
pncCCND1_B Engages an Inhibitory Protein Network to Downregulate CCND1 Expression upon DNA Damage


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Drug Treatment
2.2. Transfection Experiments
2.3. SDS–PAGE and Western Blot Analyses
2.4. Reverse Transcription and Real-Time Quantitative PCR Analyses
2.5. Immunoprecipitation
2.6. Chromatin Immunoprecipitation
2.7. In Vivo Crosslinking and Immunoprecipitation (CLIP)
2.8. Immunofluorescence Analysis
2.9. DNA: RNA Hybrid Immunoprecipitation (DRIP) and Chromatin-Associated RNA Immunoprecipitation (CARIP)
2.10. Statistical Analysis
3. Results
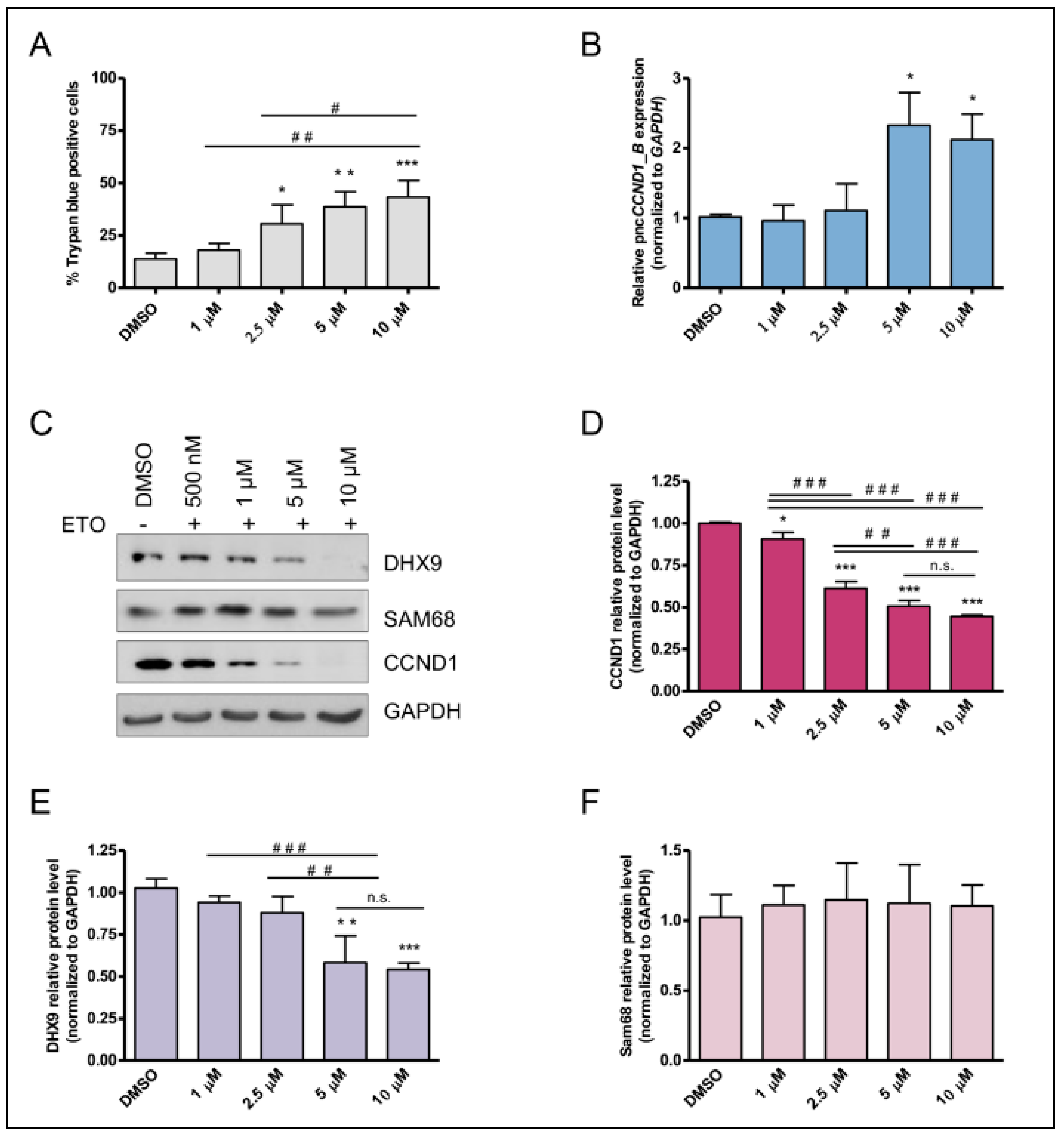
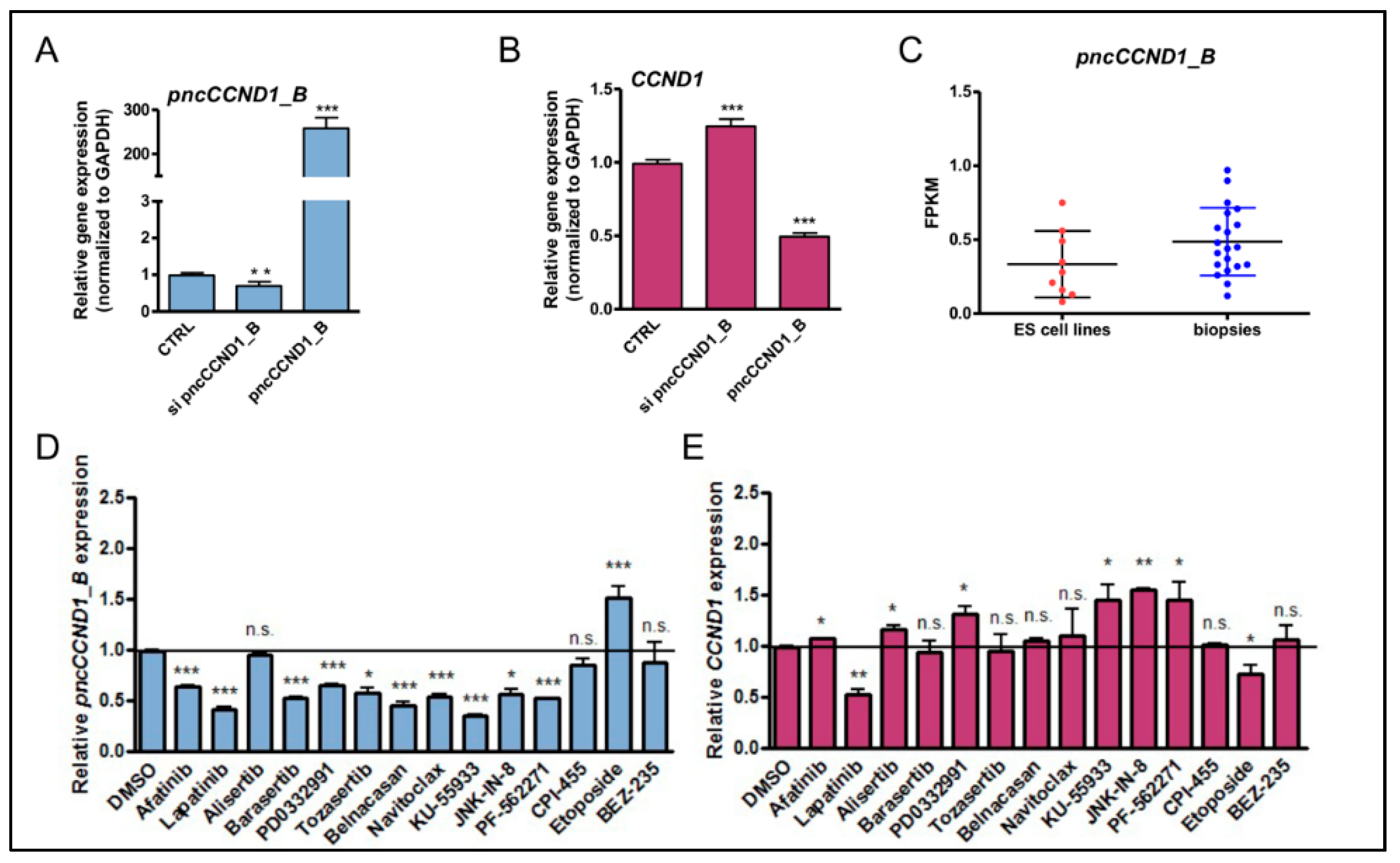
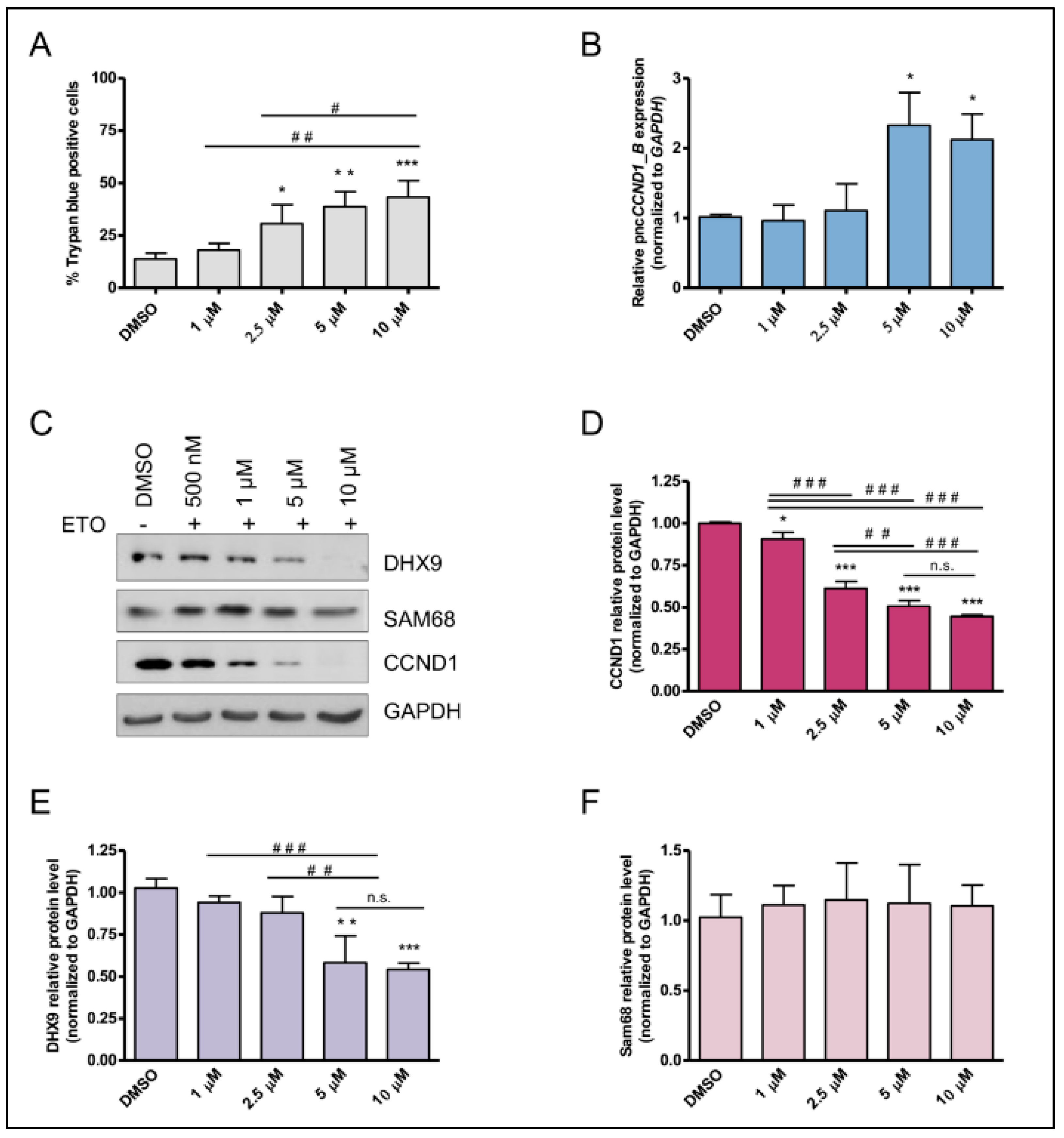
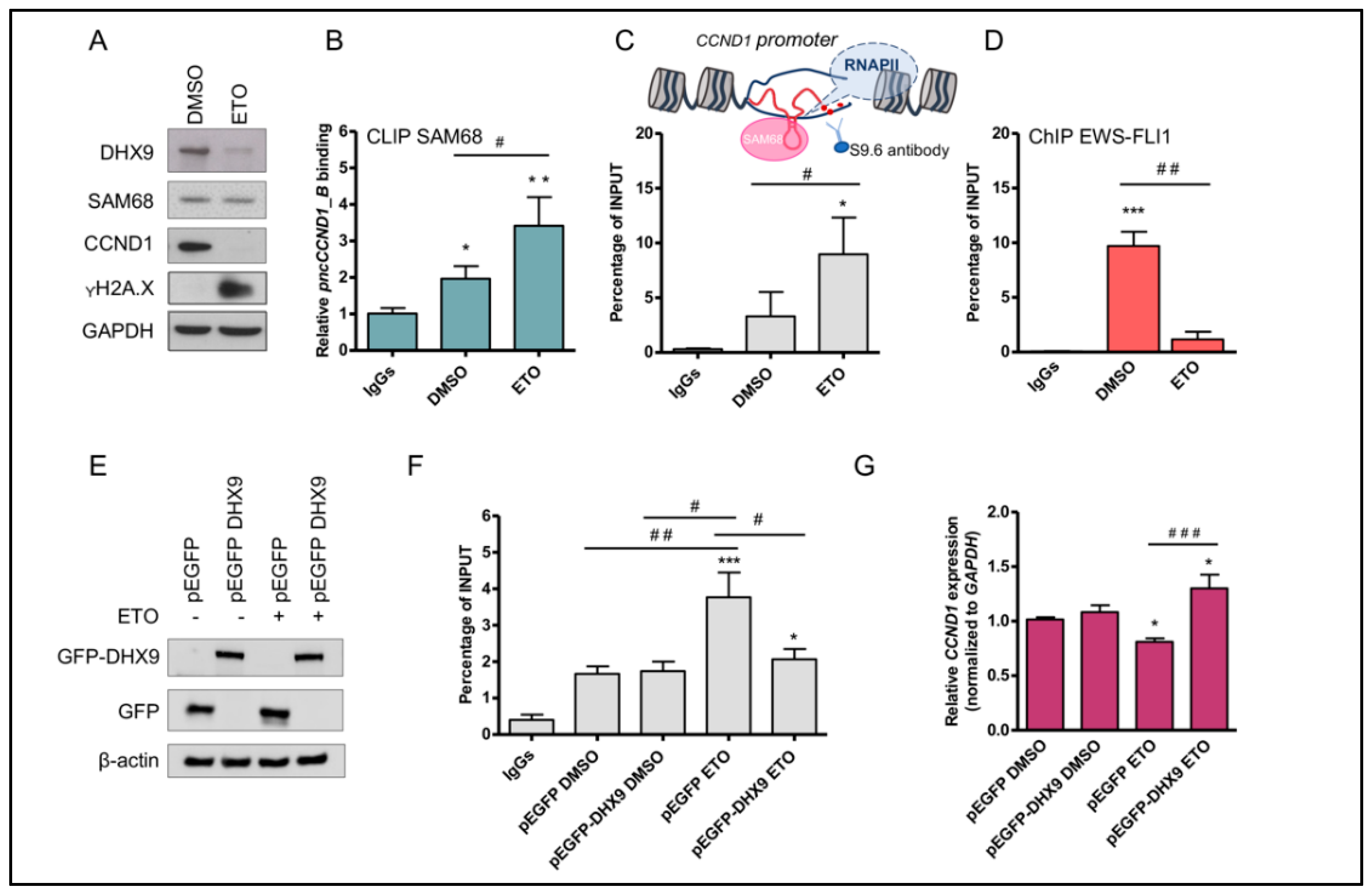
3.1. Pan-Inhibitor Screening Unveils Etoposide as a Modulator of pncCCND1_B Expression
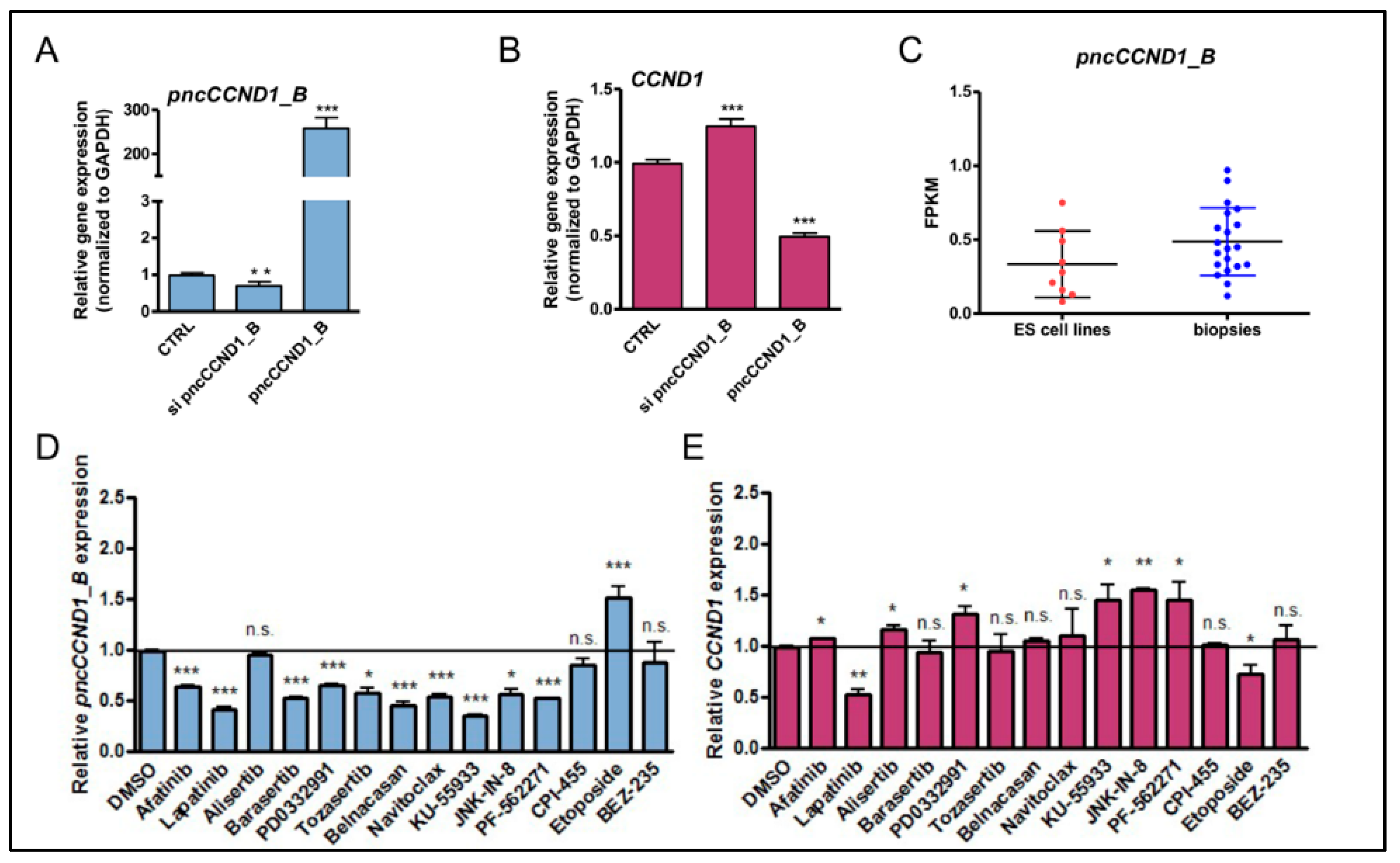
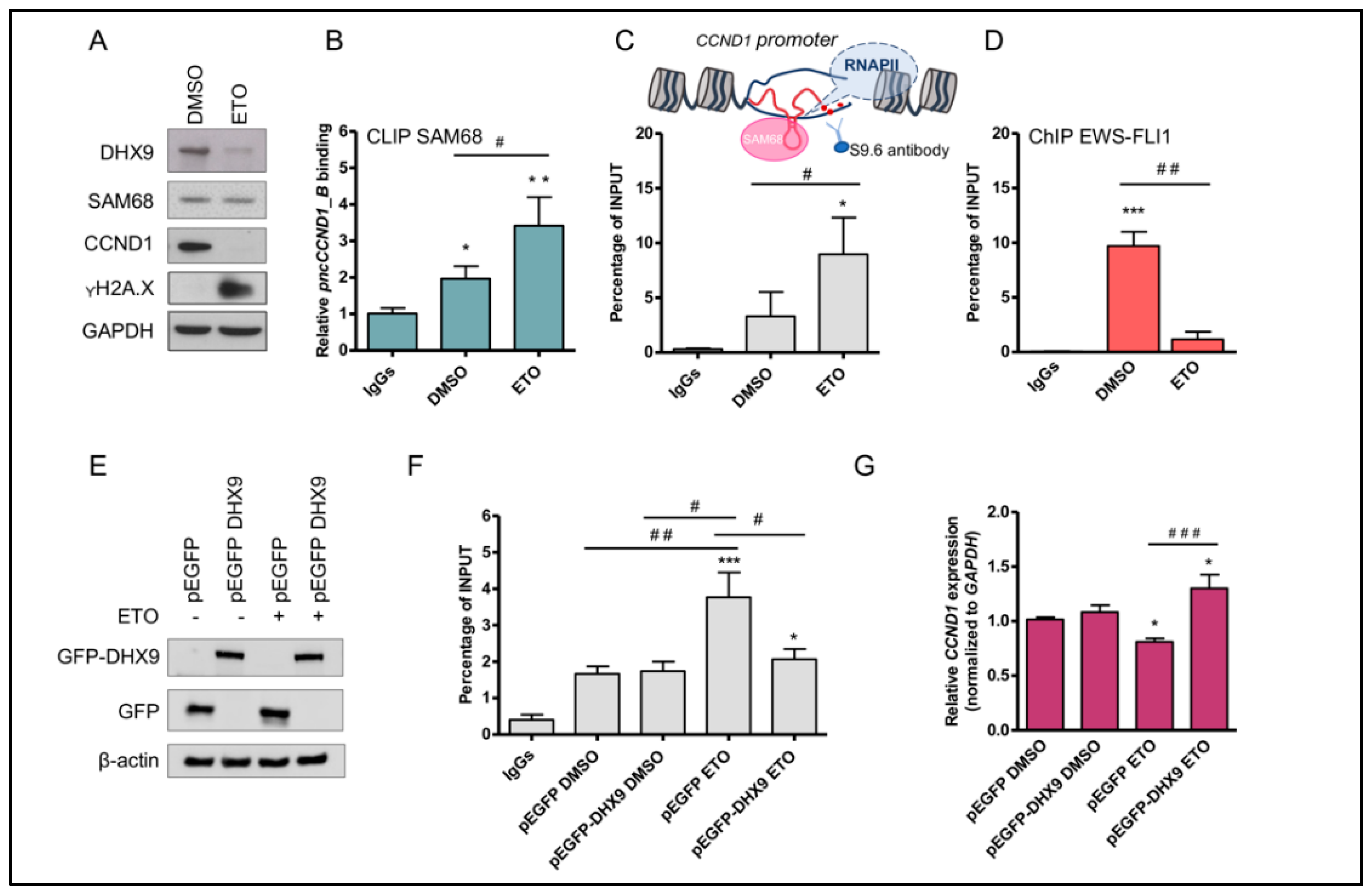
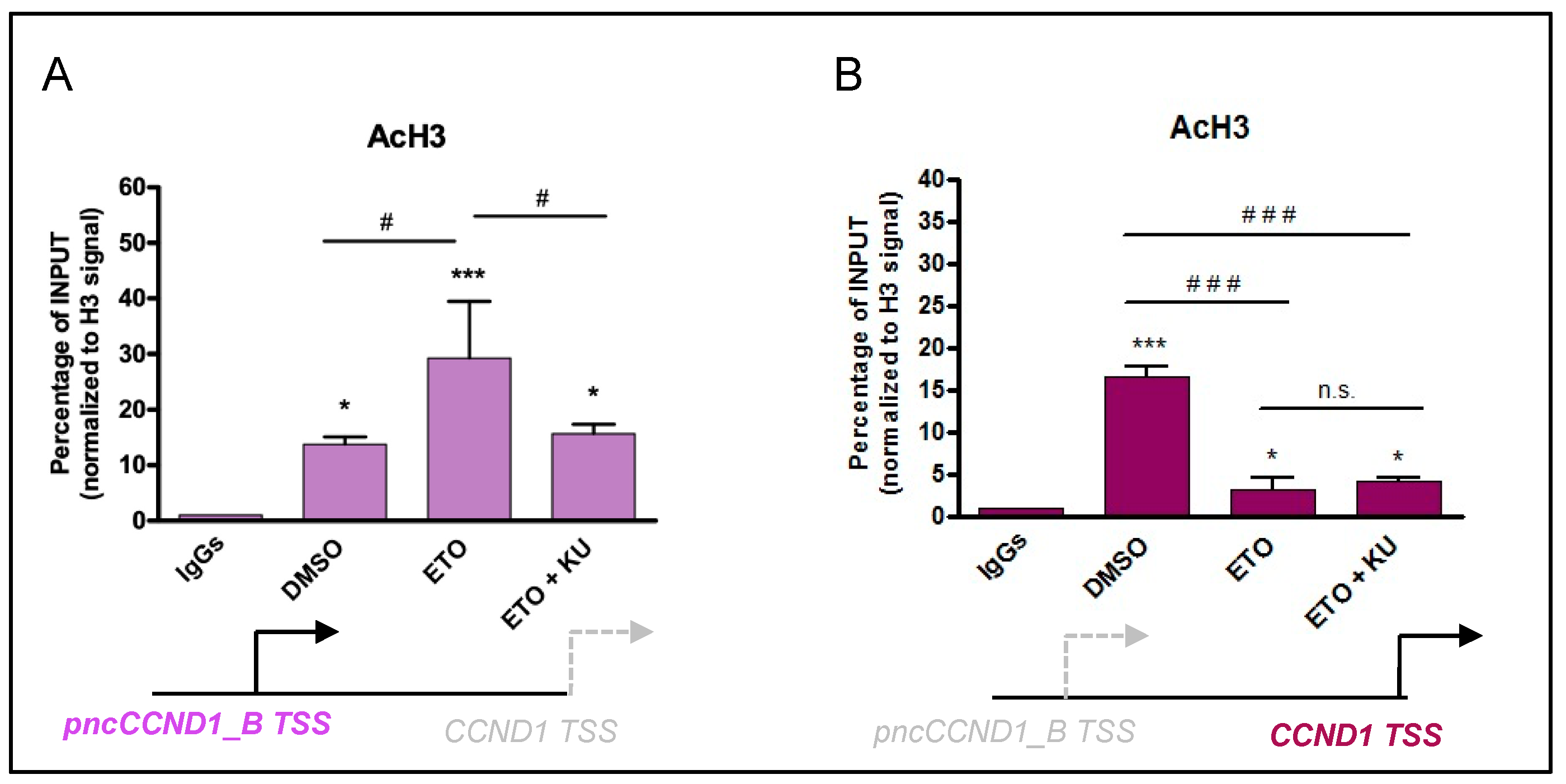
3.2. Etoposide Induces DNA:RNA Hybrids at the CCND1 Promoter
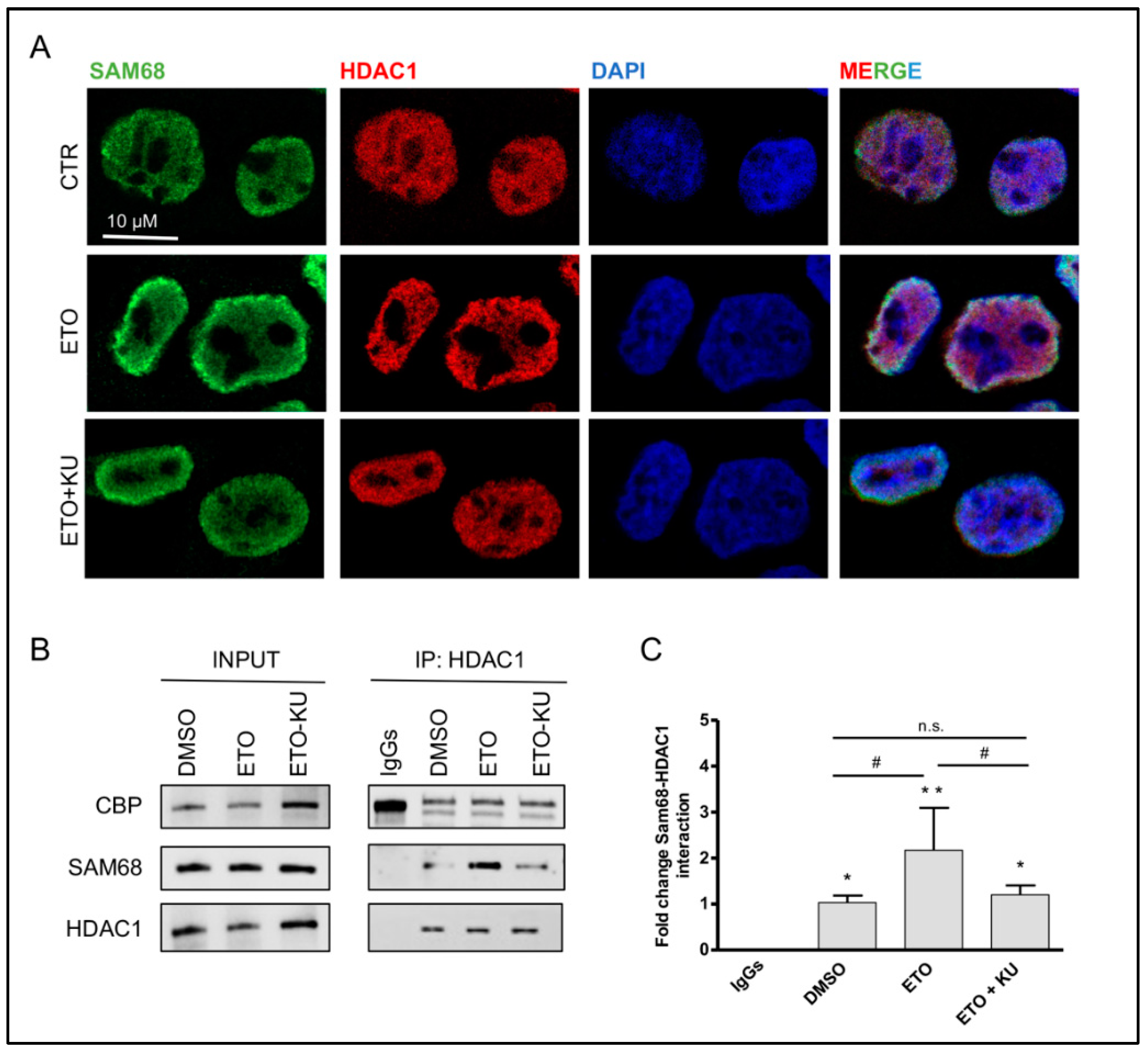
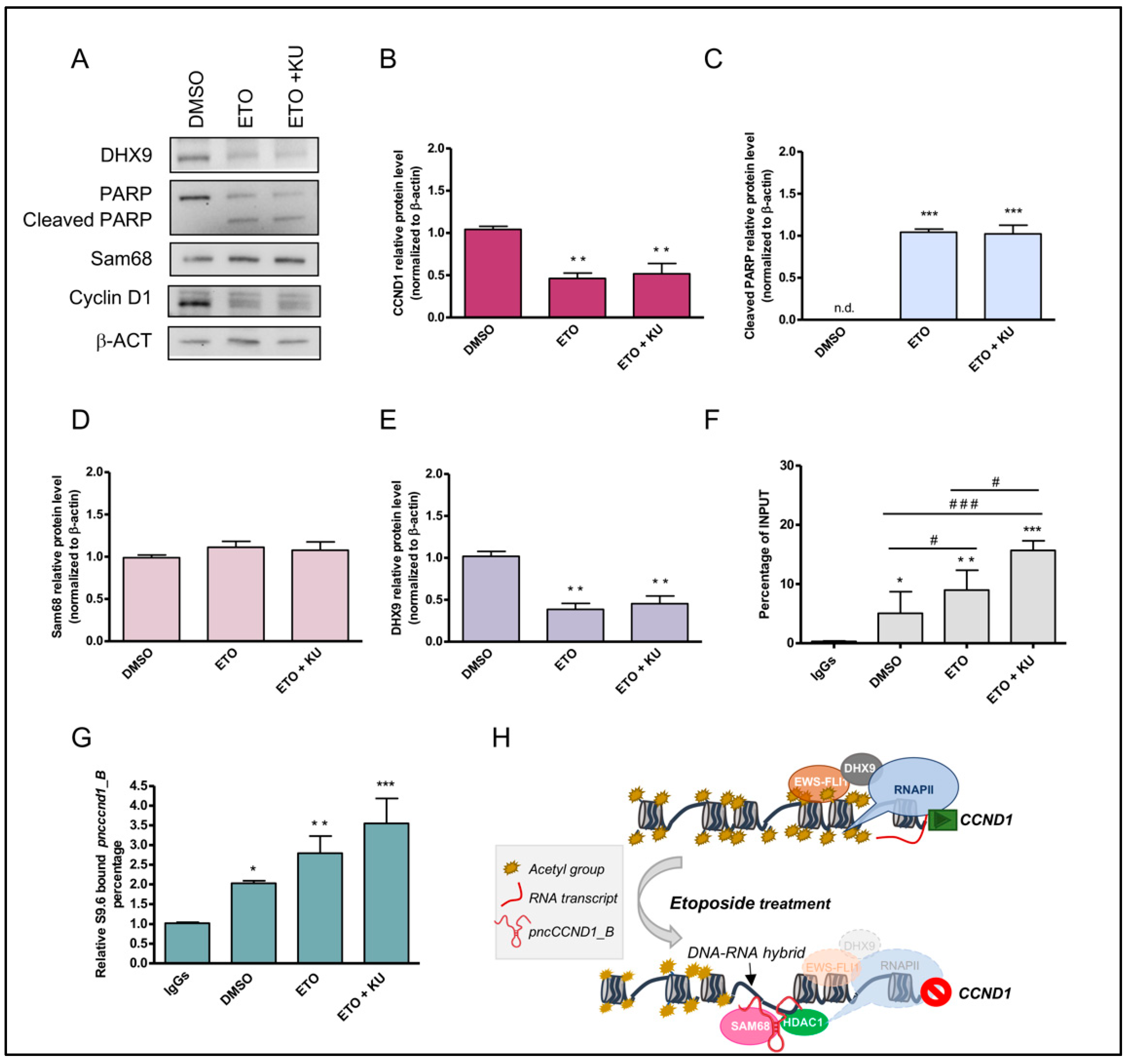
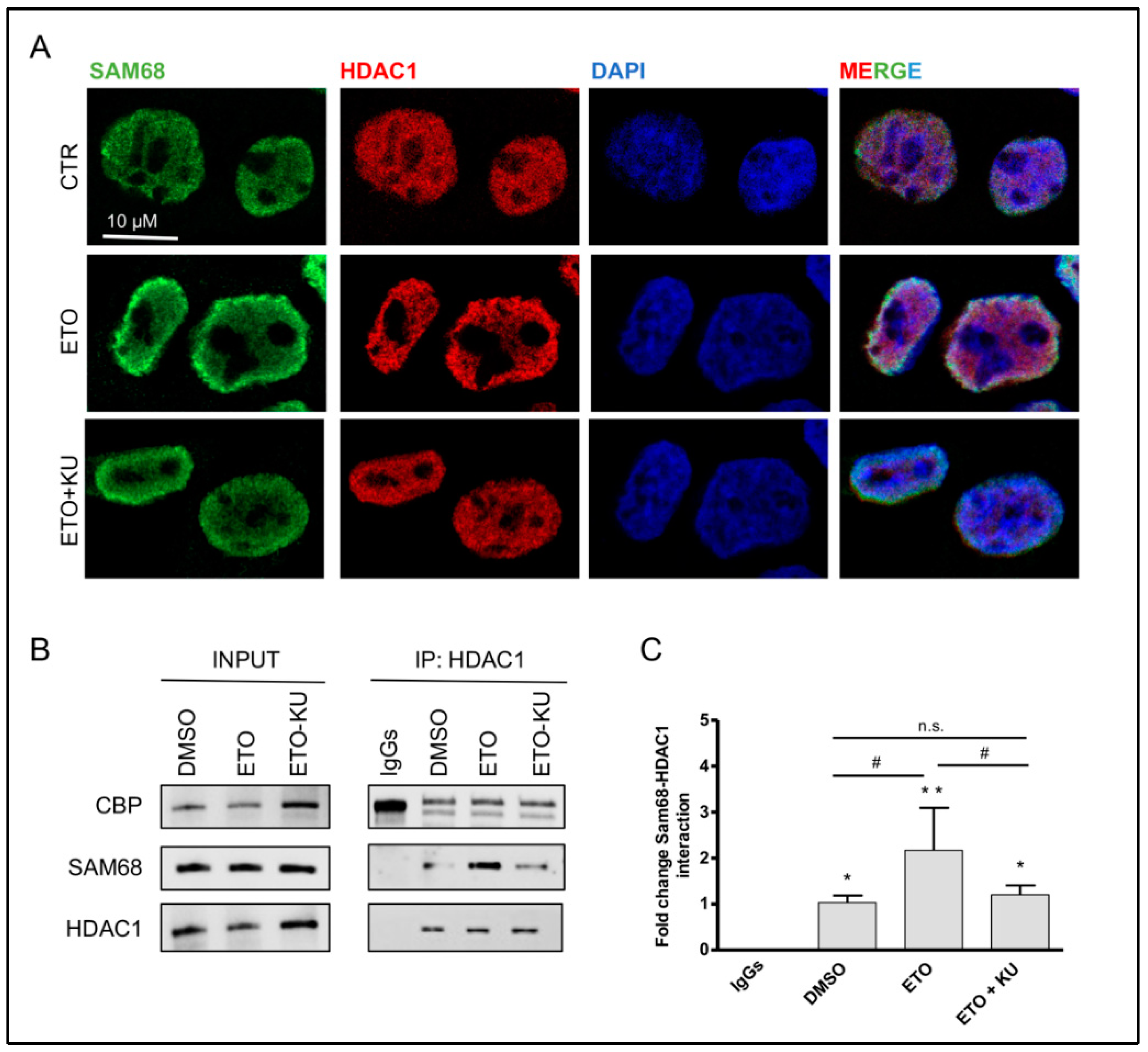
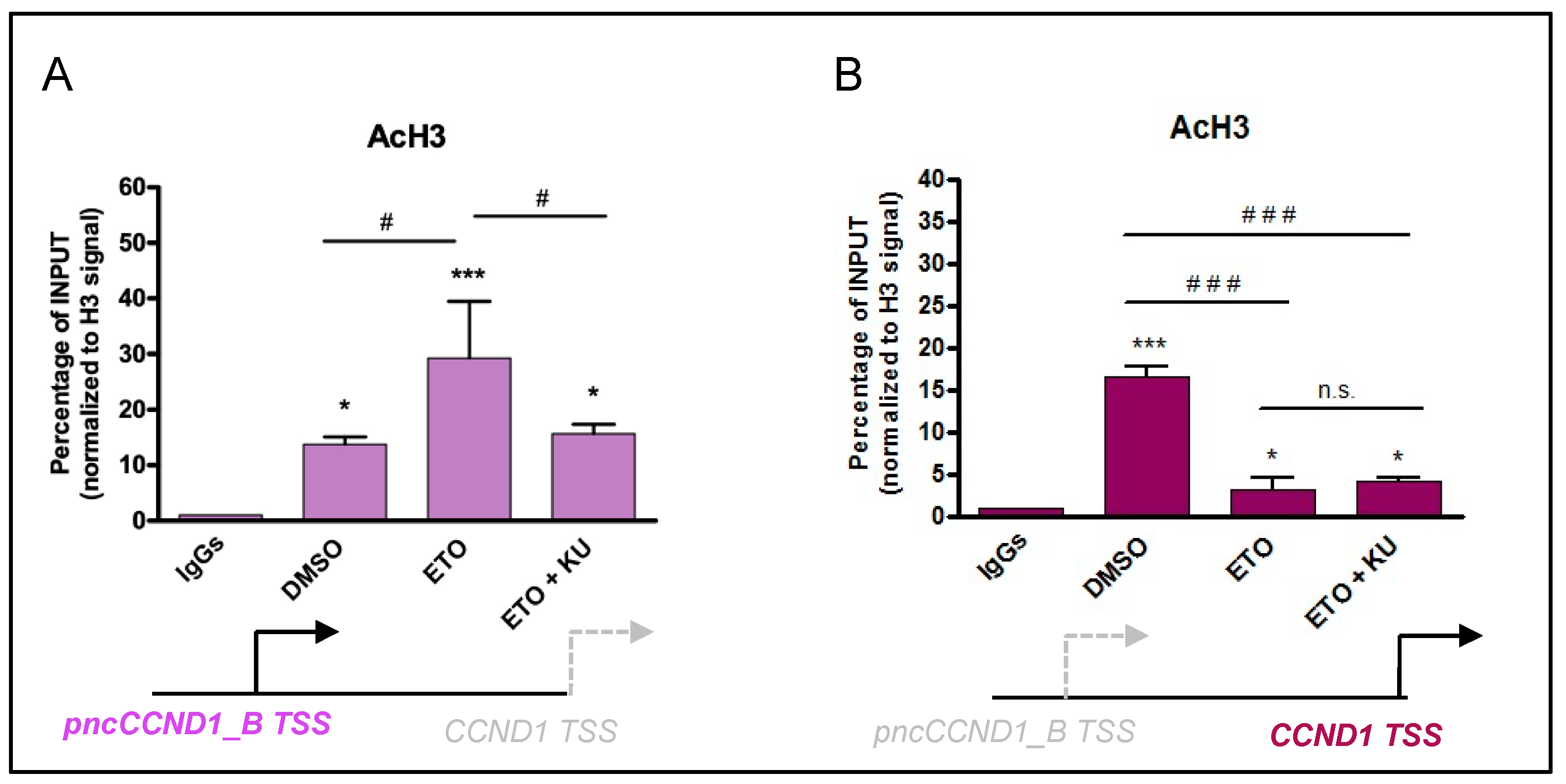
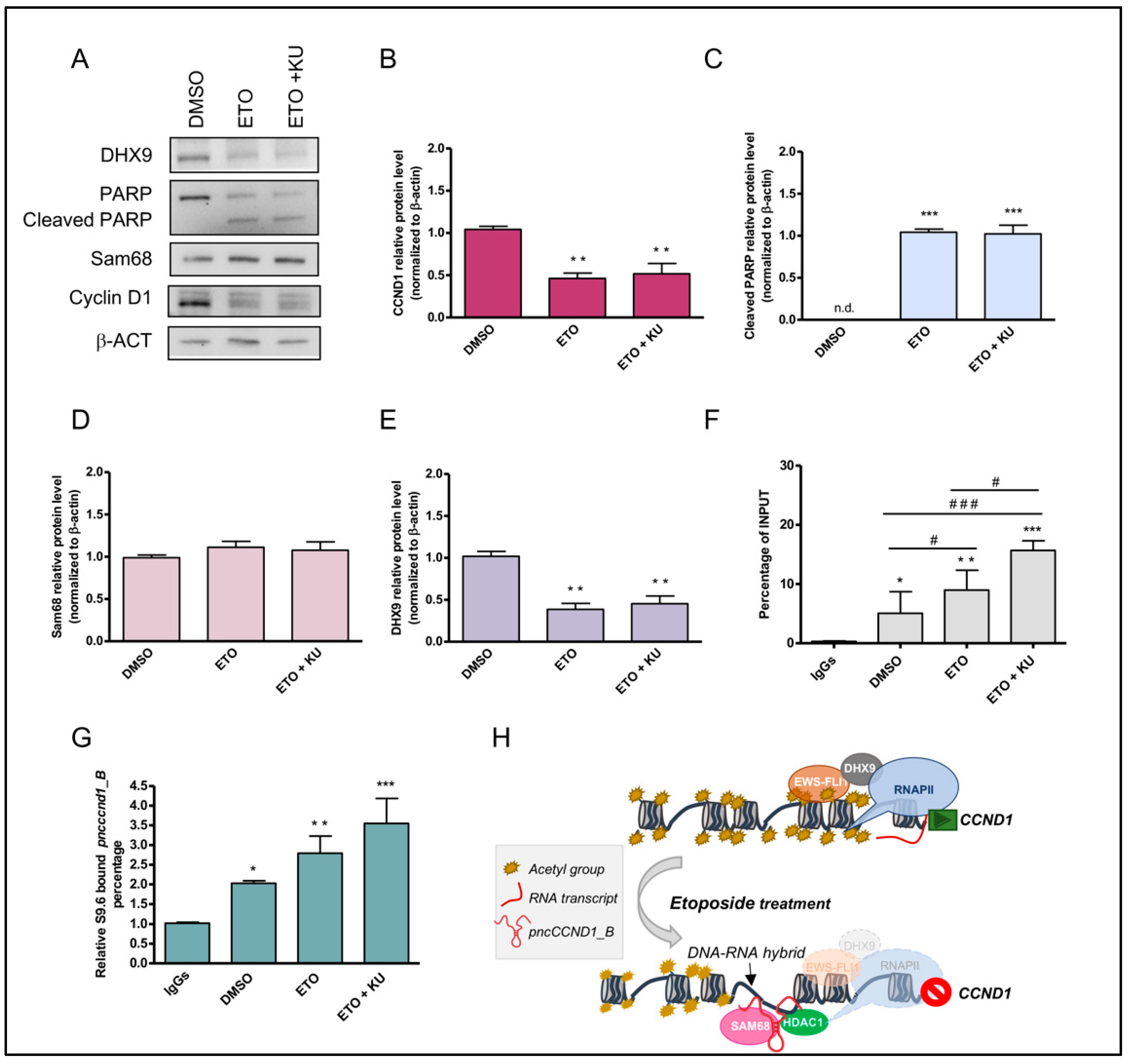
3.3. Etoposide Effect on SAM68-pncCCND1_B Complex Is Abolished by KU 55933 Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellini, L.; Frezza, V.; Paronetto, M.P. Dissecting the transcriptional regulatory networks of promoter-associated noncoding RNAs in development and cancer. J. Exp. Clin. Cancer Res. 2020, 39, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercatelli, N.; Palombo, R.; Paronetto, M.P. Emerging Contribution of PancRNAs in Cancer. Cancers 2020, 12, 2035. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef]
