
TGF-β/SMAD Pathway Is Modulated by miR-26b-5p: Another Piece in the Puzzle of Chronic Lymphocytic Leukemia Progression

 , , , , , , , , ,
, , , , , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. CLL Cohort
2.2. microRNAome Analysis
2.3. mRNA Expression Analysis
2.4. Confocal Microscopy and Quantification of Nuclear Staining
2.5. Measurements of Surface and Intracellular Antigens by Flow Cytometry
2.6. miRNA Transfections and Sorting
2.7. Statistical Analysis
2.8. NGS Data Availability
3. Results
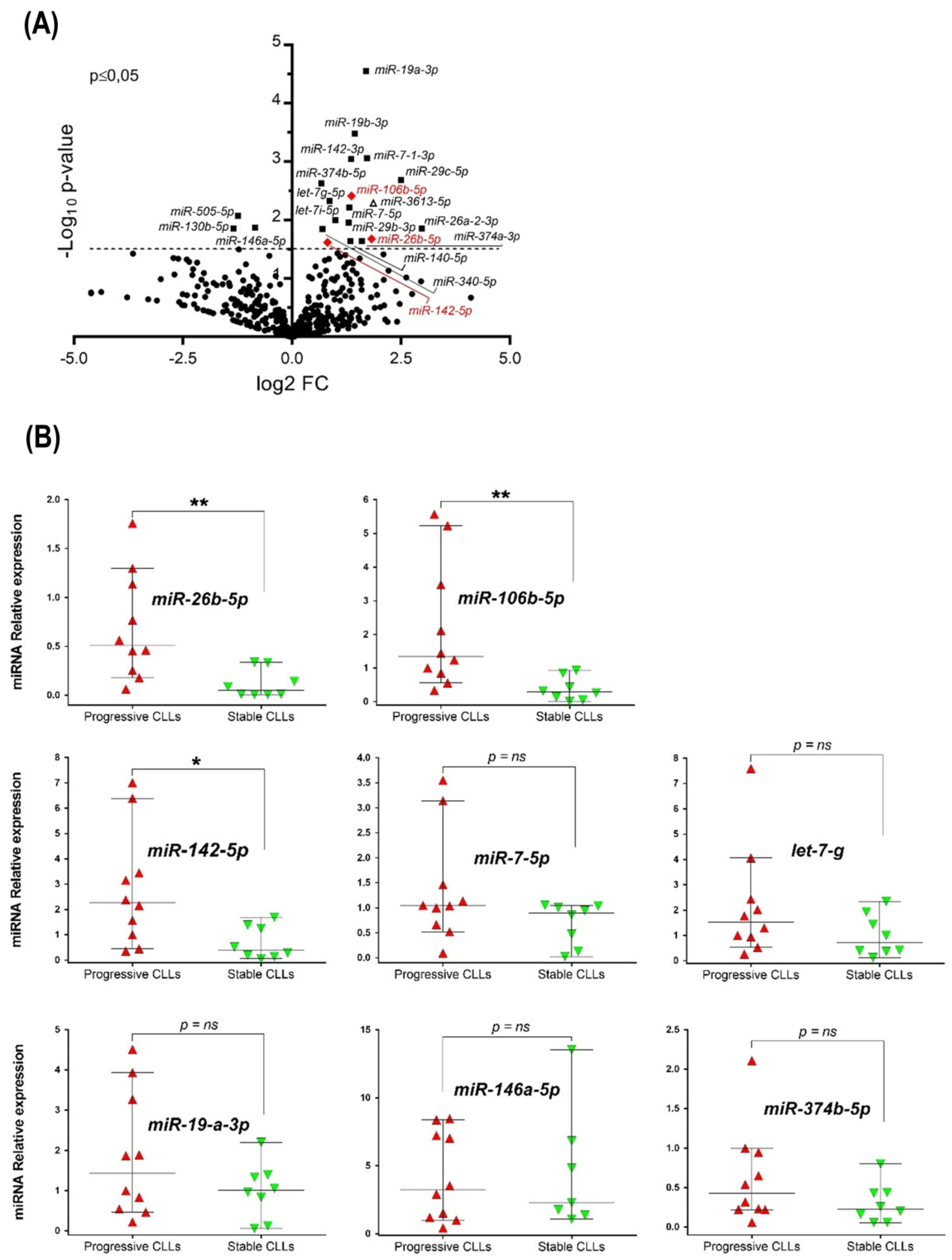
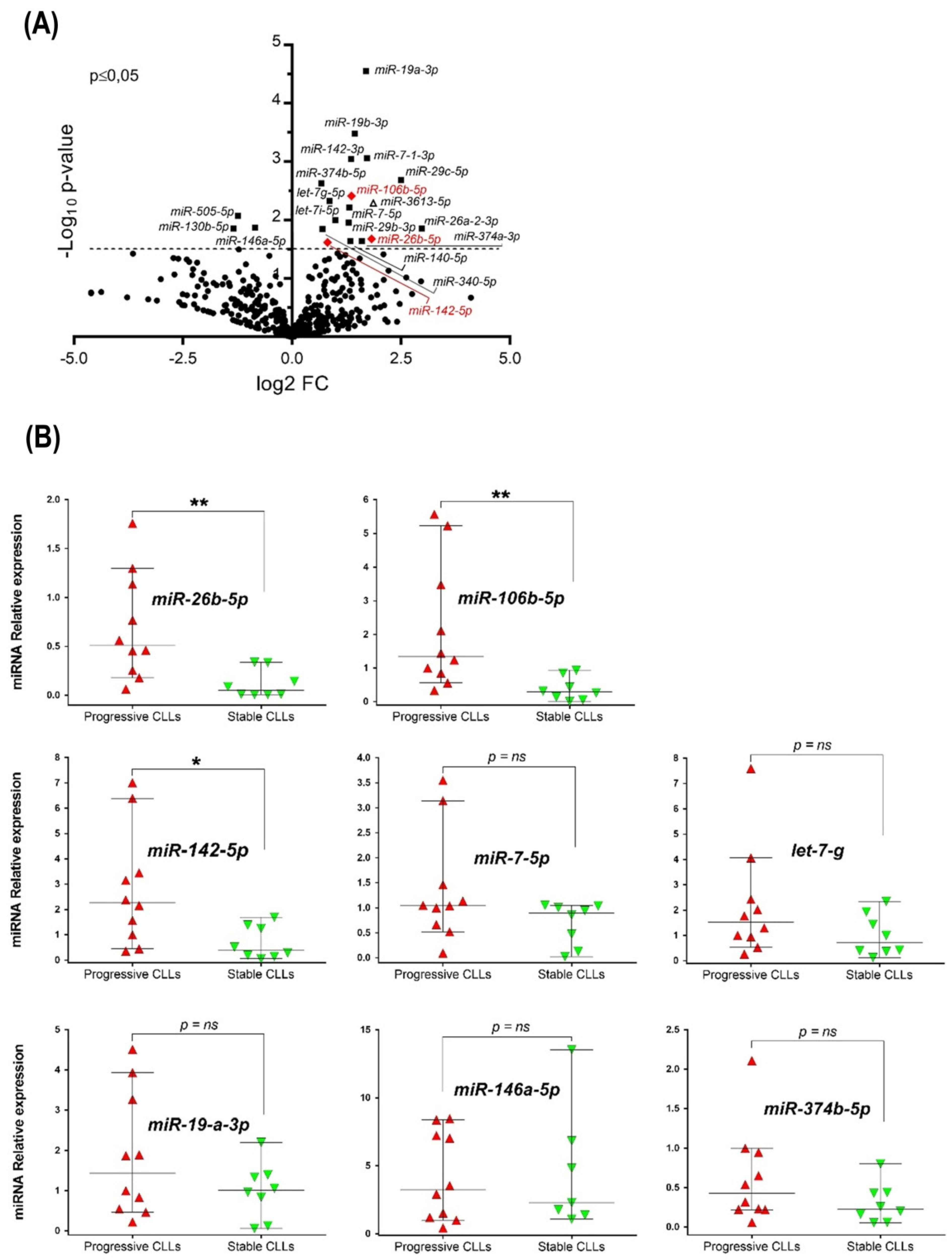
3.1. miRNAome Profile of U-CLL with Different Clinical Outcomes and Validation of Up-Regulated miRNAs in the Clinically Progressive Subgroup
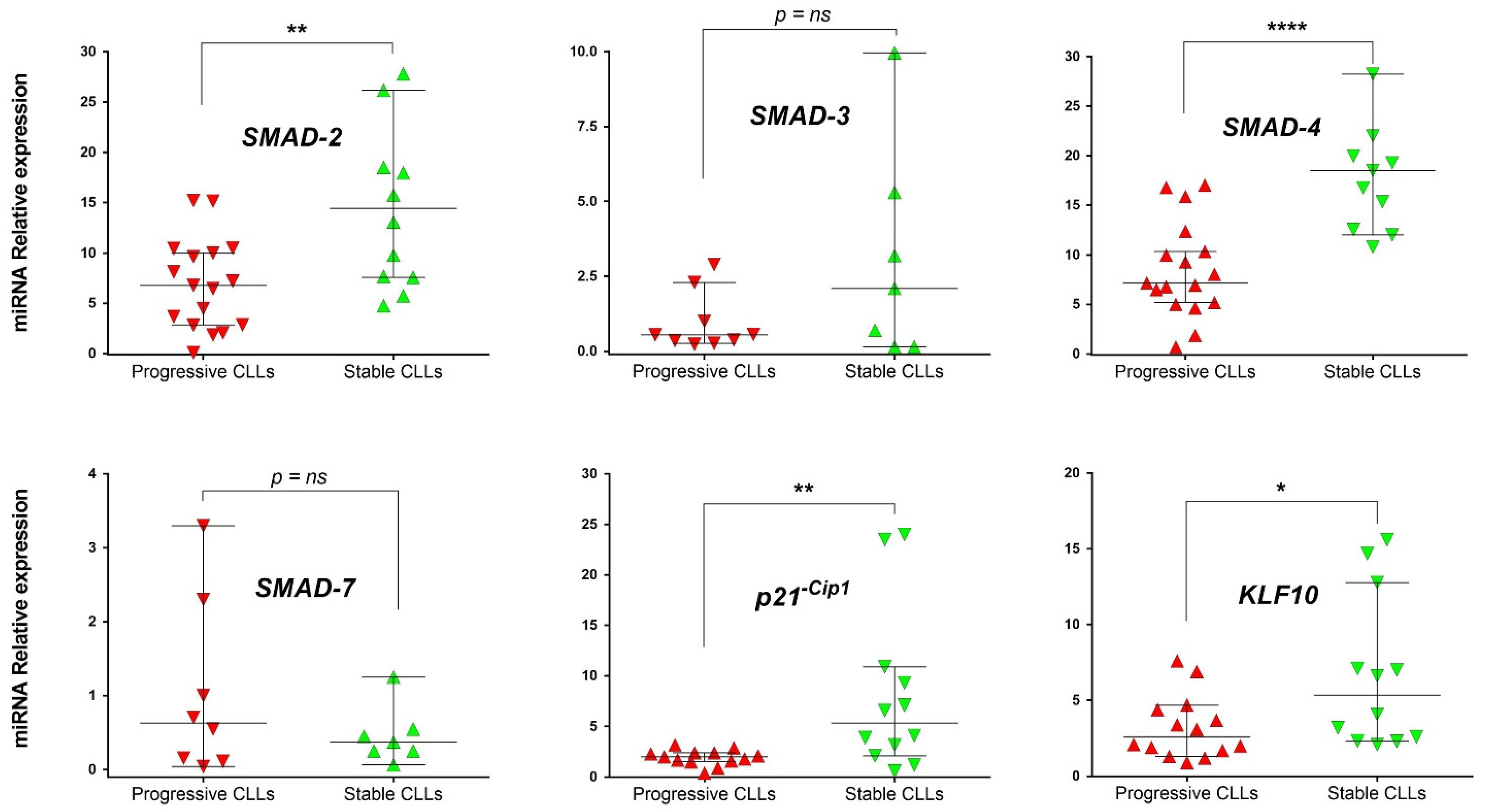
3.2. mRNA Level Expression of SMAD Proteins and Target Genes of TGF-β Pathway in Stable and Progressive U-CLL Cases
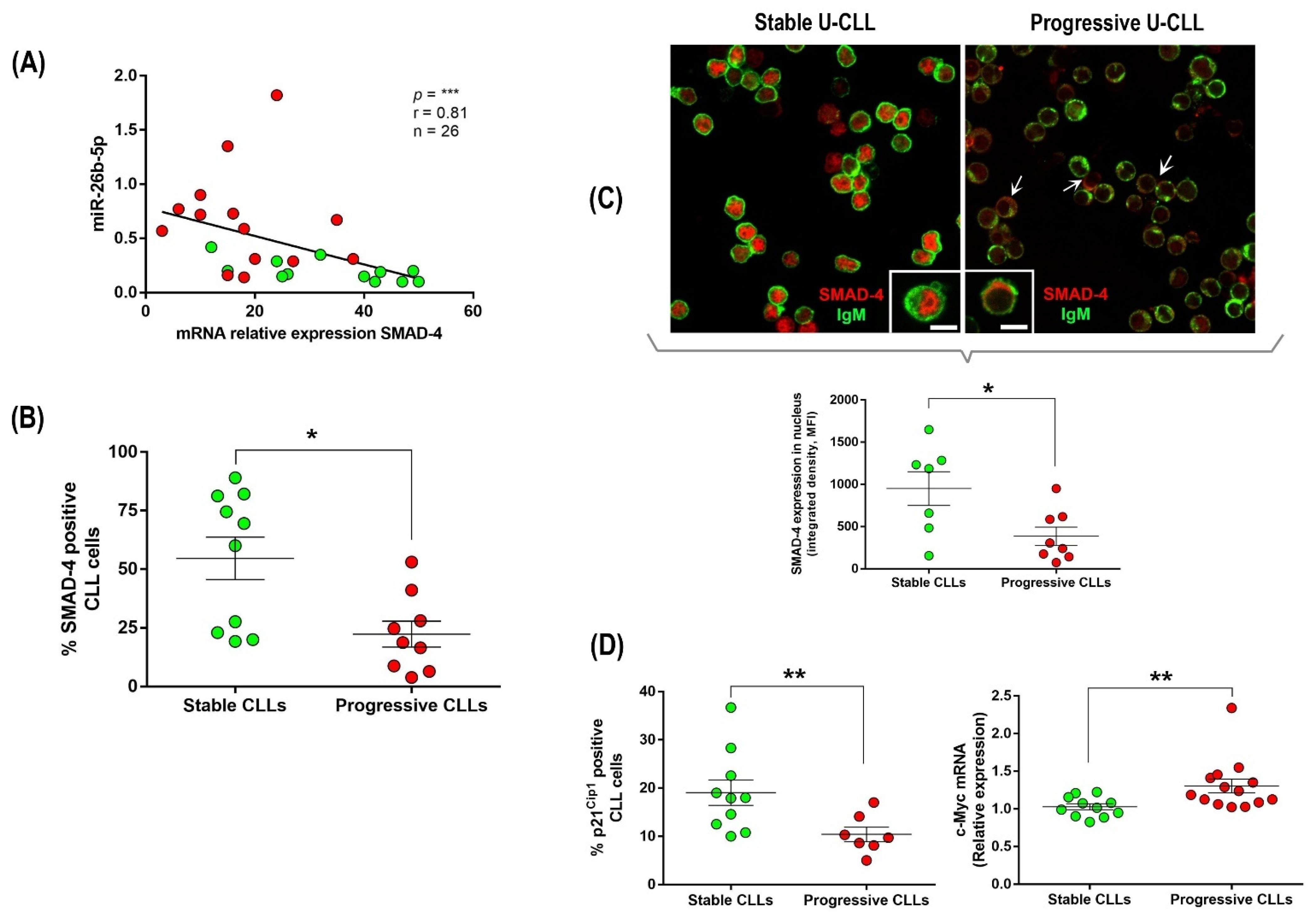
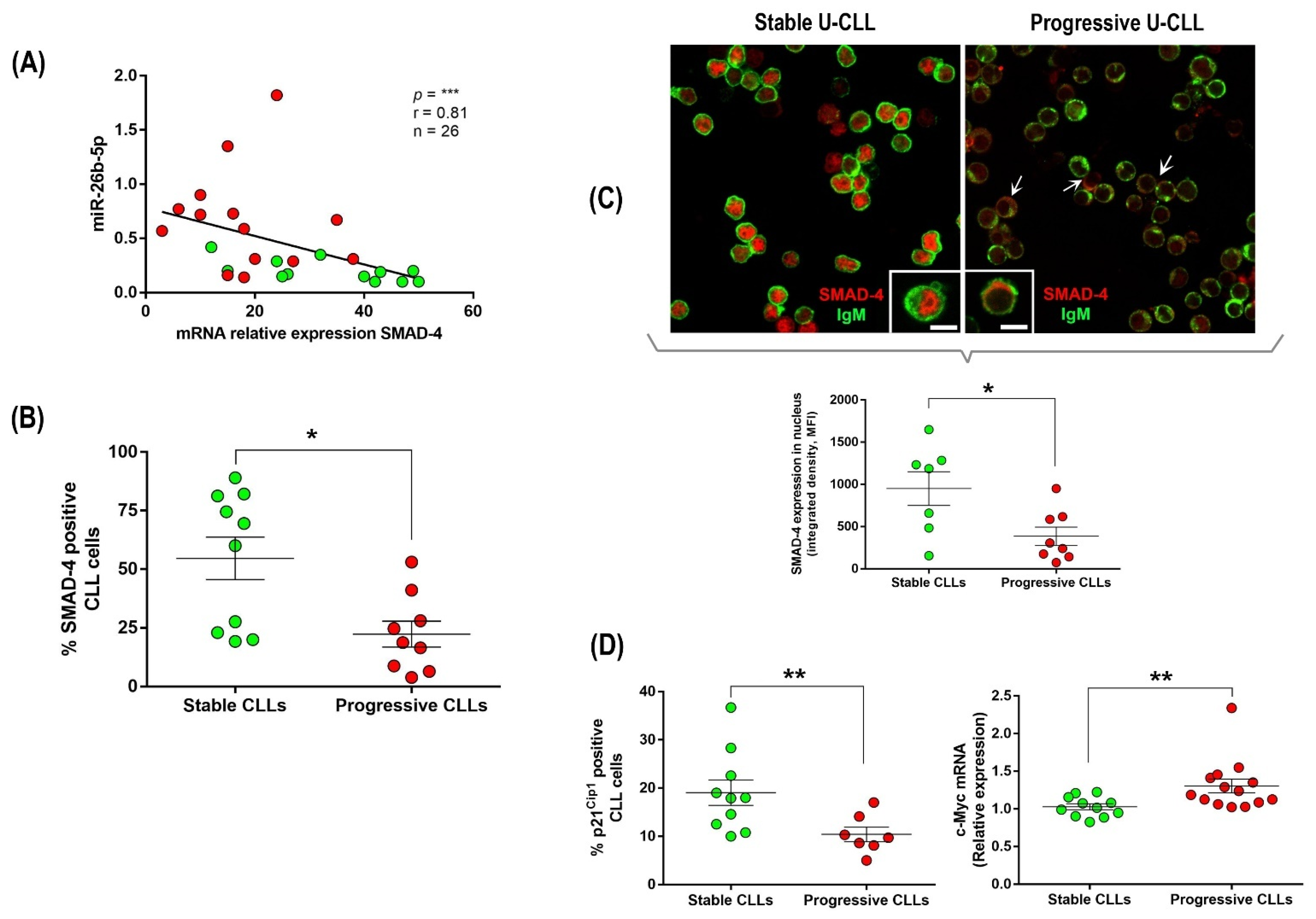
3.3. Up-Regulation of miR-26b-5p in Progressive U-CLL Is Associated with Decreased Expression and Nuclear Localization of SMAD-4, Down-Modulation of p21−Cip1 Cell Cycle Marker, and Up-Regulation of c-Myc Oncogene
