
Alternative Splicing in Cancer and Immune Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
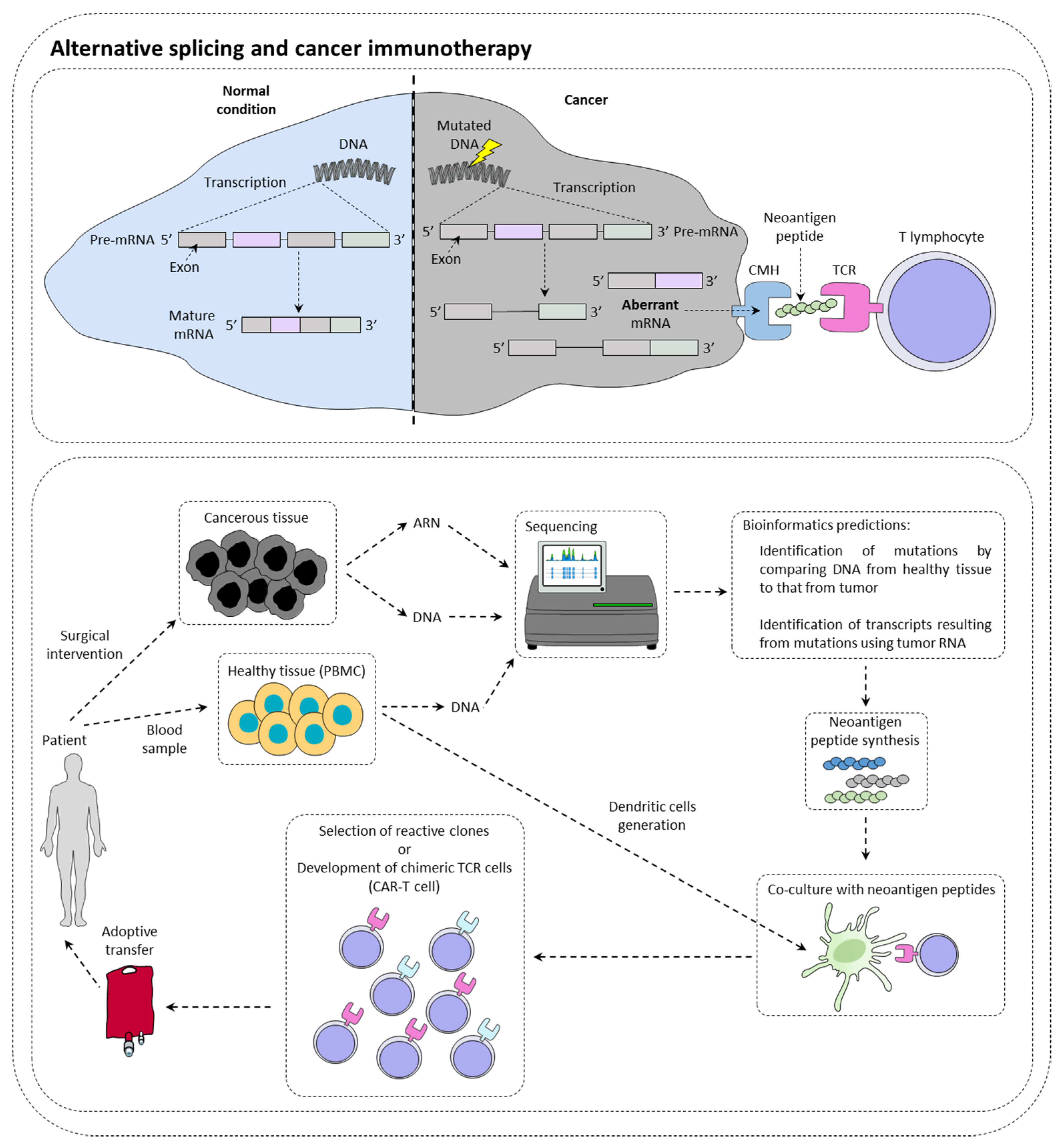
2. Alternative Splicing and Cancer Immunotherapy
2.1. Alternative Splicing Can Increase Immunotherapy Response
2.2. Alternative Splicing Generates Neoantigens
2.3. Alternative Splicing Can Limit Inhibitor of Checkpoint Inhibitor Response
2.4. Alternative Splicing May Limit the Efficiency of CAR-T Cells
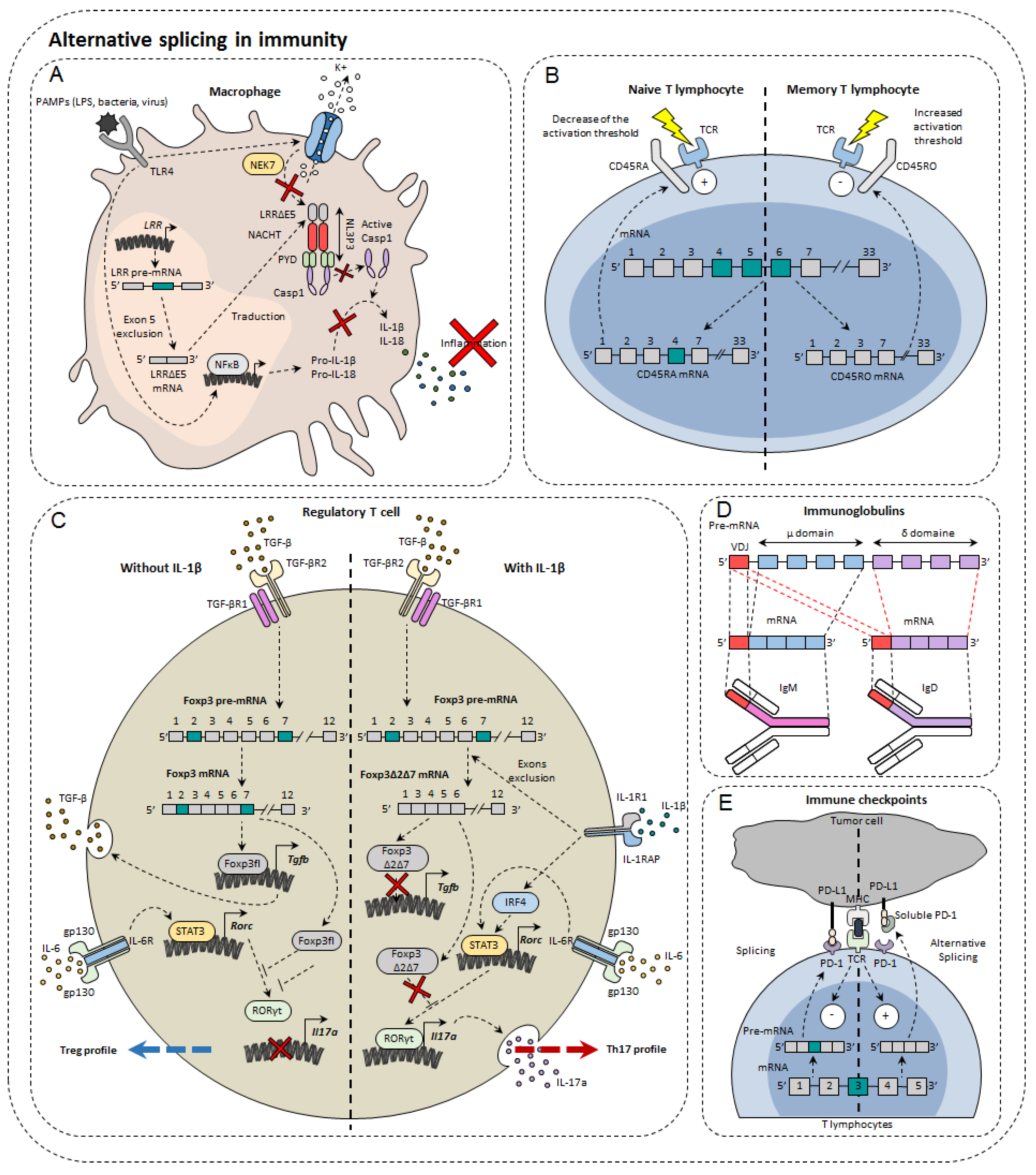
3. Alternative Splicing in Immunity Modulation
3.1. Immunoglobulins and B Cells
3.2. Immune Cell Receptors
3.3. Transcription Factors
3.4. Immune Checkpoints
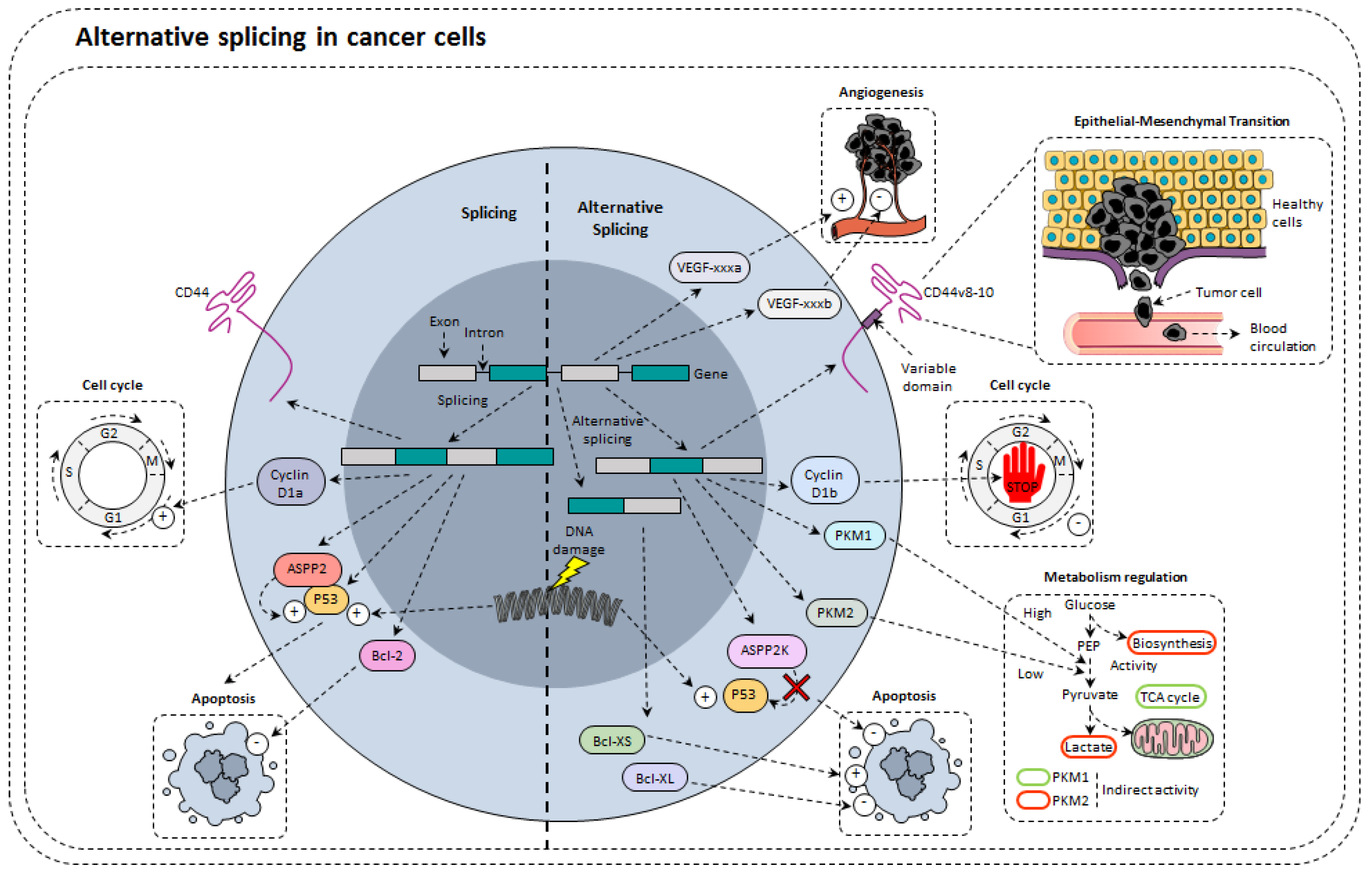
4. Alternative Splicing in Cancer Cells
4.1. Alternative Splicing of Tumor Suppressors
4.2. Alternative Splicing Modulates Proliferation
4.3. Alternative Splicing of Apoptosis Related Genes
4.4. Alternative Splicing and Angiogenesis
4.5. Alternative Splicing and Metabolism Regulation
4.6. Alternative Splicing and Epithelial-Mesenchymal Transformation
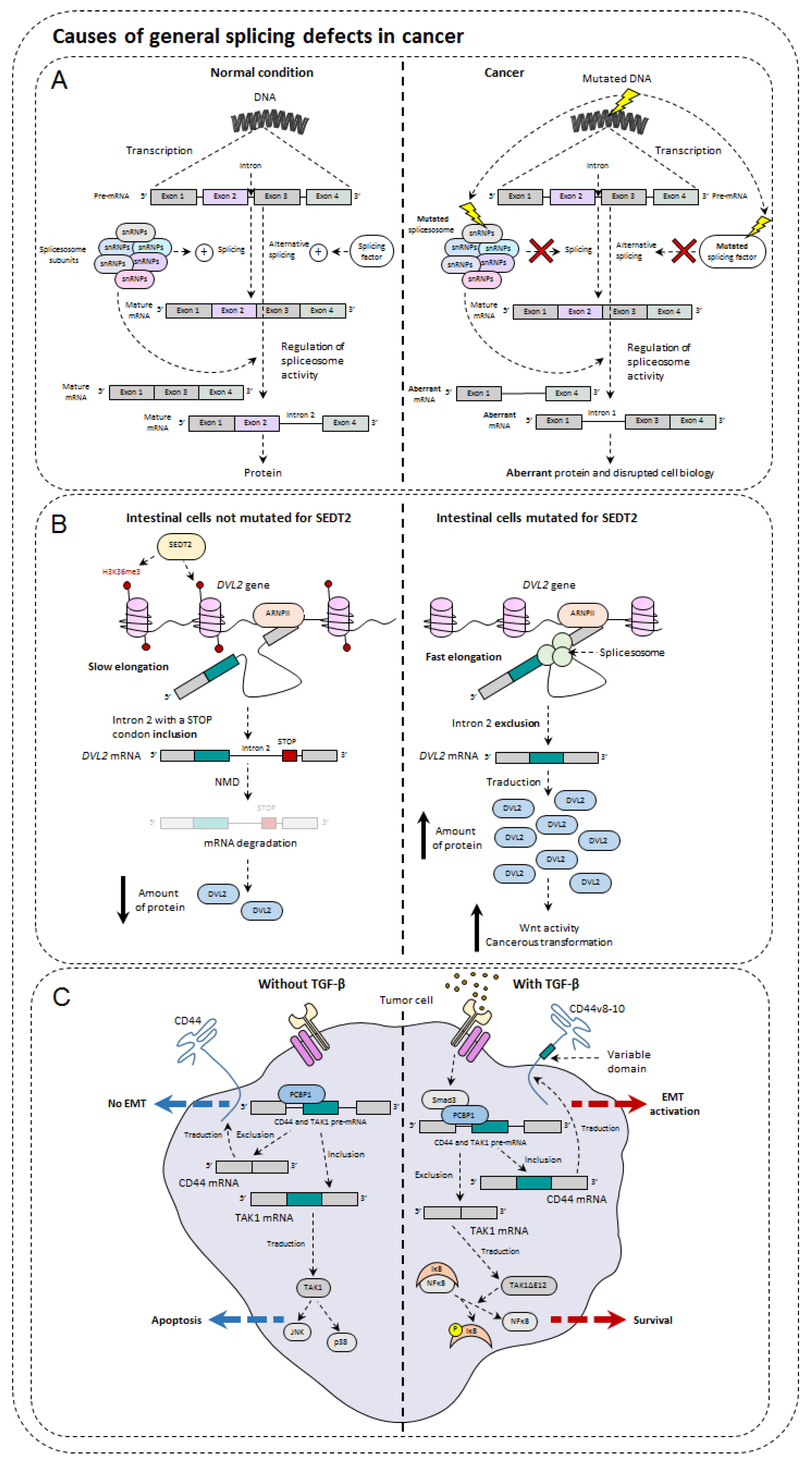
5. Causes of General Splicing Defects in Cancer
5.1. Mutations of Spliceosome Actors
5.2. Splice Factor Mutations
5.3. Impact of Epigenetics on Alternative Splicing and Cancer
5.4. Influence of TGF-β in Alternative Splicing
5.5. Metabolism and Hypoxia
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research, N.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e216. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Zhang, Z.; Pan, Z.; Ying, Y.; Xie, Z.; Adhikari, S.; Phillips, J.; Carstens, R.P.; Black, D.L.; Wu, Y.; Xing, Y. Deep-learning augmented RNA-seq analysis of transcript splicing. Nat. Methods 2019, 16, 307–310. [Google Scholar] [CrossRef]
- Caron, E.; Espona, L.; Kowalewski, D.J.; Schuster, H.; Ternette, N.; Alpizar, A.; Schittenhelm, R.B.; Ramarathinam, S.H.; Lindestam Arlehamn, C.S.; Chiek Koh, C.; et al. An open-source computational and data resource to analyze digital maps of immunopeptidomes. Elife 2015, 4, e07661. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.S.; Andersen, S.R.; Hjortso, M.D.; Lyngaa, R.; Idorn, M.; Kollgard, T.M.; Met, O.; Thor Straten, P.; Hadrup, S.R. High frequency of T cells specific for cryptic epitopes in melanoma patients. Oncoimmunology 2013, 2, e25374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kobayashi, J.; Torigoe, T.; Hirohashi, Y.; Idenoue, S.; Miyazaki, A.; Yamaguchi, A.; Hiratsuka, H.; Sato, N. Comparative study on the immunogenicity between an HLA-A24-restricted cytotoxic T-cell epitope derived from survivin and that from its splice variant survivin-2B in oral cancer patients. J. Transl. Med. 2009, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Slager, E.H.; van der Minne, C.E.; Kruse, M.; Krueger, D.D.; Griffioen, M.; Osanto, S. Identification of multiple HLA-DR-restricted epitopes of the tumor-associated antigen CAMEL by CD4+ Th1/Th2 lymphocytes. J. Immunol. 2004, 172, 5095–5102. [Google Scholar] [CrossRef] [PubMed]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Hassounah, N.B.; Malladi, V.S.; Huang, Y.; Freeman, S.S.; Beauchamp, E.M.; Koyama, S.; Souders, N.; Martin, S.; Dranoff, G.; Wong, K.K.; et al. Identification and characterization of an alternative cancer-derived PD-L1 splice variant. Cancer Immunol. Immunother. 2019, 68, 407–420. [Google Scholar] [CrossRef]
