregulations Predict Adverse Prognosis in MM
2. Current Literature of Metabolic Abnormalities in MM
2.1. Physiological Role of Metabolism in Plasma Cells
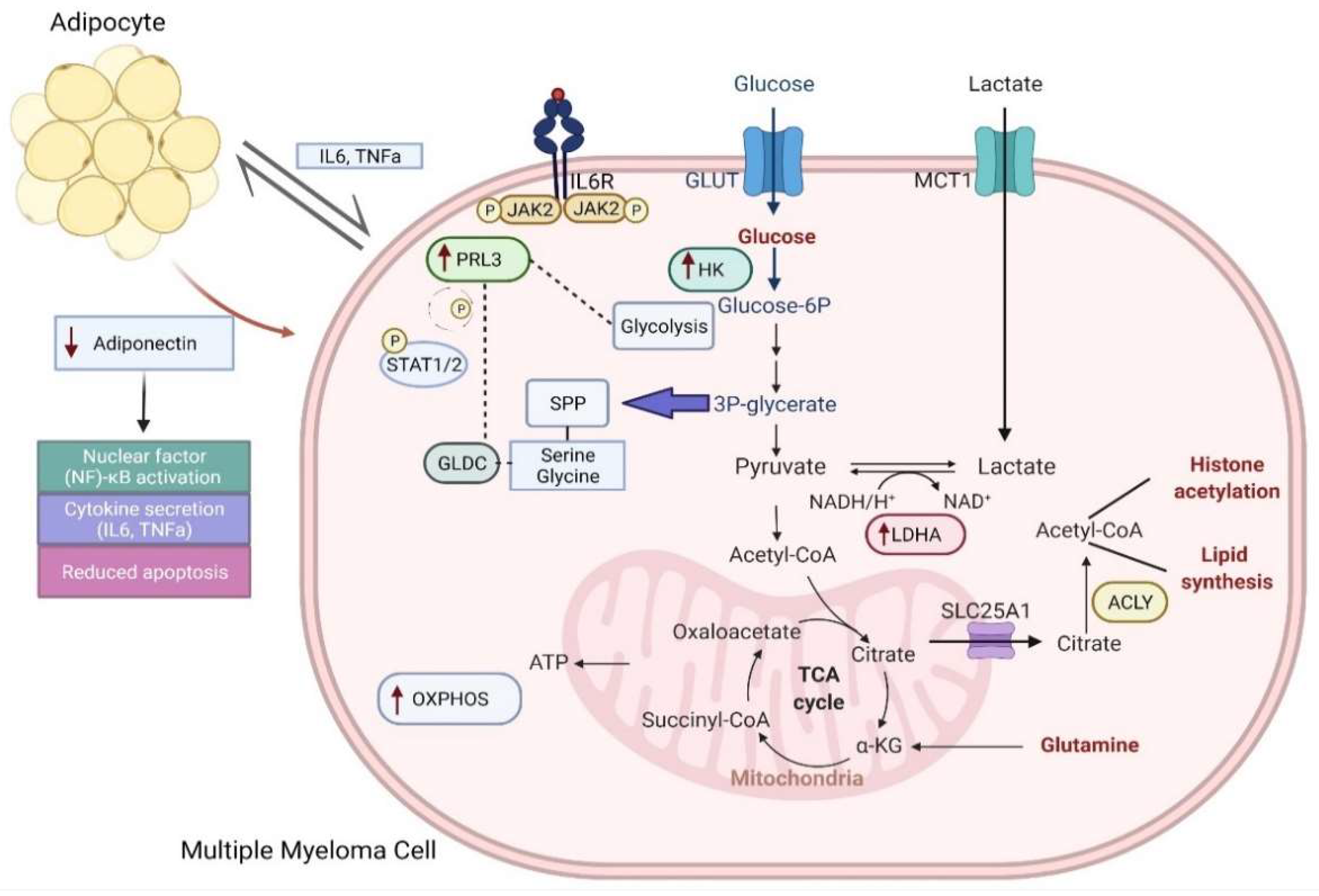
2.2. Myeloma Cells Undergo Metabolic Rewiring of Glycolysis and Mitochondria OXPHOS
2.3. Fatty Acid Metabolism Obesity as a Risk Factor in Myeloma
2.4. PRL3: An Important Metabolic Regulator of Glycolysis and Serine/Glycine Metabolism
3. Clinical Implications of Metabolic Deregulation in Myeloma
3.1. Implications of Resistance to Proteasome Inhibitors
3.2. Implications on Melphalan Resistance
3.3. Impact of Metabolism on Components of the BM Tumor Microenvironment
3.4. Metabolic Deregulation Attenuates Immunotherapy
3.4.1. Crosstalk between Metabolic Rewiring and Lenalidomide Treatment
3.4.2. Impact of Metabolic Alterations on the Regulation of T, and CAR-T Cell Functions through IMiDs
3.4.3. Impact of Adenosine Metabolic Alterations on Monoclonal CD38 Antibodies
4. Future Perspectives
4.1. Synergism between Dynamic Crosstalk of Epigenetics and Metabolism
4.2. c-MAF as a Regulator of OXPHOS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.H.; Al Hamed, R.; Malard, F.; Harousseau, J.-L.; Mohty, M. Relapsed refractory multiple myeloma: A comprehensive overview. Leukemia 2019, 33, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- El Arfani, C.; Maes, K.; Menu, E.; De Veirman, K.; De Bruyne, E. Metabolic Features of Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 1200. [Google Scholar] [CrossRef]
- Rosko, A.; Giralt, S.; Mateos, M.-V.; Dispenzieri, A. Myeloma in Elderly Patients: When Less Is More and More Is More. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Fryar, C.D.; Carroll, M.D.; Freedman, D.S.; Ogden, C.L. Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007–2008 to 2015–2016. JAMA 2018, 319, 1723–1725. [Google Scholar] [CrossRef]
- Zanwar, S.; Abeykoon, J.P.; Kapoor, P. Challenges and Strategies in the Management of Multiple Myeloma in the Elderly Population. Curr. Hematol. Malig. Rep. 2019, 14, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Fukudome, K.; Kato, J.; Ohashi, T.; Yamamoto, Y.; Eto, T. Hyperlipidemia Associated with Multiple Myeloma. Intern. Med. 1996, 35, 337–340. [Google Scholar] [CrossRef]
- Carson, K.R.; Bates, M.; Tomasson, M. The skinny on obesity and plasma cell myeloma: A review of the literature. Bone Marrow Transplant. 2014, 49, 1009–1015. [Google Scholar] [CrossRef]
- Chang, S.-H.; Luo, S.; O’Brian, K.K.; Thomas, T.S.; A Colditz, G.; Carlsson, N.P.; Carson, K.R. Association between metformin use and progression of monoclonal gammopathy of undetermined significance to multiple myeloma in US veterans with diabetes mellitus: A population-based retrospective cohort study. Lancet Haematol. 2015, 2, e30–e36. [Google Scholar] [CrossRef]
- Sanfilippo, K.M.; Keller, J.; Gage, B.F.; Luo, S.; Wang, T.-F.; Moskowitz, G.; Gumbel, J.; Blue, B.; O’Brian, K.; Carson, K.R. Statins Are Associated With Reduced Mortality in Multiple Myeloma. J. Clin. Oncol. 2016, 34, 4008–4014. [Google Scholar] [CrossRef]