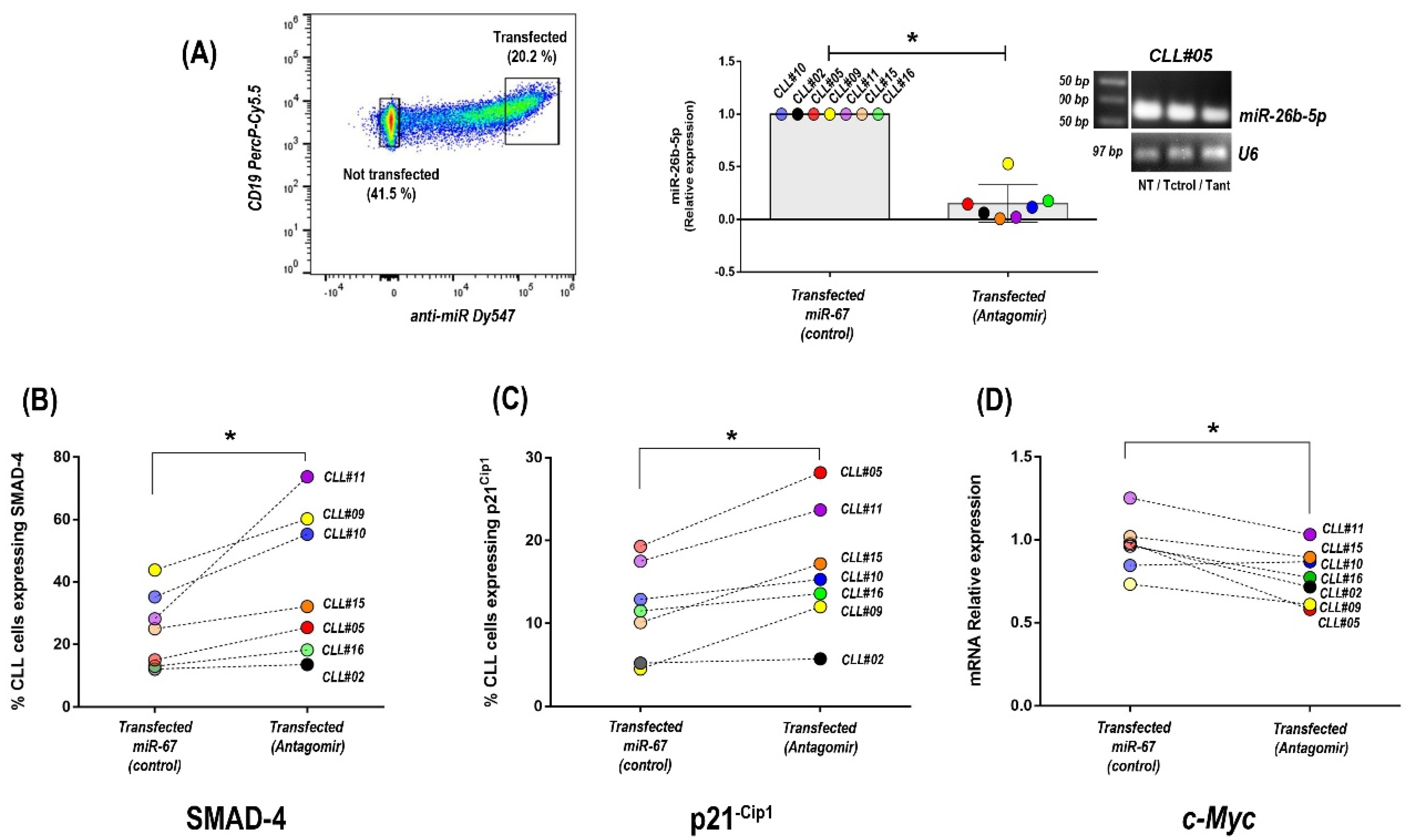
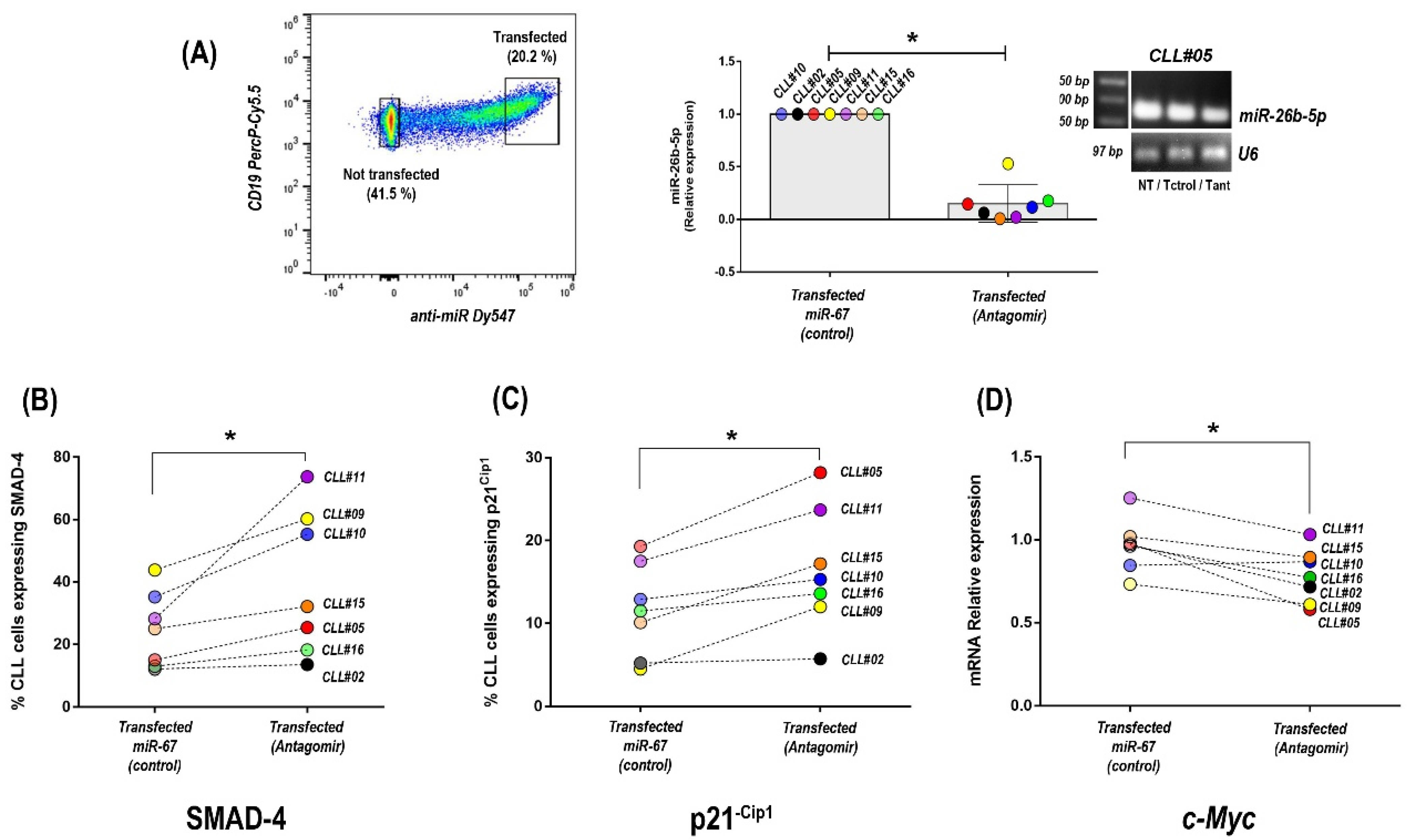
3.4. Inhibition of miR-26b-5p Recovers TGF-β/SMAD Function Regulating Cell Cycle Progression Molecules in Primary CLL Cells of Progressive Unmutated Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hallek, M. Chronic lymphocytic leukemia: 2013 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2013, 88, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dighiero, G.; Binet, J.L. When and how to treat chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1799–1801. [Google Scholar]
- Schroeder, H.W., Jr.; Dighiero, G. The pathogenesis of chronic lymphocytic leukemia: Analysis of the antibody repertoire. Immunol. Today 1994, 15, 288–294. [Google Scholar] [PubMed]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [PubMed]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [PubMed]
- Chiorazzi, N.; Stevenson, F.K. Celebrating 20 Years of IGHV Mutation Analysis in CLL. Hemasphere 2020, 4, e334. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiorazzi, N.; Ferrarini, M. Cellular origin(s) of chronic lymphocytic leukemia: Cautionary notes and additional considerations and possibilities. Blood 2010, 117, 1781–1791. [Google Scholar] [PubMed]