- Oaks, M.K.; Hallett, K.M.; Penwell, R.T.; Stauber, E.C.; Warren, S.J.; Tector, A.J. A native soluble form of CTLA-4. Cell. Immunol. 2000, 201, 144–153. [Google Scholar] [CrossRef]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Fala, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef]
- Laurent, S.; Queirolo, P.; Boero, S.; Salvi, S.; Piccioli, P.; Boccardo, S.; Minghelli, S.; Morabito, A.; Fontana, V.; Pietra, G.; et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-alpha production. J. Transl. Med. 2013, 11, 108. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef]
- Topp, M.S.; Gokbuget, N.; Zugmaier, G.; Klappers, P.; Stelljes, M.; Neumann, S.; Viardot, A.; Marks, R.; Diedrich, H.; Faul, C.; et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 4134–4140. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Schaub, A.; Glasmacher, E. Splicing in immune cells-mechanistic insights and emerging topics. Int. Immunol. 2017, 29, 173–181. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Danhorn, T.; De Arras, L.; Flatley, B.R.; Marcus, R.A.; Farias-Hesson, E.; Leach, S.M.; Alper, S. Regulation of toll-like receptor signaling by the SF3a mRNA splicing complex. PLoS Genet. 2015, 11, e1004932. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef]
- Butte, M.J.; Lee, S.J.; Jesneck, J.; Keir, M.E.; Haining, W.N.; Sharpe, A.H. CD28 costimulation regulates genome-wide effects on alternative splicing. PLoS ONE 2012, 7, e40032. [Google Scholar] [CrossRef]
- Martinez, N.M.; Pan, Q.; Cole, B.S.; Yarosh, C.A.; Babcock, G.A.; Heyd, F.; Zhu, W.; Ajith, S.; Blencowe, B.J.; Lynch, K.W. Alternative splicing networks regulated by signaling in human T cells. RNA 2012, 18, 1029–1040. [Google Scholar] [CrossRef]
- Toung, J.M.; Morley, M.; Li, M.; Cheung, V.G. RNA-sequence analysis of human B-cells. Genome Res. 2011, 21, 991–998. [Google Scholar] [CrossRef]
- Maki, T.; Okazaki, H.; Wood, M.L.; Monaco, A.P. Suppressor cells in mice bearing intact skin allografts after blood transfusions. Transplantation 1981, 32, 463–466. [Google Scholar] [CrossRef]
- Enders, M.; Hunjet, A.; Gleich, M.; Imdahl, R.; Muhlbacher, A.; Schennach, H.; Chaiwong, K.; Sakuldamrongpanich, T.; Turhan, A.; Sertoz, R.; et al. Performance evaluation of the Elecsys syphilis assay for the detection of total antibodies to Treponema pallidum. Clin. Vaccine Immunol. 2015, 22, 17–26. [Google Scholar] [CrossRef]
- Saxon, A.; Diaz-Sanchez, D.; Zhang, K. Regulation of the expression of distinct human secreted IgE proteins produced by alternative RNA splicing. Biochem. Soc. Trans. 1997, 25, 383–387. [Google Scholar] [CrossRef]
- Iwami, K.I.; Matsuguchi, T.; Masuda, A.; Kikuchi, T.; Musikacharoen, T.; Yoshikai, Y. Cutting edge: Naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol. 2000, 165, 6682–6686. [Google Scholar] [CrossRef] [PubMed]