- Gu, Y.; Yuan, Y.-H.; Xu, J.; Shi, Q.-L.; Qu, X.-Y.; Guo, R.; Bai, H.; Xu, J.-D.; Li, J.-Y.; Chen, L.-J. High serum lactate dehydrogenase predicts an unfavorable outcome in Chinese elderly patients with multiple myeloma. Oncotarget 2017, 8, 48350–48361. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Katodritou, E.; Roussou, M.; Pouli, A.; Michalis, E.; Delimpasi, S.; Parcharidou, A.; Kartasis, Z.; Zomas, A.; Symeonidis, A.; et al. High Serum Lactate Dehydrogenase Adds Prognostic Value to the International Myeloma Staging System even in the Era of Novel Agents. Eur. J. Haematol. 2010, 85, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2008, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jia, X.; Duan, Y.; Xiao, H.; Sundqvist, K.-G.; Permert, J.; Wang, F. Excess glucose induces hypoxia-inducible factor-1α in pancreatic cancer cells and stimulates glucose metabolism and cell migration. Cancer Biol. Ther. 2013, 14, 428–435. [Google Scholar] [CrossRef][Green Version]
- Huang, H.-Y.; Wang, Y.; Wang, W.-D.; Wei, X.-L.; Gale, R.P.; Li, J.-Y.; Zhang, Q.-Y.; Shu, L.-L.; Li, L.; Li, J.; et al. A prognostic survival model based on metabolism-related gene expression in plasma cell myeloma. Leukemia 2021, 35, 3212–3222. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Halliley, J.L.; Tipton, C.M.; Liesveld, J.; Rosenberg, A.F.; Darce, J.; Gregoretti, I.V.; Popova, L.; Kaminiski, D.; Fucile, C.F.; Albizua, I.; et al. Long-Lived Plasma Cells Are Contained within the CD19−CD38hiCD138+ Subset in Human Bone Marrow. Immunity 2015, 43, 132–145. [Google Scholar] [CrossRef]
- Doughty, C.A.; Bleiman, B.F.; Wagner, D.J.; Dufort, F.J.; Mataraza, J.M.; Roberts, M.F.; Chiles, T.C. Antigen receptor–mediated changes in glucose metabolism in B lymphocytes: Role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 2006, 107, 4458–4465. [Google Scholar] [CrossRef]
- Lam, W.Y.; Jash, A.; Yao, C.; D’Souza, L.; Wong, R.; Nunley, R.M.; Meares, G.P.; Patti, G.J.; Bhattacharya, D. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep. 2018, 24, 2479–2492.e6. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, W.Y.; McGee, S.; Connor, T.; Mottram, B.; Wilkinson, A.; Whitehead, J.; Vuckovic, S.; Catley, L. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br. J. Cancer 2013, 108, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic Signature Identifies Novel Targets for Drug Resistance in Multiple Myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef]
- Fujiwara, S.; Wada, N.; Kawano, Y.; Okuno, Y.; Kikukawa, Y.; Endo, S.; Nishimura, N.; Ueno, N.; Mitsuya, H.; Hata, H. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp. Hematol. Oncol. 2015, 4, 12. [Google Scholar] [CrossRef]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.; Raje, N.S.; Rosen, S.T.; et al. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin. Cancer Res. 2014, 21, 1161–1171. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.M.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2017, 43, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Falank, C.; Fairfield, H.; Reagan, M.R. Signaling Interplay between Bone Marrow Adipose Tissue and Multiple Myeloma cells. Front. Endocrinol. 2016, 7, 67. [Google Scholar] [CrossRef]
- McDonald, M.M.; Fairfield, H.; Falank, C.; Reagan, M.R. Adipose, Bone, and Myeloma: Contributions from the Microenvironment. Calcif. Tissue Res. 2016, 100, 433–448. [Google Scholar] [CrossRef]