- Kipps, T.J. Mining the Microenvironment for Therapeutic Targets in Chronic Lymphocytic Leukemia. Cancer J. 2021, 27, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Liu, C.G.; Sevignani, C.; Ferracin, M.; Felli, N.; Dumitru, C.D.; Shimizu, M.; Cimmino, A.; Zupo, S.; Dono, M.; et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. USA 2004, 101, 11755–11760. [Google Scholar]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [PubMed]
- Calin, G.A.; Garzon, R.; Cimmino, A.; Fabbri, M.; Croce, C.M. MicroRNAs and leukemias: How strong is the connection? Leuk. Res. 2006, 30, 653–655. [Google Scholar]
- Negrini, M.; Cutrona, G.; Bassi, C.; Fabris, S.; Zagatti, B.; Colombo, M.; Ferracin, M.; D’Abundo, L.; Saccenti, E.; Matis, S.; et al. microRNAome expression in chronic lymphocytic leukemia: Comparison with normal B-cell subsets and correlations with prognostic and clinical parameters. Clin. Cancer Res. 2014, 20, 4141–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabris, L.; Juracek, J.; Calin, G. Non-Coding RNAs as Cancer Hallmarks in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2020, 21, 6720. [Google Scholar] [CrossRef]
- Moussay, E.; Palissot, V.; Vallar, L.; Poirel, H.A.; Wenner, T.; El Khoury, V.; Aouali, N.; Van Moer, K.; Leners, B.; Bernardin, F.; et al. Determination of genes and microRNAs involved in the resistance to fludarabine in vivo in chronic lymphocytic leukemia. Mol. Cancer 2010, 9, 115. [Google Scholar]
- Ruiz-Lafuente, N.; Alcaraz-Garcia, M.J.; Sebastian-Ruiz, S.; Garcia-Serna, A.M.; Gomez-Espuch, J.; Moraleda, J.M.; Minguela, A.; Garcia-Alonso, A.M.; Parrado, A. IL-4 Up-Regulates MiR-21 and the MiRNAs Hosted in the CLCN5 Gene in Chronic Lymphocytic Leukemia. PLoS ONE 2015, 10, e0124936. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Chen, L.; Zhang, S.; Mraz, M.; Fecteau, J.F.; Yu, J.; Ghia, E.M.; Zhang, L.; Bao, L.; Rassenti, L.Z.; et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014, 124, 546–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekarsky, Y.; Croce, C.M. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget 2010, 1, 224–227. [Google Scholar] [PubMed]
