
Aspirin-Triggered Resolvin D1 Reduces Chronic Dust-Induced Lung Pathology without Altering Susceptibility to Dust-Enhanced Carcinogenesis

 , ,
, , 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals & Reagents
2.2. Preparation of Hog Dust Extract
2.3. Animal Housing & Care
2.4. Chronic Dust Exposure Model
2.5. Lung Histopathology Scoring
2.6. AT-RvD1 Treatment Study
2.7. Oxylipin Analysis
2.8. NanoString Gene Expression Anlyses
2.9. Culturing of A549 Lung Cells
2.10. Western Blotting
2.11. Bioinformatics Data Processing and Analyses
2.12. Statistical Analyses
3. Results
3.1. AT-RvD1 Reduces Cellular Recruitment into the Airways of Dust-Exposed Mice
3.2. Fatty Acid Levels in Murine Lung Tissues
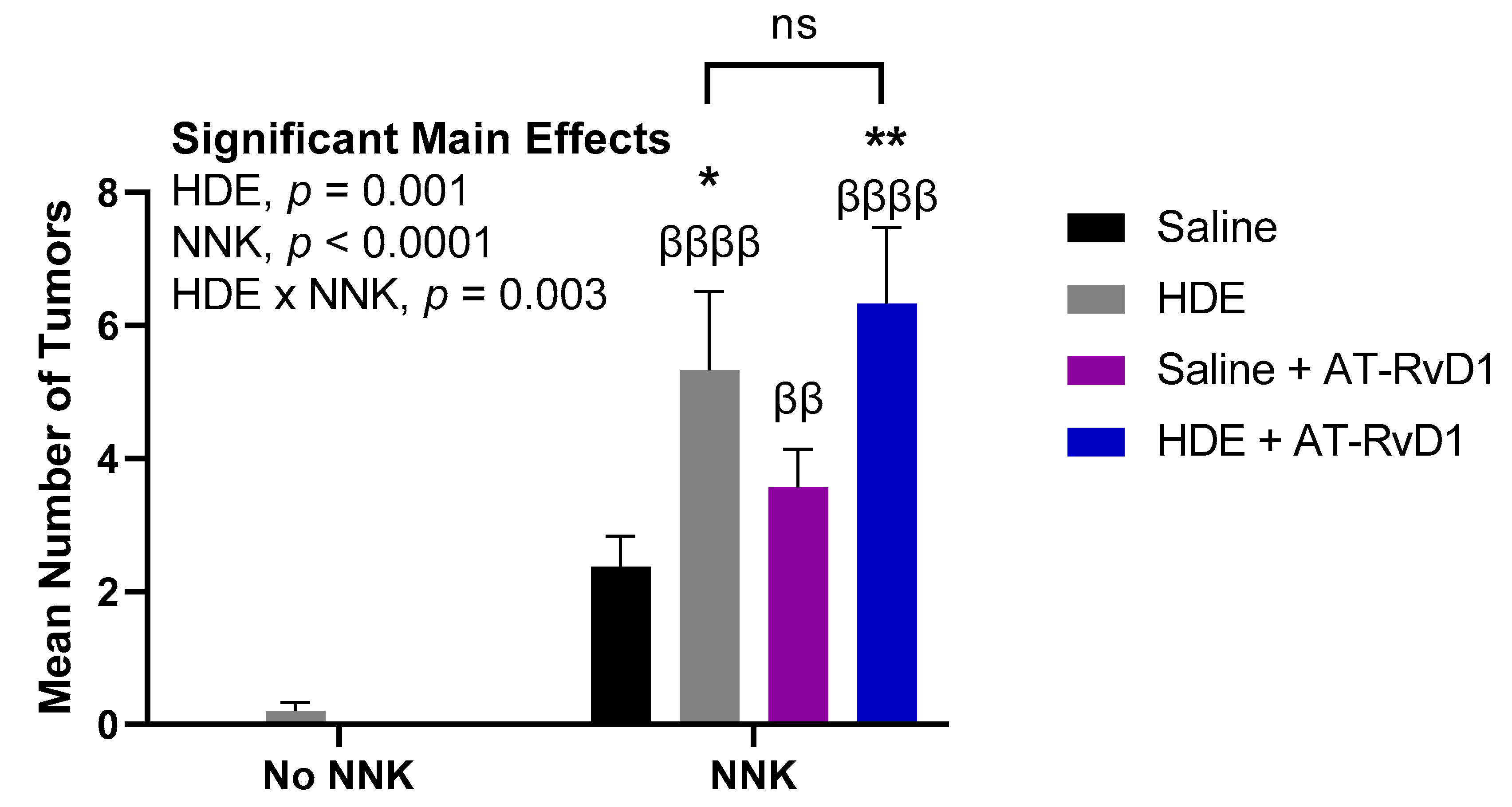
3.3. Chronic Dust Exposure Enhances Lung Tumorigenesis in Mice
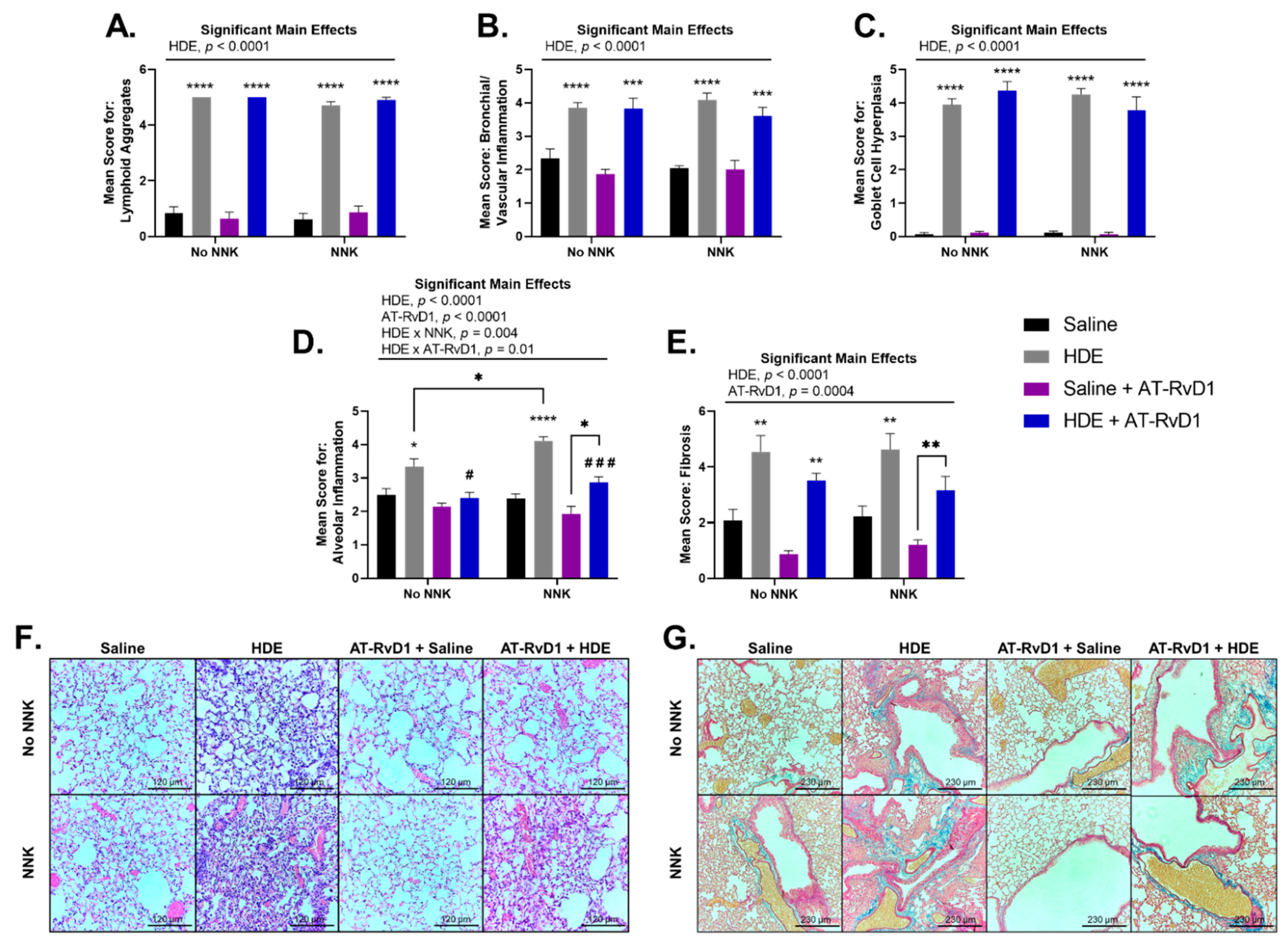
3.4. AT-RvD1 Alters Dust-Induced Lung Histopathology Outcomes
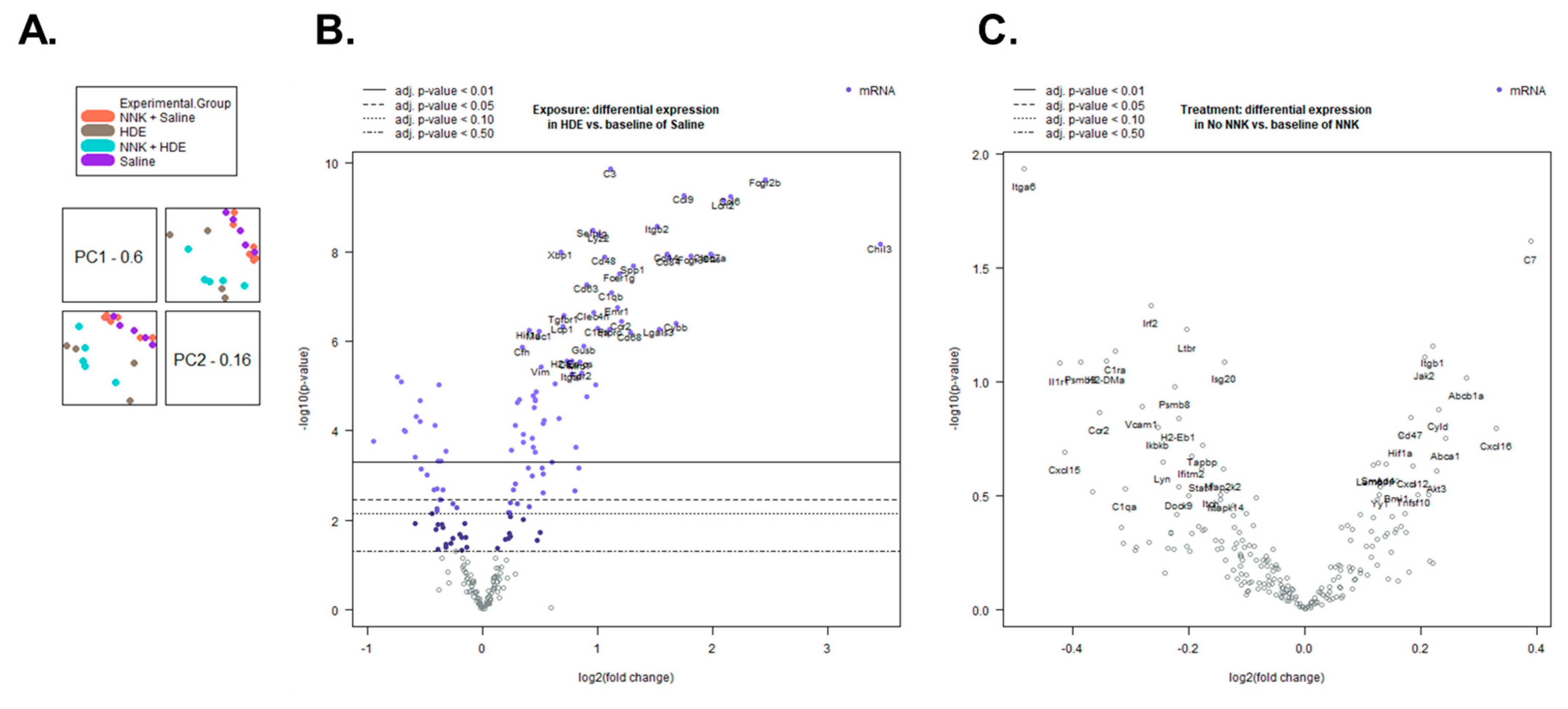
3.5. HDE Drives Increased Inflammation- and Cancer-Related Gene Expression
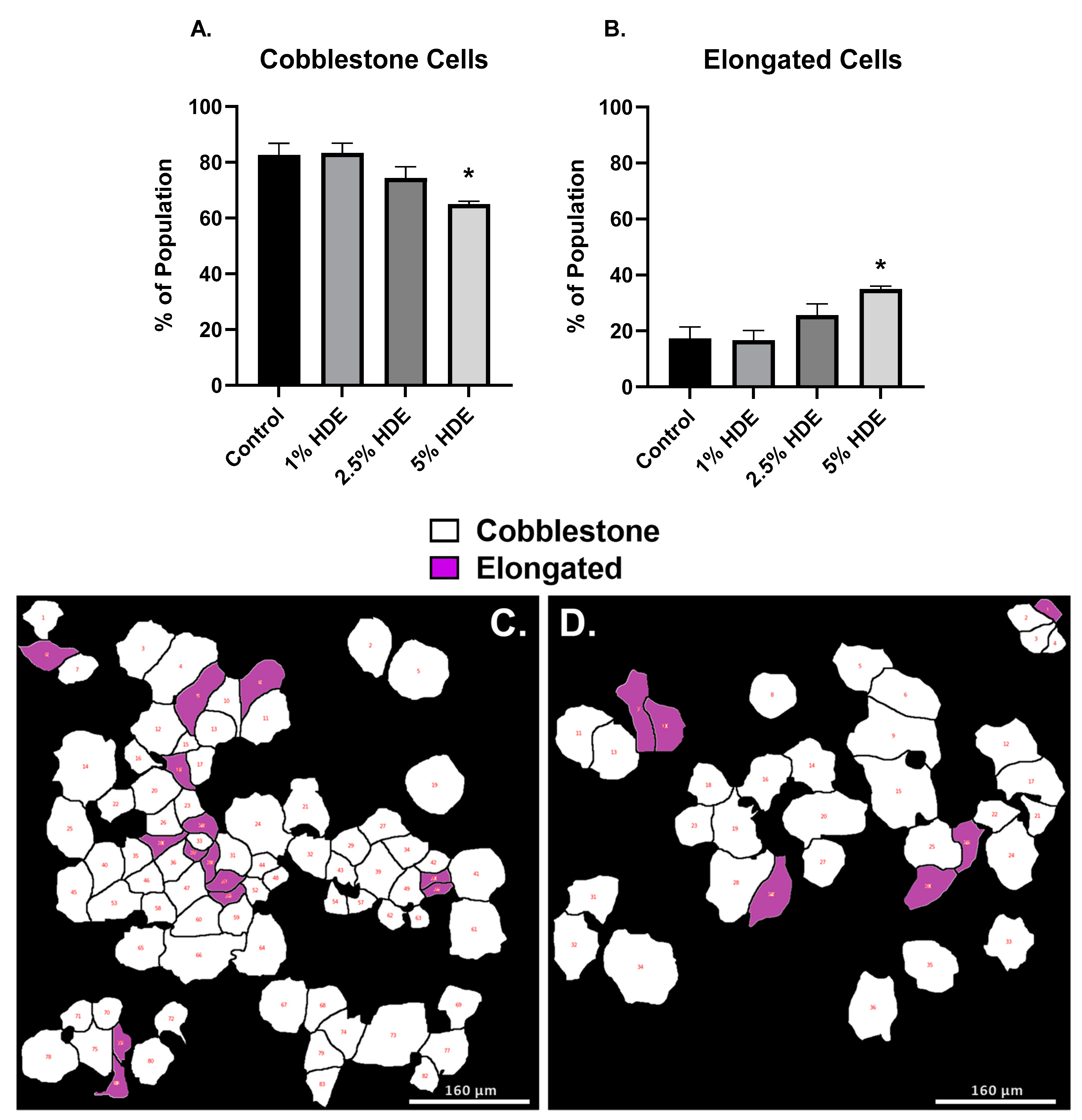
3.6. Agricultural Dust Exposure Induces EMT in Human Lung Adenocarcinoma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, T.P.; Beirão, B.C.B.; Pang, L.Y.; Queiroga, F.L.; Argyle, D.J. Inflammation and cancer: Till death tears them apart. Vet. J. 2015, 205, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Boissy, R.J.; Romberger, D.J.; Roughead, W.A.; Weissenburger-Moser, L.; Poole, J.A.; LeVan, T.D. Shotgun pyrosequencing metagenomic analyses of dusts from swine confinement and grain facilities. PLoS ONE 2014, 9, e95578. [Google Scholar] [CrossRef]
- Romberger, D.J.; Bodlak, V.; Essen, S.G.V.; Mathisen, T.; Wyatt, T.A. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J. Appl. Physiol. 2002, 93, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romberger, D.J.; Heires, A.J.; Nordgren, T.M.; Souder, C.P.; West, W.; Liu, X.-d.; Poole, J.A.; Toews, M.L.; Wyatt, T.A. Proteases in agricultural dust induce lung inflammation through PAR-1 and PAR-2 activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L388–L399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebers, V.; Brüning, T.; Raulf-Heimsoth, M. Occupational endotoxin-exposure and possible health effects on humans (review). Am. J. Ind. Med. 2006, 49, 474–491. [Google Scholar] [CrossRef]