- Fowler, J.A.; Lwin, S.T.; Drake, M.T.; Edwards, J.R.; Kyle, R.A.; Mundy, G.R.; Edwards, C. Host-derived adiponectin is tumor-suppressive and a novel therapeutic target for multiple myeloma and the associated bone disease. Blood 2011, 118, 5872–5882. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; Learman, B.S.; Parlee, S.D.; Simon, B.R.; Mori, H.; Ning, X.; Bree, A.J.; Schell, B.; Broome, D.T.; et al. Bone Marrow Adipose Tissue Is an Endocrine Organ that Contributes to Increased Circulating Adiponectin during Caloric Restriction. Cell Metab. 2014, 20, 368–375. [Google Scholar] [CrossRef]
- Masarwi, M.; DeSchiffart, A.; Ham, J.; Reagan, M.R. Multiple Myeloma and Fatty Acid Metabolism: Lipid metabolism in multiple myeloma: Cell growth, survival, drug resistance. JBMR Plus 2019, 3, e10173. [Google Scholar] [CrossRef]
- Schneiderova, P.; Pika, T.; Gajdos, P.; Fillerova, R.; Kromer, P.; Kudelka, M.; Minarik, J.; Papajik, T.; Scudla, V.; Kriegova, E. Serum protein fingerprinting by PEA immunoassay coupled with a pattern-recognition algorithms distinguishes MGUS and multiple myeloma. Oncotarget 2016, 8, 69408–69421. [Google Scholar] [CrossRef]
- Tirado-Velez, J.; Joumady, I.; Saez-Benito, A.; Cozar-Castellano, I.; Perdomo, G. Inhibition of Fatty Acid Metabolism Reduces Human Myeloma Cells Proliferation. PLoS ONE 2012, 7, e46484. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.; Smirnov, P.; Li, Z.; Branchard, E.; van Leeuwen, J.E.; Licht, J.D.; Haibe-Kains, B.; Andrews, D.W.; Keats, J.J.; Pugh, T.J.; et al. The mevalonate pathway is an actionable vulnerability of t(4;14)-positive multiple myeloma. Leukemia 2020, 35, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Ragbourne, S.C.; Maghsoodi, N.; Streetly, M.; Crook, M.A. The Association between Metabolic Syndrome and Multiple Myeloma. Acta Haematol. 2020, 144, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Wallin, A.; Larsson, S.C. Body mass index and risk of multiple myeloma: A meta-analysis of prospective studies. Eur. J. Cancer 2011, 47, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.N.; Moore, S.C.; Lim, U.; Park, Y.; Baris, D.; Hollenbeck, A.R.; Matthews, C.E.; Gibson, T.M.; Hartge, P.; Purdue, M.P. Body Mass Index and Physical Activity at Different Ages and Risk of Multiple Myeloma in the NIH-AARP Diet and Health Study. Am. J. Epidemiology 2013, 177, 776–786. [Google Scholar] [CrossRef]
- Teras, L.R.; Kitahara, C.M.; Birmann, B.; Hartge, P.A.; Wang, S.S.; Robien, K.; Patel, A.V.; Adami, H.-O.; Weiderpass, E.; Giles, G.; et al. Body size and multiple myeloma mortality: A pooled analysis of 20 prospective studies. Br. J. Haematol. 2014, 166, 667–676. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Freund, G.G.; Kulas, D.T.; A Mooney, R. Insulin and IGF-1 increase mitogenesis and glucose metabolism in the multiple myeloma cell line, RPMI 8226. J. Immunol. 1993, 151, 1811–1820. [Google Scholar]
- Hofmann, J.N.; Liao, L.; Pollak, M.N.; Wang, Y.; Pfeiffer, R.M.; Baris, D.; Andreotti, G.; Lan, Q.; Landgren, O.; Rothman, N.; et al. A prospective study of circulating adipokine levels and risk of multiple myeloma. Blood 2012, 120, 4418–4420. [Google Scholar] [CrossRef]
- Dalamaga, M.; Karmaniolas, K.; Panagiotou, A.; Hsi, A.; Chamberland, J.; Dimas, C.; Lekka, A.; Mantzoros, C.S. Low circulating adiponectin and resistin, but not leptin, levels are associated with multiple myeloma risk: A case–control study. Cancer Causes Control 2008, 20, 193–199. [Google Scholar] [CrossRef]