- Rassenti, L.Z.; Balatti, V.; Ghia, E.M.; Palamarchuk, A.; Tomasello, L.; Fadda, P.; Pekarsky, Y.; Widhopf, G.F., 2nd; Kipps, T.J.; Croce, C.M. MicroRNA dysregulation to identify therapeutic target combinations for chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2017, 114, 10731–10736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrocca, F.; Visone, R.; Onelli, M.R.; Shah, M.H.; Nicoloso, M.S.; de Martino, I.; Iliopoulos, D.; Pilozzi, E.; Liu, C.G.; Negrini, M.; et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 2008, 13, 272–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Liu, T.; Huang, W.; Liu, H.; Zhang, H.M.; Li, Q.; Chen, Z.; Guo, A.Y. MicroRNA regulatory pathway analysis identifies miR-142-5p as a negative regulator of TGF-beta pathway via targeting SMAD3. Oncotarget 2016, 7, 71504–71513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Wolfraim, L.A.; Fernandez, T.M.; Mamura, M.; Fuller, W.L.; Kumar, R.; Cole, D.E.; Byfield, S.; Felici, A.; Flanders, K.C.; Walz, T.M.; et al. Loss of Smad3 in acute T-cell lymphoblastic leukemia. N. Engl. J. Med. 2004, 351, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Lagneaux, L.; Delforge, A.; Bernier, M.; Stryckmans, P.; Bron, D. TGF-beta activity and expression of its receptors in B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 1998, 31, 99–106. [Google Scholar] [PubMed]