- Yoneda, R.; Suzuki, S.; Mashima, T.; Kondo, K.; Nagata, T.; Katahira, M.; Kurokawa, R. The binding specificity of Translocated in LipoSarcoma/FUsed in Sarcoma with lncRNA transcribed from the promoter region of cyclin D1. Cell Biosci. 2016, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Yoneda, R.; Ueda, N.; Kurokawa, R. Arginine methylation of translocated in liposarcoma (TLS) inhibits its binding to long noncoding RNA, abrogating TLS-mediated repression of CBP/p300 activity. J. Biol. Chem. 2018, 293, 10937–10948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palombo, R.; Frisone, P.; Fidaleo, M.; Mercatelli, N.; Sette, C.; Paronetto, M.P. The Promoter-Associated Noncoding RNA. Cancer Res. 2019, 79, 3570–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisone, P.; Pradella, D.; Di Matteo, A.; Belloni, E.; Ghigna, C.; Paronetto, M.P. SAM68: Signal Transduction and RNA Metabolism in Human Cancer. Biomed Res. Int. 2015, 2015, 528954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielli, P.; Busa, R.; Paola Paronetto, M.; Sette, C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr.-Relat. Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batsché, E.; Yaniv, M.; Muchardt, C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat. Struct. Mol. Biol. 2006, 13, 22–29. [Google Scholar] [CrossRef]
- Codony-Servat, C.; Codony-Servat, J.; Karachaliou, N.; Molina, M.A.; Chaib, I.; Ramirez, J.L.; de Los Llanos Gil, M.; Solca, F.; Bivona, T.G.; Rosell, R. Activation of signal transducer and activator of transcription 3 (STAT3) signaling in EGFR mutant non-small-cell lung cancer (NSCLC). Oncotarget 2017, 8, 47305–47316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.R.; Soneru, C.; Liu, J.; Grushko, T.A.; Hardeman, A.; Olopade, O.I.; Baum, A.; Solca, F.; Cohen, E.E. Afatinib efficacy against squamous cell carcinoma of the head and neck cell lines in vitro and in vivo. Target. Oncol. 2015, 10, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorlick, R.; Kolb, E.A.; Houghton, P.J.; Morton, C.L.; Phelps, D.; Schaiquevich, P.; Stewart, C.; Keir, S.T.; Lock, R.; Carol, H.; et al. Initial testing (stage 1) of lapatinib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2009, 53, 594–598. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Wu, J.; et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr. Blood Cancer 2010, 55, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Polley, E.; Kunkel, M.; Evans, D.; Silvers, T.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Selby, M.; et al. Sarcoma Cell Line Screen of Oncology Drugs and Investigational Agents Identifies Patterns Associated with Gene and microRNA Expression. Mol. Cancer Ther. 2015, 14, 2452–2462. [Google Scholar] [CrossRef] [Green Version]
- Guenther, L.M.; Dharia, N.V.; Ross, L.; Conway, A.; Robichaud, A.L.; Catlett, J.L.; Wechsler, C.S.; Frank, E.S.; Goodale, A.; Church, A.J.; et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 1343–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.E.; Rix, U.; Lissat, A.; Stukalov, A.; Müllner, M.K.; Bennett, K.L.; Colinge, J.; Nijman, S.M.; Kubicek, S.; Kovar, H.; et al. An integrated chemical biology approach identifies specific vulnerability of Ewing’s sarcoma to combined inhibition of Aurora kinases A and B. Mol. Cancer Ther. 2011, 10, 1846–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, X.; Wang, Q.; Yang, X. Roles of the pyroptosis signaling pathway in a sepsis-associated encephalopathy cell model. J. Int. Med. Res. 2020, 48, 300060520949767. [Google Scholar] [CrossRef]
- Heisey, D.A.R.; Lochmann, T.L.; Floros, K.V.; Coon, C.M.; Powell, K.M.; Jacob, S.; Calbert, M.L.; Ghotra, M.S.; Stein, G.T.; Maves, Y.K.; et al. The Ewing Family of Tumors Relies on BCL-2 and BCL-X. Clin. Cancer Res. 2019, 25, 1664–1675. [Google Scholar] [CrossRef] [Green Version]
- Marx, C.; Schaarschmidt, M.U.; Kirkpatrick, J.; Marx-Blümel, L.; Halilovic, M.; Westermann, M.; Hoelzer, D.; Meyer, F.B.; Geng, Y.; Buder, K.; et al. Cooperative treatment effectiveness of ATR and HSP90 inhibition in Ewing’s sarcoma cells. Cell. Biosci. 2021, 11, 57. [Google Scholar] [CrossRef]