- Baumann, P.; Mandl-Weber, S.; Emmerich, B.; Straka, C.; Schmidmaier, R. Activation of adenosine monophosphate activated protein kinase inhibits growth of multiple myeloma cells. Exp. Cell Res. 2007, 313, 3592–3603. [Google Scholar] [CrossRef] [PubMed]
- Ajuwon, K.M.; Spurlock, M.E. Adiponectin inhibits LPS-induced NF-κB activation and IL-6 production and increases PPARγ2 expression in adipocytes. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R1220–R1225. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The Role of Adiponectin in Cancer: A Review of Current Evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [PubMed]
- Al-Aidaroos, A.Q.O.; Zeng, Q. PRL-3 phosphatase and cancer metastasis. J. Cell. Biochem. 2010, 111, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- McParland, V.; Varsano, G.; Li, X.; Thornton, J.; Baby, J.; Aravind, A.; Meyer, C.; Pavic, K.; Rios, P.; Köhn, M. The Metastasis-Promoting Phosphatase PRL-3 Shows Activity toward Phosphoinositides. Biochemistry 2011, 50, 7579–7590. [Google Scholar] [CrossRef]
- Wang, H.; Vardy, L.A.; Tan, C.P.; Loo, J.M.; Guo, K.; Li, J.; Lim, S.G.; Zhou, J.; Chng, W.J.; Ng, S.B.; et al. PCBP1 Suppresses the Translation of Metastasis-Associated PRL-3 Phosphatase. Cancer Cell 2010, 18, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Guzińska-Ustymowicz, K.; Pryczynicz, A. PRL-3, an emerging marker of carcinogenesis, is strongly associated with poor prognosis. Anti-Cancer Agents Med. Chem. 2011, 11, 99–108. [Google Scholar] [CrossRef]
- Yang, Y.; Lian, S.; Meng, L.; Qu, L.; Shou, C. Antibody Array Revealed PRL-3 Affects Protein Phosphorylation and Cytokine Secretion. PLoS ONE 2017, 12, e0169665. [Google Scholar] [CrossRef]
- Fagerli, U.-M.; Holt, R.U.; Holien, T.; Vaatsveen, T.K.; Zhan, F.; Egeberg, K.W.; Barlogie, B.; Waage, A.; Aarset, H.; Dai, H.Y.; et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood 2008, 111, 806–815. [Google Scholar] [CrossRef]
- Slørdahl, T.S.; Abdollahi, P.; Vandsemb, E.N.; Rampa, C.; Misund, K.; Baranowska, K.A.; Westhrin, M.; Waage, A.; Rø, T.B.; Børset, M. The phosphatase of regenerating liver-3 (PRL-3) is important for IL-6-mediated survival of myeloma cells. Oncotarget 2016, 7, 27295–27306. [Google Scholar] [CrossRef]
- Chong, P.S.Y.; Zhou, J.; Lim, J.S.L.; Hee, Y.T.; Chooi, J.-Y.; Chung, T.-H.; Tan, Z.T.; Zeng, Q.; Waller, D.D.; Sebag, M.; et al. IL6 Promotes a STAT3-PRL3 Feedforward Loop via SHP2 Repression in Multiple Myeloma. Cancer Res. 2019, 79, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, P.; Vandsemb, E.N.; Børset, M. Phosphatases of regenerating liver are key regulators of metabolism in cancer cells—Role of Serine/Glycine metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2021, 25, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef]
- Xu, H.; Zeng, Y.; Liu, L.; Gao, Q.; Jin, S.; Lan, Q.; Lai, W.; Luo, X.; Wu, H.; Huang, Y.; et al. PRL-3 improves colorectal cancer cell proliferation and invasion through IL-8 mediated glycolysis metabolism. Int. J. Oncol. 2017, 51, 1271–1279. [Google Scholar] [CrossRef]
- Cancer Metabolism in Nutrient Replete Conditions, by BioRender.com. 2022. Available online: https://app.biorender.com/biorender-templates (accessed on 15 February 2022).