- Romano, S.; Mallardo, M.; Chiurazzi, F.; Bisogni, R.; D’Angelillo, A.; Liuzzi, R.; Compare, G.; Romano, M.F. The effect of FK506 on transforming growth factor beta signaling and apoptosis in chronic lymphocytic leukemia B cells. Haematologica 2008, 93, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.L.; Phyliky, R.L.; Li, C.Y. B-cell chronic lymphocytic leukemia: Correlation of clinical stages with angiogenic cytokine expression. Appl. Immunohistochem. Mol. Morphol. 2006, 14, 154–160. [Google Scholar] [CrossRef]
- Witkowska, M.; Majchrzak, A.; Cebula-Obrzut, B.; Wawrzyniak, E.; Robak, T.; Smolewski, P. The distribution and potential prognostic value of SMAD protein expression in chronic lymphocytic leukemia. Tumour Biol. 2017, 39, 1010428317694551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, R.S.; Capocasale, R.J.; Lamb, R.J.; Nowell, P.C.; Moore, J.S. Chronic lymphocytic leukemia B cells are resistant to the apoptotic effects of transforming growth factor-beta. Blood 1997, 89, 941–947. [Google Scholar] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]
- Palacios, F.; Moreno, P.; Morande, P.; Abreu, C.; Correa, A.; Porro, V.; Landoni, A.I.; Gabus, R.; Giordano, M.; Dighiero, G.; et al. High expression of AID and active class switch recombination might account for a more aggressive disease in unmutated CLL patients: Link with an activated microenvironment in CLL disease. Blood 2010, 115, 4488–4496. [Google Scholar] [PubMed] [Green Version]
- Palacios, F.; Abreu, C.; Prieto, D.; Morande, P.; Ruiz, S.; Fernandez-Calero, T.; Naya, H.; Libisch, G.; Robello, C.; Landoni, A.I.; et al. Activation of the PI3K/AKT pathway by microRNA-22 results in CLL B-cell proliferation. Leukemia 2015, 29, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Xu, B.; Benya, S.R.; Tang, X. MiRNA-26b inhibits the proliferation, migration, and epithelial-mesenchymal transition of lens epithelial cells. Mol. Cell. Biochem. 2014, 396, 229–238. [Google Scholar] [CrossRef]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [Green Version]
- Marton, S.; Garcia, M.R.; Robello, C.; Persson, H.; Trajtenberg, F.; Pritsch, O.; Rovira, C.; Naya, H.; Dighiero, G.; Cayota, A. Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia 2008, 22, 330–338. [Google Scholar]