- Satterfield, L.; Shuck, R.; Kurenbekova, L.; Allen-Rhoades, W.; Edwards, D.; Huang, S.; Rajapakshe, K.; Coarfa, C.; Donehower, L.A.; Yustein, J.T. miR-130b directly targets ARHGAP1 to drive activation of a metastatic CDC42-PAK1-AP1 positive feedback loop in Ewing sarcoma. Int. J. Cancer 2017, 141, 2062–2075. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Hwang, E.E.; Guha, R.; O’Neill, A.F.; Melong, N.; Veinotte, C.J.; Conway Saur, A.; Wuerthele, K.; Shen, M.; McKnight, C.; et al. High-throughput Chemical Screening Identifies Focal Adhesion Kinase and Aurora Kinase B Inhibition as a Synergistic Treatment Combination in Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 4552–4566. [Google Scholar] [CrossRef] [Green Version]
- McCann, T.S.; Parrish, J.K.; Hsieh, J.; Sechler, M.; Sobral, L.M.; Self, C.; Jones, K.L.; Goodspeed, A.; Costello, J.C.; Jedlicka, P. KDM5A and PHF2 positively control expression of pro-metastatic genes repressed by EWS/Fli1, and promote growth and metastatic properties in Ewing sarcoma. Oncotarget 2020, 11, 3818–3831. [Google Scholar] [CrossRef]
- May, W.A.; Grigoryan, R.S.; Keshelava, N.; Cabral, D.J.; Christensen, L.L.; Jenabi, J.; Ji, L.; Triche, T.J.; Lawlor, E.R.; Reynolds, C.P. Characterization and drug resistance patterns of Ewing’s sarcoma family tumor cell lines. PLoS ONE 2013, 8, e80060. [Google Scholar] [CrossRef]
- Fidaleo, M.; Svetoni, F.; Volpe, E.; Miñana, B.; Caporossi, D.; Paronetto, M.P. Genotoxic stress inhibits Ewing sarcoma cell growth by modulating alternative pre-mRNA processing of the RNA helicase DHX9. Oncotarget 2015, 6, 31740–31757. [Google Scholar] [CrossRef] [Green Version]
- Passacantilli, I.; Frisone, P.; De Paola, E.; Fidaleo, M.; Paronetto, M.P. hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 2017, 45, 12270–12284. [Google Scholar] [CrossRef]