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2006, 109, 3177–3188. [Google Scholar] [CrossRef]
- Cao, X.; Fang, L.; Gibbs, S.; Huang, Y.; Dai, Z.; Wen, P.; Zheng, X.; Sadee, W.; Sun, D. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother. Pharmacol. 2006, 59, 495–505. [Google Scholar] [CrossRef]
- Wei, C.; Bajpai, R.; Sharma, H.; Heitmeier, M.; Jain, A.D.; Matulis, S.M.; Nooka, A.K.; Mishra, R.K.; Hruz, P.; Schiltz, G.E.; et al. Development of GLUT4-selective antagonists for multiple myeloma therapy. Eur. J. Med. Chem. 2017, 139, 573–586. [Google Scholar] [CrossRef]
- Kurtoglu, M.; Gao, N.; Shang, J.; Maher, J.C.; Lehrman, M.A.; Wangpaichitr, M.; Savaraj, N.; Lane, A.N.; Lampidis, T.J. Under normoxia, 2-deoxy-d-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer Ther. 2007, 6, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.C.; Krishan, A.; Lampidis, T.J. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-d-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother. Pharmacol. 2003, 53, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate dehydrogenase a in cancer: A promising target for diagnosis and therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Myeloma today: Disease definitions and treatment advances. Am. J. Hematol. 2015, 91, 90–100. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef]
- Starheim, K.K.; Holien, T.; Misund, K.; Johansson, I.; A Baranowska, K.; Sponaas, A.-M.; Hella, H.; Buene, G.; Waage, A.; Sundan, A.; et al. Intracellular glutathione determines bortezomib cytotoxicity in multiple myeloma cells. Blood Cancer J. 2016, 6, e446. [Google Scholar] [CrossRef]
- Zheng, Z.; Fan, S.; Zheng, J.; Huang, W.; Gasparetto, C.; Chao, N.J.; Hu, J.; Kang, Y. Inhibition of thioredoxin activates mitophagy and overcomes adaptive bortezomib resistance in multiple myeloma. J. Hematol. Oncol. 2018, 11, 29. [Google Scholar] [CrossRef]
- Besse, L.; Besse, A.; Mendez-Lopez, M.; Vasickova, K.; Sedlackova, M.; Vanhara, P.; Kraus, M.; Bader, J.; Ferreira, R.B.; Castellano, R.K.; et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica 2019, 104, e415–e419. [Google Scholar] [CrossRef] [PubMed]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; Van Den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Ishigaki, S.; Merksamer, P.I.; Papa, F.R.; Urano, F. Establishment of a system for monitoring endoplasmic reticulum redox state in mammalian cells. Lab. Investig. 2013, 93, 1254–1258. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2018, 33, 1011–1022. [Google Scholar] [CrossRef]
- Willenbacher, E.; Balog, A.; Willenbacher, W. Short overview on the current standard of treatment in newly diagnosed multiple myeloma. memo-Mag. Eur. Med. Oncol. 2018, 11, 59–64. [Google Scholar] [CrossRef]
- Al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.-L.; Mohty, M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Osborne, M.; Lawley, P. Alkylation of DNA by melphalan with special reference to adenine derivatives and adenine-guanine cross-linking. Chem. Interact. 1993, 89, 49–60. [Google Scholar] [CrossRef]
- Polavarapu, A.; Stillabower, J.A.; Stubblefield, S.G.W.; Taylor, W.M.; Baik, M.-H. The Mechanism of Guanine Alkylation by Nitrogen Mustards: A Computational Study. J. Org. Chem. 2012, 77, 5914–5921. [Google Scholar] [CrossRef] [PubMed]
- Povirk, L.F.; Shuker, D.E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. Genet. Toxicol. 1994, 318, 205–226. [Google Scholar] [CrossRef]
- Koomen, D.C.; Meads, M.B.; Magaletti, D.M.; Guingab-Cagmat, J.D.; Oliveira, P.S.; Fang, B.; Liu, M.; Welsh, E.A.; Meke, L.E.; Jiang, Z.; et al. Metabolic Changes Are Associated with Melphalan Resistance in Multiple Myeloma. J. Proteome Res. 2021, 20, 3134–3149. [Google Scholar] [CrossRef]