- Lundin, J.I.; Checkoway, H. Endotoxin and cancer. Env. Health Perspect 2009, 117, 1344–1350. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, G.; Marzia, V.; Marcer, G. Reduced lung cancer mortality in dairy farmers: Is endotoxin exposure the key factor? Am. J. Ind. Med. 1996, 30, 601–609. [Google Scholar] [CrossRef]
- Peters, S.; Kromhout, H.; Olsson, A.C.; Wichmann, H.-E.; Brüske, I.; Consonni, D.; Landi, M.T.; Caporaso, N.; Siemiatycki, J.; Richiardi, L.; et al. Occupational exposure to organic dust increases lung cancer risk in the general population. Thorax 2012, 67, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Nordgren, T.M.; Heires, A.J.; Bailey, K.L.; Katafiasz, D.M.; Toews, M.L.; Wichman, C.S.; Romberger, D.J. Docosahexaenoic acid enhances amphiregulin-mediated bronchial epithelial cell repair processes following organic dust exposure. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L421–L431. [Google Scholar] [CrossRef]
- Poole, J.A.; Wyatt, T.A.; Oldenburg, P.J.; Elliott, M.K.; West, W.W.; Sisson, J.H.; Von Essen, S.G.; Romberger, D.J. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L1085–L1095. [Google Scholar] [CrossRef] [PubMed]
- Salerno, C.; Sacco, S.; Panella, M.; Berchialla, P.; Vanhaecht, K.; Palin, L.A. Cancer risk among farmers in the Province of Vercelli (Italy) from 2002 to 2005: An ecological study. Ann. Ig 2014, 26, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.L.; Meza, J.L.; Smith, L.M.; Von Essen, S.G.; Romberger, D.J. Agricultural exposures in patients with COPD in health systems serving rural areas. J. Agromedicine 2007, 12, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Guillien, A.; Puyraveau, M.; Soumagne, T.; Guillot, S.; Rannou, F.; Marquette, D.; Berger, P.; Jouneau, S.; Monnet, E.; Mauny, F.; et al. Prevalence and risk factors for COPD in farmers: A cross-sectional controlled study. Eur. Respir. J. 2016, 47, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgren, T.M.; Bailey, K.L. Pulmonary health effects of agriculture. Curr. Opin. Pulm. Med. 2016, 22, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.C.; Schenker, M.B. Protection against breathing dust: Behavior over time in Californian farmers. J. Agric. Saf. Health 2008, 14, 189–203. [Google Scholar] [CrossRef]