- De Arras, L.; Laws, R.; Leach, S.M.; Pontis, K.; Freedman, J.H.; Schwartz, D.A.; Alper, S. Comparative genomics RNAi screen identifies Eftud2 as a novel regulator of innate immunity. Genetics 2014, 197, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, B.P.; Toy, D.Y.; Swart, D.A.; Smithgall, M.D.; Smith, D. Deletion of the ST2 proximal promoter disrupts fibroblast-specific expression but does not reduce the amount of soluble ST2 in circulation. Eur. J. Immunol. 2012, 42, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Kim, S.; Lin, P.C. Interleukin-33 and ST2 Signaling in Tumor Microenvironment. J. Interferon Cytokine Res. 2019, 39, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, M.; Takenaga, K. Role of the IL-33/ST2L axis in colorectal cancer progression. Cell. Immunol. 2019, 343, 103740. [Google Scholar] [CrossRef]
- Akimoto, M.; Maruyama, R.; Takamaru, H.; Ochiya, T.; Takenaga, K. Soluble IL-33 receptor sST2 inhibits colorectal cancer malignant growth by modifying the tumour microenvironment. Nat. Commun. 2016, 7, 13589. [Google Scholar] [CrossRef]
- Hoss, F.; Mueller, J.L.; Rojas Ringeling, F.; Rodriguez-Alcazar, J.F.; Brinkschulte, R.; Seifert, G.; Stahl, R.; Broderick, L.; Putnam, C.D.; Kolodner, R.D.; et al. Alternative splicing regulates stochastic NLRP3 activity. Nat. Commun. 2019, 10, 3238. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- McNeill, L.; Cassady, R.L.; Sarkardei, S.; Cooper, J.C.; Morgan, G.; Alexander, D.R. CD45 isoforms in T cell signalling and development. Immunol. Lett. 2004, 92, 125–134. [Google Scholar] [CrossRef]
- Tong, A.; Nguyen, J.; Lynch, K.W. Differential expression of CD45 isoforms is controlled by the combined activity of basal and inducible splicing-regulatory elements in each of the variable exons. J. Biol. Chem. 2005, 280, 38297–38304. [Google Scholar] [CrossRef]
- DeGrendele, H.C.; Estess, P.; Siegelman, M.H. Requirement for CD44 in activated T cell extravasation into an inflammatory site. Science 1997, 278, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Matter, N.; Herrlich, P.; Konig, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Pure, E.; Cuff, C.A. A crucial role for CD44 in inflammation. Trends Mol. Med. 2001, 7, 213–221. [Google Scholar] [CrossRef]
- Mailer, R.K.; Falk, K.; Rotzschke, O. Absence of leucine zipper in the natural FOXP3Delta2Delta7 isoform does not affect dimerization but abrogates suppressive capacity. PLoS ONE 2009, 4, e6104. [Google Scholar] [CrossRef]
- Harbuz, R.; Lespinasse, J.; Boulet, S.; Francannet, C.; Creveaux, I.; Benkhelifa, M.; Jouk, P.S.; Lunardi, J.; Ray, P.F. Identification of new FOXP3 mutations and prenatal diagnosis of IPEX syndrome. Prenat. Diagn. 2010, 30, 1072–1078. [Google Scholar] [CrossRef]
- Ueno, A.; Jeffery, L.; Kobayashi, T.; Hibi, T.; Ghosh, S.; Jijon, H. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 2018, 87, 38–49. [Google Scholar] [CrossRef]
- Kano, S.; Sato, K.; Morishita, Y.; Vollstedt, S.; Kim, S.; Bishop, K.; Honda, K.; Kubo, M.; Taniguchi, T. The contribution of transcription factor IRF1 to the interferon-gamma-interleukin 12 signaling axis and TH1 versus TH-17 differentiation of CD4+ T cells. Nat. Immunol. 2008, 9, 34–41. [Google Scholar] [CrossRef]