- A Hazlehurst, L.; A Enkemann, S.; A Beam, C.; Argilagos, R.F.; Painter, J.; Shain, K.H.; Saporta, S.; Boulware, D.; Moscinski, L.; Alsina, M.; et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res. 2003, 63, 7900–7906. [Google Scholar]
- Zub, K.A.; De Sousa, M.M.L.; Sarno, A.; Sharma, A.; Demirovic, A.; Rao, S.; Young, C.; Aas, P.A.; Ericsson, I.; Sundan, A.; et al. Modulation of Cell Metabolic Pathways and Oxidative Stress Signaling Contribute to Acquired Melphalan Resistance in Multiple Myeloma Cells. PLoS ONE 2015, 10, e0119857. [Google Scholar] [CrossRef]
- Takebe, N.; Cheng, X.; Wu, S.; Bauer, K.; Goloubeva, O.G.; Fenton, R.G.; Heyman, M.; Rapoport, A.P.; Badros, A.; Shaughnessy, J.; et al. Phase I Clinical Trial of the Inosine Monophosphate Dehydrogenase Inhibitor Mycophenolate Mofetil (Cellcept) in Advanced Multiple Myeloma Patients. Clin. Cancer Res. 2004, 10, 8301–8308. [Google Scholar] [CrossRef]
- Naffouje, R.; Grover, P.; Yu, H.; Sendilnathan, A.; Wolfe, K.; Majd, N.; Smith, E.P.; Takeuchi, K.; Senda, T.; Kofuji, S.; et al. Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers 2019, 11, 1346. [Google Scholar] [CrossRef]
- Gourzones, C.; Bellanger, C.; Lamure, S.; Gadacha, O.C.; De Paco, E.G.; Vincent, L.; Cartron, G.; Klein, B.; Moreaux, J. Antioxidant Defenses Confer Resistance to High Dose Melphalan in Multiple Myeloma Cells. Cancers 2019, 11, 439. [Google Scholar] [CrossRef]
- Görgün, G.; Calabrese, E.; Soydan, E.; Hideshima, T.; Perrone, G.; Bandi, M.; Cirstea, D.; Santo, L.; Hu, Y.; Tai, Y.-T.; et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010, 116, 3227–3237. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Akiyama, M.; Hayashi, T.; Chauhan, D.; Richardson, P.; Schlossman, R.; Podar, K.; Munshi, N.C.; Mitsiades, N.; et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003, 101, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Shima, Y.; Raje, N.; Davies, F.E.; Tai, Y.-T.; Treon, S.P.; Lin, B.; Schlossman, R.L.; Richardson, P.; et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood 2000, 96, 2943–2950. [Google Scholar] [CrossRef]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.E.; Coomber, B.L.; Bridle, B.W. Metabolic reprogramming in the tumour microenvironment: A hallmark shared by cancer cells and T lymphocytes. Immunology 2017, 152, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Y.; Liu, H.; Xu, X.; He, S.; Tang, J.; Huang, Y.; Miao, X.; Wu, Y.; Wang, Q.; et al. Pyruvate kinase isoform M2 (PKM2) participates in multiple myeloma cell proliferation, adhesion and chemoresistance. Leuk. Res. 2015, 39, 1428–1436. [Google Scholar] [CrossRef]
- Steinbrunn, T.; Stühmer, T.; Sayehli, C.; Chatterjee, M.; Einsele, H.; Bargou, R.C. Combined targeting of MEK/MAPK and PI3K/Akt signalling in multiple myeloma. Br. J. Haematol. 2012, 159, 430–440. [Google Scholar] [CrossRef]
- Qin, X.; Lin, L.; Cao, L.; Zhang, X.; Song, X.; Hao, J.; Zhang, Y.; Wei, R.; Huang, X.; Lu, J.; et al. Extracellular matrix protein Reelin promotes myeloma progression by facilitating tumor cell proliferation and glycolysis. Sci. Rep. 2017, 7, 45305. [Google Scholar] [CrossRef]
- Tamura, H. Immunopathogenesis and immunotherapy of multiple myeloma. Int. J. Hematol. 2018, 107, 278–285. [Google Scholar] [CrossRef]
- Krejcik, J.; A Frerichs, K.; Nijhof, I.S.; Van Kessel, B.; Van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.; Zweegman, S.; Van Meerloo, J.; Musters, R.J.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef] [PubMed]