- Schenker, M.B.; Orenstein, M.R.; Samuels, S.J. Use of protective equipment among California farmers. Am. J. Ind. Med. 2002, 42, 455–464. [Google Scholar] [CrossRef]
- Von Essen, S.; Fryzek, J.; Nowakowski, B.; Wampler, M. Respiratory symptoms and farming practices in farmers associated with an acute febrile illness after organic dust exposure. Chest 1999, 116, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.D.; Serhan, C.N. Resolution of acute inflammation in the lung. Annu. Rev. Physiol. 2014, 76, 467–492. [Google Scholar] [CrossRef] [Green Version]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Gilligan, M.M.; Gartung, A.; Sulciner, M.L.; Norris, P.C.; Sukhatme, V.P.; Bielenberg, D.R.; Huang, S.; Kieran, M.W.; Serhan, C.N.; Panigrahy, D. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, H.-M.; Thatcher, T.H.; Colas, R.A.; Serhan, C.N.; Phipps, R.P.; Sime, P.J. Resolvin D1 Reduces Emphysema and Chronic Inflammation. Am. J. Pathol. 2015, 185, 3189–3201. [Google Scholar] [CrossRef]
- Posso, S.V.; Quesnot, N.; Moraes, J.A.; Brito-Gitirana, L.; Kennedy-Feitosa, E.; Barroso, M.V.; Porto, L.C.; Lanzetti, M.; Valença, S.S. AT-RVD1 repairs mouse lung after cigarette smoke-induced emphysema via downregulation of oxidative stress by NRF2/KEAP1 pathway. Int. Immunopharmacol. 2018, 56, 330–338. [Google Scholar] [CrossRef]
- Dominguez, E.C.; Heires, A.J.; Pavlik, J.; Larsen, T.D.; Guardado, S.; Sisson, J.H.; Baack, M.L.; Romberger, D.J.; Nordgren, T.M. A High Docosahexaenoic Acid Diet Alters the Lung Inflammatory Response to Acute Dust Exposure. Nutrients 2020, 12, 2334. [Google Scholar] [CrossRef]
- Ulu, A.; Burr, A.; Heires, A.J.; Pavlik, J.; Larsen, T.; Perez, P.A.; Bravo, C.; DiPatrizio, N.V.; Baack, M.; Romberger, D.J.; et al. A high docosahexaenoic acid diet alters lung inflammation and recovery following repetitive exposure to aqueous organic dust extracts. J. Nutr. Biochem. 2021, 97, 108797. [Google Scholar] [CrossRef] [PubMed]