- Paronetto, M.P.; Bernardis, I.; Volpe, E.; Bechara, E.; Sebestyén, E.; Eyras, E.; Valcárcel, J. Regulation of FAS exon definition and apoptosis by the Ewing sarcoma protein. Cell Rep. 2014, 7, 1211–1226. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Minana, B.; Valcarcel, J. The Ewing Sarcoma Protein Regulates DNA Damage-Induced Alternative Splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar] [CrossRef] [Green Version]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.J.; Hamperl, S.; Swigut, T.; Cimprich, K.A. qDRIP: A method to quantitatively assess RNA-DNA hybrid formation genome-wide. Nucleic Acids Res. 2020, 48, e84. [Google Scholar] [CrossRef]
- Kidder, B.L. CARIP-Seq and ChIP-Seq: Methods to Identify Chromatin-Associated RNAs and Protein-DNA Interactions in Embryonic Stem Cells. J. Vis. Exp. 2018, 135, e57481. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. (Berl.) 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Grier, H.E.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 2003, 348, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Palombo, R.; Verdile, V.; Paronetto, M.P. Poison-Exon Inclusion in DHX9 Reduces Its Expression and Sensitizes Ewing Sarcoma Cells to Chemotherapeutic Treatment. Cells 2020, 9, 328. [Google Scholar] [CrossRef] [Green Version]
- Ip, J.Y.; Schmidt, D.; Pan, Q.; Ramani, A.K.; Fraser, A.G.; Odom, D.T.; Blencowe, B.J. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res. 2011, 21, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, A.; Biamonti, G. Cellular response to etoposide treatment. Cancer Lett. 2007, 252, 9–18. [Google Scholar] [CrossRef]
- Krietsch, J.; Caron, M.C.; Gagné, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef] [Green Version]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Busà, R.; Geremia, R.; Sette, C. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 2010, 38, 3005–3018. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fu, K.; Hodgson, A.; Wier, E.M.; Wen, M.G.; Kamenyeva, O.; Xia, X.; Koo, L.Y.; Wan, F. Sam68 Is Required for DNA Damage Responses via Regulating Poly(ADP-ribosyl)ation. PLoS Biol. 2016, 14, e1002543. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Venables, J.P.; Elliott, D.J.; Geremia, R.; Rossi, P.; Sette, C. Tr-kit promotes the formation of a multimolecular complex composed by Fyn, PLCgamma1 and Sam68. Oncogene 2003, 22, 8707–8715. [Google Scholar] [CrossRef] [Green Version]