- Unutmaz, D.; Vilcek, J. IRF1: A deus ex machina in TH1 differentiation. Nat. Immunol. 2008, 9, 9–10. [Google Scholar] [CrossRef]
- Bernard, A.; Hibos, C.; Richard, C.; Viltard, E.; Chevrier, S.; Lemoine, S.; Melin, J.; Humblin, E.; Mary, R.; Accogli, T.; et al. The Tumor Microenvironment Impairs Th1 IFNgamma Secretion through Alternative Splicing Modifications of Irf1 Pre-mRNA. Cancer Immunol. Res. 2021, 9, 324–336. [Google Scholar] [CrossRef]
- Sorensen, S.F.; Demuth, C.; Weber, B.; Sorensen, B.S.; Meldgaard, P. Increase in soluble PD-1 is associated with prolonged survival in patients with advanced EGFR-mutated non-small cell lung cancer treated with erlotinib. Lung Cancer 2016, 100, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Bai, J.; Jiang, T.; Gao, Y.; Hua, Y. Modulation of PDCD1 exon 3 splicing. RNA Biol. 2019, 16, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Ward, F.J.; Dahal, L.N.; Khanolkar, R.C.; Shankar, S.P.; Barker, R.N. Targeting the alternatively spliced soluble isoform of CTLA-4: Prospects for immunotherapy? Immunotherapy 2014, 6, 1073–1084. [Google Scholar] [CrossRef]
- Chabot, B.; Shkreta, L. Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol. 2016, 212, 13–27. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Ouyang, J.; Zhang, Y.; Xiong, F.; Zhang, S.; Gong, Z.; Yan, Q.; He, Y.; Wei, F.; Zhang, W.; Zhou, M.; et al. The role of alternative splicing in human cancer progression. Am. J. Cancer Res. 2021, 11, 4642–4667. [Google Scholar]
- Smeby, J.; Sveen, A.; Eilertsen, I.A.; Danielsen, S.A.; Hoff, A.M.; Eide, P.W.; Johannessen, B.; Hektoen, M.; Skotheim, R.I.; Guren, M.G.; et al. Transcriptional and functional consequences of TP53 splice mutations in colorectal cancer. Oncogenesis 2019, 8, 35. [Google Scholar] [CrossRef]
- Bouvard, V.; Zaitchouk, T.; Vacher, M.; Duthu, A.; Canivet, M.; Choisy-Rossi, C.; Nieruchalski, M.; May, E. Tissue and cell-specific expression of the p53-target genes: Bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene 2000, 19, 649–660. [Google Scholar] [CrossRef]
- Fei, P.; Bernhard, E.J.; El-Deiry, W.S. Tissue-specific induction of p53 targets in vivo. Cancer Res. 2002, 62, 7316–7327. [Google Scholar]
- Rohaly, G.; Chemnitz, J.; Dehde, S.; Nunez, A.M.; Heukeshoven, J.; Deppert, W.; Dornreiter, I. A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell 2005, 122, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Uthaiah, R.; Howard, J.; Herrmann, C.; Wolf, E. Crystal structure of IIGP1: A paradigm for interferon-inducible p47 resistance GTPases. Mol. Cell 2004, 15, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Fujita, T.; Kimura, Y.; Maruyama, M.; Harada, H.; Sudo, Y.; Miyata, T.; Taniguchi, T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell 1988, 54, 903–913. [Google Scholar] [CrossRef]
- Tanaka, N.; Ishihara, M.; Lamphier, M.S.; Nozawa, H.; Matsuyama, T.; Mak, T.W.; Aizawa, S.; Tokino, T.; Oren, M.; Taniguchi, T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature 1996, 382, 816–818. [Google Scholar] [CrossRef]