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S.; et al. The activated conformation of integrin β7 is a novel multiple myeloma–specific target for CAR T cell therapy. Nat. Med. 2017, 23, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, K.E.; Dudzinski, S.O.; Rathmell, J.C. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017, 35, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Noël, G.; Fontsa, M.L.; Willard-Gallo, K. The impact of tumor cell metabolism on T cell-mediated immune responses and immuno-metabolic biomarkers in cancer. Semin. Cancer Biol. 2018, 52, 66–74. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.A.; Miller, K.H.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.E.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Eichner, R.; Heider, M.; Fernández-Sáiz, V.; Van Bebber, F.; Garz, A.-K.; Lemeer, S.; Rudelius, M.; Targosz, B.-S.; Jacobs, L.; Knorn, A.-M.; et al. Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Heider, M.; Eichner, R.; Stroh, J.; Morath, V.; Kuisl, A.; Zecha, J.; Lawatscheck, J.; Baek, K.; Garz, A.-K.; Rudelius, M.; et al. The IMiD target CRBN determines HSP90 activity toward transmembrane proteins essential in multiple myeloma. Mol. Cell 2021, 81, 1170–1186.e10. [Google Scholar] [CrossRef]
- Storti, P.; Toscani, D.; Airoldi, I.; Marchica, V.; Maiga, S.; Bolzoni, M.; Fiorini, E.; Campanini, N.; Martella, E.; Mancini, C.; et al. The anti-tumoral effect of lenalidomide is increased in vivo by hypoxia-inducible factor (HIF)-1α inhibition in myeloma cells. Haematologica 2016, 101, e107–e110. [Google Scholar] [CrossRef]
- Busch, A.; Zeh, D.; Janzen, V.; Mügge, L.; Wolf, D.; Fingerhut, L.; Hahn-Ast, C.; Maurer, O.; Brossart, P.; von Lilienfeld-Toal, M. Treatment with lenalidomide induces immunoactivating and counter-regulatory immunosuppressive changes in myeloma patients. Clin. Exp. Immunol. 2014, 177, 439–453. [Google Scholar] [CrossRef]
- Wu, S.; Kuang, H.; Ke, J.; Pi, M.; Yang, D.-H. Metabolic Reprogramming Induces Immune Cell Dysfunction in the Tumor Microenvironment of Multiple Myeloma. Front. Oncol. 2021, 10, 597342. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Works, M.; Soni, N.; Hauskins, C.; Sierra, C.; Baturevych, A.; Jones, J.C.; Curtis, W.; Carlson, P.; Johnstone, T.G.; Kugler, D.; et al. Anti–B-cell Maturation Antigen Chimeric Antigen Receptor T cell Function against Multiple Myeloma Is Enhanced in the Presence of Lenalidomide. Mol. Cancer Ther. 2019, 18, 2246–2257. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 109. [Google Scholar] [CrossRef]
- Chillemi, A.; Quarona, V.; Antonioli, L.; Ferrari, D.; Horenstein, A.L.; Malavasi, F. Roles and Modalities of Ectonucleotidases in Remodeling the Multiple Myeloma Niche. Front. Immunol. 2017, 8, 305. [Google Scholar] [CrossRef]
- Giron, M.C.; Bin, A.; Brun, P.; Etteri, S.; Bolego, C.; Florio, C.; Gaion, R.M. Cyclic AMP in Rat Ileum: Evidence for the Presence of an Extracellular Cyclic AMP-Adenosine Pathway. Gastroenterology 2008, 134, 1116–1126. [Google Scholar] [CrossRef]
- Whiteside, T.L. Targeting adenosine in cancer immunotherapy: A review of recent progress. Expert Rev. Anticancer Ther. 2017, 17, 527–535. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef]
- Wang, T.; Kumru, O.S.; Yi, L.; Wang, Y.J.; Zhang, J.; Kim, J.H.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Effect of Ionic Strength and pH on the Physical and Chemical Stability of a Monoclonal Antibody Antigen-Binding Fragment. J. Pharm. Sci. 2013, 102, 2520–2537. [Google Scholar] [CrossRef] [PubMed]