- Mernitz, H.; Lian, F.; Smith, D.E.; Meydani, S.N.; Wang, X.-D. Fish Oil Supplementation Inhibits NNK-Induced Lung Carcinogenesis in the A/J Mouse. Nutr. Cancer 2009, 61, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgren, T.M.; Bauer, C.D.; Heires, A.J.; Poole, J.A.; Wyatt, T.A.; West, W.W.; Romberger, D.J. Maresin-1 reduces airway inflammation associated with acute and repetitive exposures to organic dust. Transl. Res. 2015, 166, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, J.A.; Alexis, N.E.; Parks, C.; MacInnes, A.K.; Gentry-Nielsen, M.J.; Fey, P.D.; Larsson, L.; Allen-Gipson, D.; Von Essen, S.G.; Romberger, D.J. Repetitive organic dust exposure in vitro impairs macrophage differentiation and function. J. Allergy Clin. Immunol. 2008, 122, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Biochemistry, Biology, and Carcinogenicity of Tobacco-Specific N-Nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Hecht, S.S.; Chen, C.-h.B.; Hirota, N.; Ornaf, R.M.; Tso, T.C.; Hoffmann, D. Tobacco-Specific Nitrosamines: Formation From Nicotine In Vitro and During Tobacco Curing and Carcinogenicity in Strain A Mice23. JNCI: J. Natl. Cancer Inst. 1978, 60, 819–824. [Google Scholar] [CrossRef]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Hübner, R.-H.; Gitter, W.; Eddine El Mokhtari, N.; Mathiak, M.; Both, M.; Bolte, H.; Freitag-Wolf, S.; Bewig, B. Standardized quantification of pulmonary fibrosis in histological samples. BioTechniques 2008, 44, 507–517. [Google Scholar] [CrossRef]
- Sun, Y.-P.; Oh, S.F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S.P.; Petasis, N.A.; Serhan, C.N. Resolvin D1 and its aspirin-triggered 17R epimer stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 2007, 282, 9323–9334. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, H.-M.; Sapinoro, R.E.; Thatcher, T.H.; Croasdell, A.; Levy, E.P.; Fulton, R.A.; Olsen, K.C.; Pollock, S.J.; Serhan, C.N.; Phipps, R.P.; et al. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS ONE 2013, 8, e58258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strassburg, K.; Huijbrechts, A.M.L.; Kortekaas, K.A.; Lindeman, J.H.; Pedersen, T.L.; Dane, A.; Berger, R.; Brenkman, A.; Hankemeier, T.; van Duynhoven, J.; et al. Quantitative profiling of oxylipins through comprehensive LC-MS/MS analysis: Application in cardiac surgery. Anal. Bioanal. Chem. 2012, 404, 1413–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgren, T.M.; Heires, A.J.; Wyatt, T.A.; Poole, J.A.; LeVan, T.D.; Cerutis, D.R.; Romberger, D.J. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respir. Res. 2013, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Zahedi, A.; Phandthong, R.; Chaili, A.; Remark, G.; Talbot, P. Epithelial-to-mesenchymal transition of A549 lung cancer cells exposed to electronic cigarettes. Lung Cancer 2018, 122, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yuan, X.; Li, W.; Cao, Q.; Shu, Y. Aspirin-triggered resolvin D1 inhibits TGF-β1-induced EMT through the inhibition of the mTOR pathway by reducing the expression of PKM2 and is closely linked to oxidative stress. Int. J. Mol. Med. 2016, 38, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgren, T.M.; Friemel, T.D.; Heires, A.J.; Poole, J.A.; Wyatt, T.A.; Romberger, D.J. The Omega-3 Fatty Acid Docosahexaenoic Acid Attenuates Organic Dust-Induced Airway Inflammation. Nutrients 2014, 6, 5434–5452. [Google Scholar] [CrossRef] [Green Version]
- Bailey, K.L.; Poole, J.A.; Mathisen, T.L.; Wyatt, T.A.; Von Essen, S.G.; Romberger, D.J. Toll-like receptor 2 is upregulated by hog confinement dust in an IL-6-dependent manner in the airway epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1049–L1054. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, H.; Patel, S.B.; Pastar, I. The role of TGFβ signaling in wound epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linaker, C.; Smedley, J. Respiratory illness in agricultural workers. Occup. Med. 2002, 52, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Essen, S.; Romberger, D. The respiratory inflammatory response to the swine confinement building environment: The adaptation to respiratory exposures in the chronically exposed worker. J. Agric. Saf. Health 2003, 9, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, B.; Burdick, M.D.; Strieter, R.M. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2009, 41, 1708–1718. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 2004, 114, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Strieter, R.M.; Keeley, E.C.; Hughes, M.A.; Burdick, M.D.; Mehrad, B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J. Leukoc. Biol. 2009, 86, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Chang, E.H.; Chen, Y.; Lu, W.; Kim, M.M.; Niihori, M.; Hecker, L.; Kim, K.C. MUC1 contributes to goblet cell metaplasia and MUC5AC expression in response to cigarette smoke in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L82–L90. [Google Scholar] [CrossRef]
- Lange, J.H. Reduced cancer rates in agricultural workers: A benefit of environmental and occupational endotoxin exposure. Med. Hypotheses 2000, 55, 383–385. [Google Scholar] [CrossRef]
- Liebers, V.; Raulf-Heimsoth, M.; Bruning, T. Health effects due to endotoxin inhalation (review). Arch. Toxicol. 2008, 82, 203–210. [Google Scholar] [CrossRef]
- Ben Khedher, S.; Neri, M.; Guida, F.; Matrat, M.; Cenée, S.; Sanchez, M.; Menvielle, G.; Molinié, F.; Luce, D.; Stücker, I. Occupational exposure to endotoxins and lung cancer risk: Results of the ICARE Study. Occup. Environ. Med. 2017, 74, 667. [Google Scholar] [CrossRef]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. In Lung Cancer and Personalized Medicine: Current Knowledge and Therapies; Ahmad, A., Gadgeel, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–19. [Google Scholar] [CrossRef]
- Vannitamby, A.; Saad, M.I.; Aloe, C.; Wang, H.; Kumar, B.; Vlahos, R.; Selemidis, S.; Irving, L.; Steinfort, D.; Jenkins, B.J.; et al. Aspirin-Triggered Resolvin D1 Reduces Proliferation and the Neutrophil to Lymphocyte Ratio in a Mutant KRAS-Driven Lung Adenocarcinoma Model. Cancers Basel 2021, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biol. Basel 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Levy, B. Resolvin D1 and Resolvin E1 Promote the Resolution of Allergic Airway Inflammation via Shared and Distinct Molecular Counter-Regulatory Pathways. Front. Immunol. 2012, 3, 390. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Krishnamoorthy, S.; Recchiuti, A.; Chiang, N. Novel anti-inflammatory--pro-resolving mediators and their receptors. Curr. Top. Med. Chem. 2011, 11, 629–647. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Murthy, S.; Larson-Casey, J.L.; Ryan, A.J.; He, C.; Kobzik, L.; Carter, A.B. Alternative activation of macrophages and pulmonary fibrosis are modulated by scavenger receptor, macrophage receptor with collagenous structure. FASEB J. 2015, 29, 3527–3536. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Louie, M.C.; Vannella, K.M.; Wilke, C.A.; LeVine, A.M.; Moore, B.B.; Shanley, T.P. New concepts of IL-10-induced lung fibrosis: Fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L341–L353. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Makita, N.; Hizukuri, Y.; Yamashiro, K.; Murakawa, M.; Hayashi, Y. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. Int. Immunol. 2014, 27, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, B.B.; Kolodsick, J.E.; Thannickal, V.J.; Cooke, K.; Moore, T.A.; Hogaboam, C.; Wilke, C.A.; Toews, G.B. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 2005, 166, 675–684. [Google Scholar] [CrossRef] [Green Version]
- He, H.-Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Pirault, J.; Bäck, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Shen, W.; Peng, H.; Li, Y.; Chen, F.; Zheng, L.; Xu, J.; Jia, L. Fibronectin 1 promotes melanoma proliferation and metastasis by inhibiting apoptosis and regulating EMT. Onco Targets 2019, 12, 3207–3221. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Park, M.K.; Lee, E.J.; Lee, C.H. Resolvin D1 inhibits TGF-β1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int. J. Biochem. Cell Biol. 2013, 45, 2801–2807. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
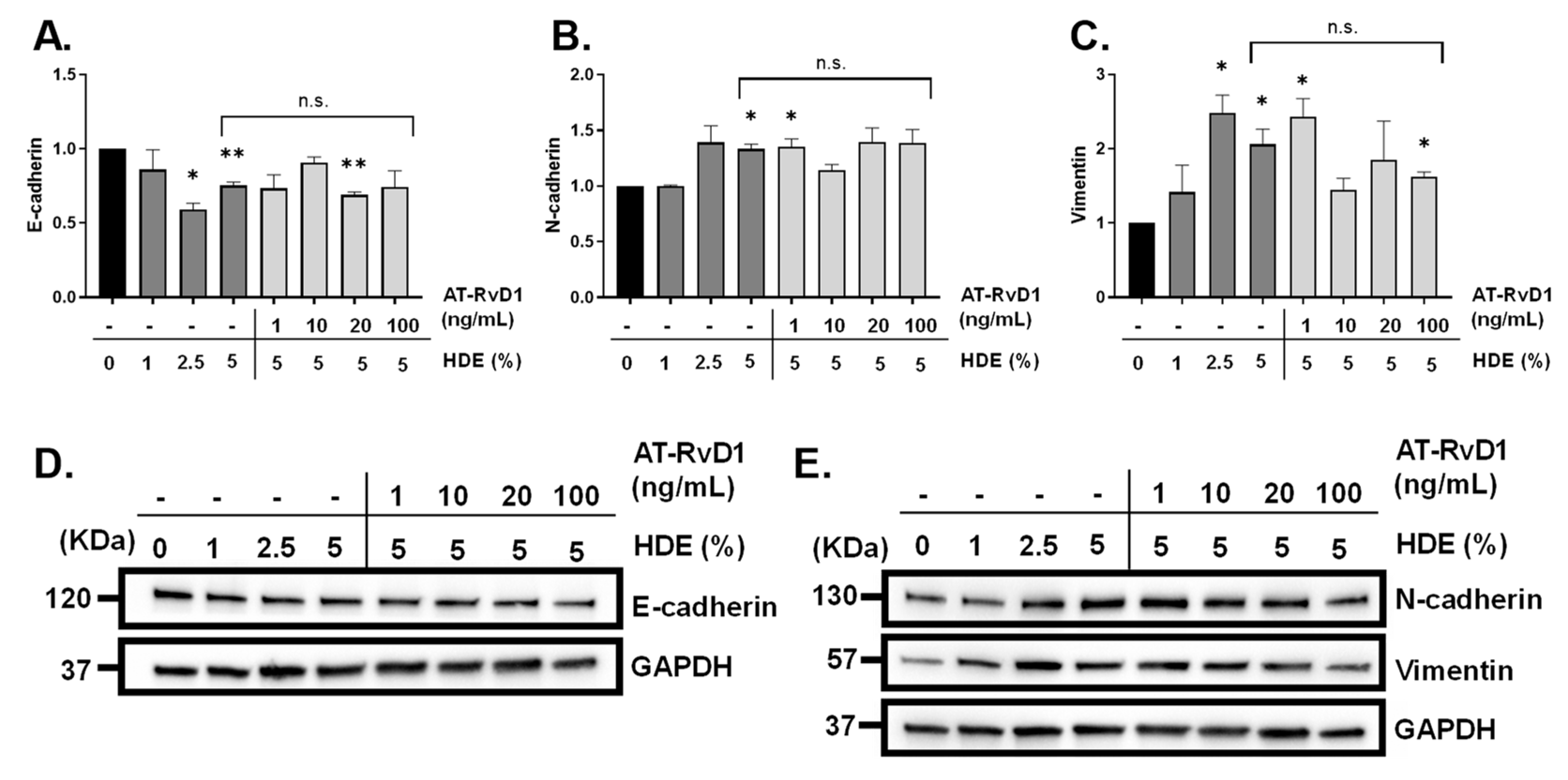{kind=link}
| Experimental Group | % of Mice with Lung Tumor a |
Lung Tumor/ Mouse ± SE a |
Confidence Interval (95%) |
|---|---|---|---|
| Saline Only | 0 | 0 | (0.00–0.00) |
| Saline + AT-RvD1 | 0 | 0 | (0.00–0.00) |
| HDE Only | 20 | 0.20 + 0.13 | (0.00–0.50) |
| HDE + AT-RvD1 | 0 | 0 | (0.00–0.00) |
| Saline + NNK | 100 | 2.38 ± 0.46 | (1.29–3.46) |
| Saline + NNK + AT-RvD1 | 100 | 3.57 ± 0.57 | (2.17–4.97) |