- Milon, B.C.; Cheng, H.; Tselebrovsky, M.V.; Lavrov, S.A.; Nenasheva, V.V.; Mikhaleva, E.A.; Shevelyov, Y.Y.; Nurminsky, D.I. Role of histone deacetylases in gene regulation at nuclear lamina. PLoS ONE 2012, 7, e49692. [Google Scholar] [CrossRef]
- Toomey, E.C.; Schiffman, J.D.; Lessnick, S.L. Recent advances in the molecular pathogenesis of Ewing’s sarcoma. Oncogene 2010, 29, 4504–4516. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Messina, V.; Bianchi, E.; Barchi, M.; Vogel, G.; Moretti, C.; Palombi, F.; Stefanini, M.; Geremia, R.; Richard, S.; et al. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. J. Cell Biol. 2009, 185, 235–249. [Google Scholar] [CrossRef] [Green Version]
- De Paola, E.; Forcina, L.; Pelosi, L.; Pisu, S.; La Rosa, P.; Cesari, E.; Nicoletti, C.; Madaro, L.; Mercatelli, N.; Biamonte, F.; et al. Sam68 splicing regulation contributes to motor unit establishment in the postnatal skeletal muscle. Life Sci. Alliance 2020, 3, e201900637. [Google Scholar] [CrossRef]
- Lukong, K.E.; Richard, S. Sam68, the KH domain-containing superSTAR. Biochim. Biophys. Acta 2003, 1653, 73–86. [Google Scholar] [CrossRef]
- Gagné, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976. [Google Scholar] [CrossRef] [Green Version]
- Busà, R.; Paronetto, M.P.; Farini, D.; Pierantozzi, E.; Botti, F.; Angelini, D.F.; Attisani, F.; Vespasiani, G.; Sette, C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene 2007, 26, 4372–4382. [Google Scholar] [CrossRef] [Green Version]