- Yanai, H.; Negishi, H.; Taniguchi, T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Oncoimmunology 2012, 1, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Um, S.J.; Rhyu, J.W.; Kim, E.J.; Jeon, K.C.; Hwang, E.S.; Park, J.S. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Tzoanopoulos, D.; Speletas, M.; Arvanitidis, K.; Veiopoulou, C.; Kyriaki, S.; Thyphronitis, G.; Sideras, P.; Kartalis, G.; Ritis, K. Low expression of interferon regulatory factor-1 and identification of novel exons skipping in patients with chronic myeloid leukaemia. Br. J. Haematol. 2002, 119, 46–53. [Google Scholar] [CrossRef]
- Lee, E.J.; Jo, M.; Park, J.; Zhang, W.; Lee, J.H. Alternative splicing variants of IRF-1 lacking exons 7, 8, and 9 in cervical cancer. Biochem. Biophys. Res. Commun. 2006, 347, 882–888. [Google Scholar] [CrossRef]
- Schittenhelm, M.M.; Walter, B.; Tsintari, V.; Federmann, B.; Bajrami Saipi, M.; Akmut, F.; Illing, B.; Mau-Holzmann, U.; Fend, F.; Lopez, C.D.; et al. Alternative splicing of the tumor suppressor ASPP2 results in a stress-inducible, oncogenic isoform prevalent in acute leukemia. EBioMedicine 2019, 42, 340–351. [Google Scholar] [CrossRef]
- Betticher, D.C.; Thatcher, N.; Altermatt, H.J.; Hoban, P.; Ryder, W.D.; Heighway, J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene 1995, 11, 1005–1011. [Google Scholar] [PubMed]
- Paronetto, M.P.; Cappellari, M.; Busa, R.; Pedrotti, S.; Vitali, R.; Comstock, C.; Hyslop, T.; Knudsen, K.E.; Sette, C. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010, 70, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Comstock, C.E.; Augello, M.A.; Benito, R.P.; Karch, J.; Tran, T.H.; Utama, F.E.; Tindall, E.A.; Wang, Y.; Burd, C.J.; Groh, E.M.; et al. Cyclin D1 splice variants: Polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin. Cancer Res. 2009, 15, 5338–5349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dean, J.L.; Millar, E.K.; Tran, T.H.; McNeil, C.M.; Burd, C.J.; Henshall, S.M.; Utama, F.E.; Witkiewicz, A.; Rui, H.; et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008, 68, 5628–5638. [Google Scholar] [CrossRef]
- Zhu, J.; Sen, S.; Wei, C.; Frazier, M.L. Cyclin D1b represses breast cancer cell growth by antagonizing the action of cyclin D1a on estrogen receptor alpha-mediated transcription. Int. J. Oncol. 2010, 36, 39–48. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Han, Z.; Hendrickson, E.A.; Bremner, T.A.; Wyche, J.H. A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J. Biol. Chem. 1997, 272, 13432–13436. [Google Scholar] [CrossRef]
- Huang, Y.; Shin, N.H.; Sun, Y.; Wang, K.K. Molecular cloning and characterization of a novel caspase-3 variant that attenuates apoptosis induced by proteasome inhibition. Biochem. Biophys. Res. Commun. 2001, 283, 762–769. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R.; Solary, E.; Lizard-Nacol, S. A short caspase-3 isoform inhibits chemotherapy-induced apoptosis by blocking apoptosome assembly. PLoS ONE 2011, 6, e29058. [Google Scholar] [CrossRef]