| HDE + NNK | 100 | 5.33 ± 1.18 | (2.62–8.054) |
| HDE + NNK + AT-RvD1 | 100 | 6.33 ± 1.15 | (3.39–9.28) |
| Gene Symbol | Fold Change (HDE vs. Saline) |
|---|---|
| CCL6 | 4.43 ↑ |
| CCL9 | 3.37 ↑ |
| CCR2 | 2.30 ↑ |
| CD14 | 3.03 ↑ |
| CD68 | 2.44 ↑ |
| CD84 | 3.08 ↑ |
| CHIL3 | 10.9 ↑ |
| CLEC7a | 3.95 ↑ |
| CSF1R | 1.23 ↑ |
| CXCR4 | 1.18 ↑ |
| FPR2 | 1.81 ↑ |
| IL13Ra1 | 1.35 ↑ |
| JAK2 | 1.21 ↑ |
| LCN2 | 4.26 ↑ |
| MRC1 | 1.79 ↑ |
| STAT1 | 1.25 ↓ |
| TLR4 | 1.35 ↑ |
| Gene Symbol | Fold Change (HDE vs. Saline) |
|---|---|
| CD274 | 1.87 ↑ |
| CD9 | 1.14 ↑ |
| CDH1 | 1.12 ↓ |
| FN1 | 1.44 ↑ |
| KDR | 1.63 ↓ |
| LAMP1 | 1.24 ↑ |
| MUC1 | 1.40 ↑ |
| TGFB1 | 1.38 ↑ |
| TGFBR1 | 1.63 ↑ |
| VEGFA | 1.68 ↓ |
| VIM | 1.42 ↑ |
| Gene Symbol | Fold Change (HDE vs. HDE + NNK) |
|---|---|
| C7 | 1.40 ↓ |
| ITGA6 | 1.31 ↑ |
| IRF2 | 1.20 ↓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dominguez, E.C.; Phandthong, R.; Nguyen, M.; Ulu, A.; Guardado, S.; Sveiven, S.; Talbot, P.; Nordgren, T.M. Aspirin-Triggered Resolvin D1 Reduces Chronic Dust-Induced Lung Pathology without Altering Susceptibility to Dust-Enhanced Carcinogenesis. Cancers 2022, 14, 1900. https://doi.org/10.3390/cancers14081900
Dominguez EC, Phandthong R, Nguyen M, Ulu A, Guardado S, Sveiven S, Talbot P, Nordgren TM. Aspirin-Triggered Resolvin D1 Reduces Chronic Dust-Induced Lung Pathology without Altering Susceptibility to Dust-Enhanced Carcinogenesis. Cancers. 2022; 14(8):1900. https://doi.org/10.3390/cancers14081900
Chicago/Turabian StyleDominguez, Edward C., Rattapol Phandthong, Matthew Nguyen, Arzu Ulu, Stephanie Guardado, Stefanie Sveiven, Prue Talbot, and Tara M. Nordgren. 2022. "Aspirin-Triggered Resolvin D1 Reduces Chronic Dust-Induced Lung Pathology without Altering Susceptibility to Dust-Enhanced Carcinogenesis" Cancers 14, no. 8: 1900. https://doi.org/10.3390/cancers14081900
APA StyleDominguez, E. C., Phandthong, R., Nguyen, M., Ulu, A., Guardado, S., Sveiven, S., Talbot, P., & Nordgren, T. M. (2022). Aspirin-Triggered Resolvin D1 Reduces Chronic Dust-Induced Lung Pathology without Altering Susceptibility to Dust-Enhanced Carcinogenesis. Cancers, 14(8), 1900. https://doi.org/10.3390/cancers14081900