- Zink, D.; Amaral, M.D.; Englmann, A.; Lang, S.; Clarke, L.A.; Rudolph, C.; Alt, F.; Luther, K.; Braz, C.; Sadoni, N.; et al. Transcription-dependent spatial arrangements of CFTR and adjacent genes in human cell nuclei. J. Cell Biol. 2004, 166, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Zuleger, N.; Robson, M.I.; Schirmer, E.C. The nuclear envelope as a chromatin organizer. Nucleus 2011, 2, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Cremer, T.; Cremer, C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef]
- Fidaleo, M.; De Paola, E.; Paronetto, M.P. The RNA helicase A in malignant transformation. Oncotarget 2016, 7, 28711–28723. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Roth, A.; Yu, M.; Morris, R.; Bersani, F.; Rivera, M.N.; Lu, J.; Shioda, T.; Vasudevan, S.; Ramaswamy, S.; et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013, 27, 2543–2548. [Google Scholar] [CrossRef] [Green Version]
- Myöhänen, S.; Baylin, S.B. Sequence-specific DNA binding activity of RNA helicase A to the p16INK4a promoter. J. Biol. Chem. 2001, 276, 1634–1642. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Uchida, C.; Anderson, S.F.; Lee, C.G.; Hurwitz, J.; Parvin, J.D.; Montminy, M. RNA helicase A mediates association of CBP with RNA polymerase II. Cell 1997, 90, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Erkizan, H.V.; Schneider, J.A.; Sajwan, K.; Graham, G.T.; Griffin, B.; Chasovskikh, S.; Youbi, S.E.; Kallarakal, A.; Chruszcz, M.; Padmanabhan, R.; et al. RNA helicase A activity is inhibited by oncogenic transcription factor EWS-FLI1. Nucleic Acids Res. 2015, 43, 1069–1080. [Google Scholar] [CrossRef] [Green Version]
- Toretsky, J.A.; Erkizan, V.; Levenson, A.; Abaan, O.D.; Parvin, J.D.; Cripe, T.P.; Rice, A.M.; Lee, S.B.; Uren, A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006, 66, 5574–5581. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; Grosse, F. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair. (Amst.) 2011, 10, 654–665. [Google Scholar] [CrossRef]