- Bernard, A.; Chevrier, S.; Beltjens, F.; Dosset, M.; Viltard, E.; Lagrange, A.; Derangere, V.; Oudot, A.; Ghiringhelli, F.; Collin, B.; et al. Cleaved Caspase-3 Transcriptionally Regulates Angiogenesis-Promoting Chemotherapy Resistance. Cancer Res. 2019, 79, 5958–5970. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.A.; Li, S.D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, P.G.; Tsiakanikas, P.; Adam, E.E.; Scorilas, A. Unraveling novel survivin mRNA transcripts in cancer cells using an in-house developed targeted high-throughput sequencing approach. Genomics 2021, 113, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Sharp, T.V.; Koumi, A.; Koentges, G.; Boshoff, C. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi’s sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 2002, 21, 2602–2615. [Google Scholar] [CrossRef]
- Krieg, A.; Mahotka, C.; Krieg, T.; Grabsch, H.; Muller, W.; Takeno, S.; Suschek, C.V.; Heydthausen, M.; Gabbert, H.E.; Gerharz, C.D. Expression of different survivin variants in gastric carcinomas: First clues to a role of survivin-2B in tumour progression. Br. J. Cancer 2002, 86, 737–743. [Google Scholar] [CrossRef]
- Caldas, H.; Honsey, L.E.; Altura, R.A. Survivin 2alpha: A novel Survivin splice variant expressed in human malignancies. Mol. Cancer 2005, 4, 11. [Google Scholar] [CrossRef]
- Vegran, F.; Mary, R.; Gibeaud, A.; Mirjolet, C.; Collin, B.; Oudot, A.; Charon-Barra, C.; Arnould, L.; Lizard-Nacol, S.; Boidot, R. Survivin-3B potentiates immune escape in cancer but also inhibits the toxicity of cancer chemotherapy. Cancer Res. 2013, 73, 5391–5401. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R. Survivin-3B promotes chemoresistance and immune escape by inhibiting caspase-8 and -6 in cancer cells. Oncoimmunology 2013, 2, e26328. [Google Scholar] [CrossRef][Green Version]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Ferrara, N.; Frantz, G.; LeCouter, J.; Dillard-Telm, L.; Pham, T.; Draksharapu, A.; Giordano, T.; Peale, F. Differential expression of the angiogenic factor genes vascular endothelial growth factor (VEGF) and endocrine gland-derived VEGF in normal and polycystic human ovaries. Am. J. Pathol. 2003, 162, 1881–1893. [Google Scholar] [CrossRef]
- Albuquerque, R.J.; Hayashi, T.; Cho, W.G.; Kleinman, M.E.; Dridi, S.; Takeda, A.; Baffi, J.Z.; Yamada, K.; Kaneko, H.; Green, M.G.; et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 2009, 15, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Goldman, C.K.; Kendall, R.L.; Cabrera, G.; Soroceanu, L.; Heike, Y.; Gillespie, G.Y.; Siegal, G.P.; Mao, X.; Bett, A.J.; Huckle, W.R.; et al. Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc. Natl. Acad. Sci. USA 1998, 95, 8795–8800. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Manley, J.L. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010, 70, 8977–8980. [Google Scholar] [CrossRef]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Yan, S.; Yang, X.F.; Liu, H.L.; Fu, N.; Ouyang, Y.; Qing, K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J. Gastroenterol. 2015, 21, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Soupene, E.; Kuypers, F.A. Mammalian long-chain acyl-CoA synthetases. Exp. Biol. Med. (Maywood) 2008, 233, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Zhao, P.; Damerow, M.S.; Stern, P.; Liu, A.H.; Sweet-Cordero, A.; Siziopikou, K.; Neilson, J.R.; Sharp, P.A.; Cheng, C. CD44 promotes Kras-dependent lung adenocarcinoma. Oncogene 2013, 32, 5186–5190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prochazka, L.; Tesarik, R.; Turanek, J. Regulation of alternative splicing of CD44 in cancer. Cell. Signal. 