- Latypov, R.F.; Hogan, S.; Lau, H.; Gadgil, H.; Liu, D. Elucidation of Acid-induced Unfolding and Aggregation of Human Immunoglobulin IgG1 and IgG2 Fc. J. Biol. Chem. 2012, 287, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody Structure, Instability, and Formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Rashid, M.B.M.A.; Toh, T.B.; Hooi, L.; Silva, A.; Zhang, Y.; Tan, P.F.; Teh, A.L.; Karnani, N.; Jha, S.; Ho, C.-M.; et al. Optimizing drug combinations against multiple myeloma using a quadratic phenotypic optimization platform (QPOP). Sci. Transl. Med. 2018, 10, eaan0941. [Google Scholar] [CrossRef]
- Heuck, C.J.; Mehta, J.; Bhagat, T.; Gundabolu, K.; Yu, Y.; Khan, S.; Chrysofakis, G.; Schinke, C.; Tariman, J.D.; Vickrey, E.; et al. Myeloma Is Characterized by Stage-Specific Alterations in DNA Methylation That Occur Early during Myelomagenesis. J. Immunol. 2013, 190, 2966–2975. [Google Scholar] [CrossRef]
- Nervi, C.; De Marinis, E.; Codacci-Pisanelli, G. Epigenetic treatment of solid tumours: A review of clinical trials. Clin. Epigenetics 2015, 7, 1–20. [Google Scholar] [CrossRef]
- Qiang, Y.-W.; Ye, S.; Chen, Y.; Buros, A.F.; Edmonson, R.; Van Rhee, F.; Barlogie, B.; Epstein, J.; Morgan, G.; Davies, F. MAF protein mediates innate resistance to proteasome inhibition therapy in multiple myeloma. Blood 2016, 128, 2919–2930. [Google Scholar] [CrossRef]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.L.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.; Staudt, L.M. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef]
- Mattioli, M.; Agnelli, L.; Fabris, S.; Baldini, L.; Morabito, F.; Bicciato, S.; Verdelli, D.; Intini, D.; Nobili, L.; Cro, L.; et al. Gene expression profiling of plasma cell dyscrasias reveals molecular patterns associated with distinct IGH translocations in multiple myeloma. Oncogene 2005, 24, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tong, Z.; Ding, C.; Luo, F.; Wu, S.; Wu, C.; Albeituni, S.; He, L.; Hu, X.; Tieri, D.; et al. Transcription factor c-Maf is a checkpoint that programs macrophages in lung cancer. J. Clin. Investig. 2020, 130, 2081–2096. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-T.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Anti-Myeloma Agent Mechanism of Action | Resistance Mechanism | Combination Treatment to Reduce Resistance |
|---|---|---|
| Bortezomib Proteasome inhibitor | Glucose transporters | Phloretin daunorubicin, compound 20 and ritonavir |
| HKII | LDHA knockdown | |
| OXPHOS | Target mitochondria metabolism | |
| SSP | Serine starvation | |
| Glutaminolysis | CB-839 | |
| Protein folding disulphide bond formation | 16F16 TCyDTDO | |
| Melphalan DNA alkylating agent | PPP | 6-AN |
| Reduced guanine and guanosine | Mycophenolate mofetil | |
| GSH | Reduce oxidative stress | |
| Immunotherapy | Acidification of TME | Bicarbonate |
| Adenosine | Degrade extracellular adenosine Inhibit adenosine production Inhibit nucleotide release channels |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, J.S.L.; Chong, P.S.Y.; Chng, W.-J. Metabolic Vulnerabilities in Multiple Myeloma. Cancers 2022, 14, 1905. https://doi.org/10.3390/cancers14081905
Lim JSL, Chong PSY, Chng W-J. Metabolic Vulnerabilities in Multiple Myeloma. Cancers. 2022; 14(8):1905. https://doi.org/10.3390/cancers14081905
Chicago/Turabian StyleLim, Julia S. L., Phyllis S. Y. Chong, and Wee-Joo Chng. 2022. "Metabolic Vulnerabilities in Multiple Myeloma" Cancers 14, no. 8: 1905. https://doi.org/10.3390/cancers14081905
APA StyleLim, J. S. L., Chong, P. S. Y., & Chng, W.-J. (2022). Metabolic Vulnerabilities in Multiple Myeloma. Cancers, 14(8), 1905. https://doi.org/10.3390/cancers14081905