- Boros-Oláh, B.; Dobos, N.; Hornyák, L.; Szabó, Z.; Karányi, Z.; Halmos, G.; Roszik, J.; Székvölgyi, L. Drugging the R-loop interactome: RNA-DNA hybrid binding proteins as targets for cancer therapy. DNA Repair. (Amst.) 2019, 84, 102642. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Concentration | Biological Activity | References |
|---|---|---|---|
| AFATINIB | 1 μM | Irreversible inhibitor of EGFR, HER2 and HER4 | [11,12] |
| LAPATINIB | 5 μM | Inhibitor of EGFR and HER2 tyrosine kinase domain | [13] |
| ALISERTIB | 100 nM | Selective inhibitor of the Aurora-A kinase | [14] |
| BARASERTIB | 20 nM | Inhibitor of Aurora-B and Aurora-A kinases | [15] |
| PD0332991 | 100 nM | CDK4/6 inhibitor | [16] |
| TOZASERTIB | 100 nM | Pan-Aurora inhibitor | [17] |
| BELNACASAN | 10 μM | Selective caspase-1 inhibitor | [18] |
| NAVITOCLAX | 1 μM | High-affinity inhibitor of BCL-2 | [19] |
| KU 55933 | 10 μM | ATM inhibitor | [20] |
| JNK-IN-8 | 1 μM | Irreversible JNK inhibitor for JNK1, JNK2 and JNK3 | [21] |
| PF-562271 | 1 μM | ATP-competitive, reversible inhibitor of FAK | [22] |
| CPI-455 | 10 μM | TNF alpha inhibitor | [23] |
| ETOPOSIDE | 5 μM | Topoisomerase II inhibitor | [24,25] |
| BEZ-235 | 100 nM | Dual PI3K/mTOR inhibition | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palombo, R.; Paronetto, M.P. pncCCND1_B Engages an Inhibitory Protein Network to Downregulate CCND1 Expression upon DNA Damage. Cancers 2022, 14, 1537. https://doi.org/10.3390/cancers14061537
Palombo R, Paronetto MP. pncCCND1_B Engages an Inhibitory Protein Network to Downregulate CCND1 Expression upon DNA Damage. Cancers. 2022; 14(6):1537. https://doi.org/10.3390/cancers14061537
Chicago/Turabian StylePalombo, Ramona, and Maria Paola Paronetto. 2022. "pncCCND1_B Engages an Inhibitory Protein Network to Downregulate CCND1 Expression upon DNA Damage" Cancers 14, no. 6: 1537. https://doi.org/10.3390/cancers14061537
APA StylePalombo, R., & Paronetto, M. P. (2022). pncCCND1_B Engages an Inhibitory Protein Network to Downregulate CCND1 Expression upon DNA Damage. Cancers, 14(6), 1537. https://doi.org/10.3390/cancers14061537