2014, 26, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.; Heider, K.H.; Offerhaus, G.J.; Adolf, G.R.; van den Berg, F.M.; Ponta, H.; Herrlich, P.; Pals, S.T. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res. 1993, 53, 4754–4756. [Google Scholar] [PubMed]
- Ni, J.; Cozzi, P.J.; Hao, J.L.; Beretov, J.; Chang, L.; Duan, W.; Shigdar, S.; Delprado, W.J.; Graham, P.H.; Bucci, J.; et al. CD44 variant 6 is associated with prostate cancer metastasis and chemo-/radioresistance. Prostate 2014, 74, 602–617. [Google Scholar] [CrossRef]
- Biddle, A.; Gammon, L.; Fazil, B.; Mackenzie, I.C. CD44 staining of cancer stem-like cells is influenced by down-regulation of CD44 variant isoforms and up-regulation of the standard CD44 isoform in the population of cells that have undergone epithelial-to-mesenchymal transition. PLoS ONE 2013, 8, e57314. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan-CD44 Interactions in Cancer: Paradoxes and Possibilities. Clin. Cancer Res. 2009, 15, 7462–7468. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Gozani, O.; Feld, R.; Reed, R. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes Dev. 1996, 10, 233–243. [Google Scholar] [CrossRef]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A.; et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Investig. 2017, 127, 3557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.J.; Dai, Z.; Zhou, S.L.; Fu, X.T.; Zhao, Y.M.; Shi, Y.H.; Zhou, J.; Fan, J. Overexpression of HnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int. J. Cancer 2013, 132, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M., Jr.; Fu, X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Kornblihtt, A.R. Epigenetics at the base of alternative splicing changes that promote colorectal cancer. J. Clin. Investig. 2017, 127, 3281–3283. [Google Scholar] [CrossRef]
- Tripathi, V.; Sixt, K.M.; Gao, S.; Xu, X.; Huang, J.; Weigert, R.; Zhou, M.; Zhang, Y.E. Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-beta. Mol. Cell 2016, 64, 1010. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Arsura, M.; Panta, G.R.; Bilyeu, J.D.; Cavin, L.G.; Sovak, M.A.; Oliver, A.A.; Factor, V.; Heuchel, R.; Mercurio, F.; Thorgeirsson, S.S.; et al. Transient activation of NF-kappaB through a TAK1/IKK kinase pathway by TGF-beta1 inhibits AP-1/SMAD signaling and apoptosis: Implications in liver tumor formation. Oncogene 2003, 22, 412–425. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Shin, J.H.; Stuelten, C.H.; Zhang, Y.E. TGF-beta-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene 2019, 38, 3185–3200. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Amirkhah, R.; Naderi-Meshkin, H.; Shah, J.S.; Dunne, P.D.; Schmitz, U. The Intricate Interplay between Epigenetic Events, Alternative Splicing and Noncoding RNA Deregulation in Colorectal Cancer. Cells 2019, 8, 929. [Google Scholar] [CrossRef]
- Dhamija, S.; Diederichs, S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int. J. Cancer 2016, 139, 269–280. [Google Scholar] [CrossRef]
- Shih, J.W.; Kung, H.J. Long non-coding RNA and tumor hypoxia: New players ushered toward an old arena. J. Biomed. Sci. 2017, 24, 53. [Google Scholar] [CrossRef]
- Cho, H.S.; Han, T.S.; Hur, K.; Ban, H.S. The Roles of Hypoxia-Inducible Factors and Non-Coding RNAs in Gastrointestinal Cancer. Genes 2019, 10, 1008. [Google Scholar] [CrossRef]
- Xiao, H.; Tang, K.; Liu, P.; Chen, K.; Hu, J.; Zeng, J.; Xiao, W.; Yu, G.; Yao, W.; Zhou, H.; et al. LncRNA MALAT1 functions as a competing endogenous RNA to regulate ZEB2 expression by sponging miR-200s in clear cell kidney carcinoma. Oncotarget 2015, 6, 38005–38015. [Google Scholar] [CrossRef]
- Liu, Z.; Han, L.; Dong, Y.; Tan, Y.; Li, Y.; Zhao, M.; Xie, H.; Ju, H.; Wang, H.; Zhao, Y.; et al. EGFRvIII/integrin beta3 interaction in hypoxic and vitronectinenriching microenvironment promote GBM progression and metastasis. Oncotarget 2016, 7, 4680–4694. [Google Scholar] [CrossRef]
- Mardy, S.; Miura, Y.; Endo, F.; Matsuda, I.; Sztriha, L.; Frossard, P.; Moosa, A.; Ismail, E.A.; Macaya, A.; Andria, G.; et al. Congenital insensitivity to pain with anhidrosis: Novel mutations in the TRKA (NTRK1) gene encoding a high-affinity receptor for nerve growth factor. Am. J. Hum. Genet. 1999, 64, 1570–1579. [Google Scholar] [CrossRef]
- Riffo-Campos, A.L.; Gimeno-Valiente, F.; Rodriguez, F.M.; Cervantes, A.; Lopez-Rodas, G.; Franco, L.; Castillo, J. Role of epigenetic factors in the selection of the alternative splicing isoforms of human KRAS in colorectal cancer cell lines. Oncotarget 2018, 9, 20578–20589. [Google Scholar] [CrossRef] [PubMed]
- Voice, J.K.; Klemke, R.L.; Le, A.; Jackson, J.H. Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J. Biol. Chem. 1999, 274, 17164–17170. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Ashok, C.; Natua, S.; Pant, D.; Cherian, A.; Pandkar, M.R.; Yadav, P.; Vishnu, N.S.S.; Mishra, J.; Samaiya, A.; et al. Hypoxia-induced TGF-beta-RBFOX2-ESRP1 axis regulates human MENA alternative splicing and promotes EMT in breast cancer. NAR Cancer 2020, 2, zcaa021. [Google Scholar] [CrossRef]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Penet, M.F.; Nimmagadda, S.; Mironchik, Y.; Raman, V.; Solaiyappan, M.; Semenza, G.L.; Pomper, M.G.; Bhujwalla, Z.M. Hypoxia regulates CD44 and its variant isoforms through HIF-1alpha in triple negative breast cancer. PLoS ONE 2012, 7, e44078. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, A.; Boidot, R.; Végran, F. Alternative Splicing in Cancer and Immune Cells. Cancers 2022, 14, 1726. https://doi.org/10.3390/cancers14071726
Bernard A, Boidot R, Végran F. Alternative Splicing in Cancer and Immune Cells. Cancers. 2022; 14(7):1726. https://doi.org/10.3390/cancers14071726
Chicago/Turabian StyleBernard, Antoine, Romain Boidot, and Frédérique Végran. 2022. "Alternative Splicing in Cancer and Immune Cells" Cancers 14, no. 7: 1726. https://doi.org/10.3390/cancers14071726
APA StyleBernard, A., Boidot, R., & Végran, F. (2022). Alternative Splicing in Cancer and Immune Cells. Cancers, 14(7), 1726. https://doi.org/10.3390/cancers14071726