- Li, S.; Moffett, H.F.; Lu, J.; Werner, L.; Zhang, H.; Ritz, J.; Neuberg, D.; Wucherpfennig, K.W.; Brown, J.R.; Novina, C.D. MicroRNA Expression Profiling Identifies Activated B Cell Status in Chronic Lymphocytic Leukemia Cells. PLoS ONE 2011, 6, e16956. [Google Scholar]
- Sharma, S.; Pavlasova, G.M.; Seda, V.; Cerna, K.A.; Vojackova, E.; Filip, D.; Ondrisova, L.; Sandova, V.; Kostalova, L.; Zeni, P.F.; et al. miR-29 modulates CD40 signaling in chronic lymphocytic leukemia by targeting TRAF4: An axis affected by BCR inhibitors. Blood 2021, 137, 2481–2494. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.A.; Leof, E.B. TGF-beta signaling: A tale of two responses. J. Cell Biochem. 2007, 102, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Ranheim, E.; Kipps, T.J. Transforming growth factor beta as endogenous growth inhibitor of chronic lymphocytic leukemia B cells. J. Exp. Med. 1994, 179, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.D.; Choi, J.Y.; Lee, S.J.; Kim, J.S.; Min, B.R.; Lee, Y.I.; Kang, Y.K. TGF-beta-induced cell-cycle arrest through the p21(WAF1/CIP1)-G1 cyclin/Cdks-p130 pathway in gastric-carcinoma cells. Int. J. Cancer 1999, 83, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, M.; Harris, S.A.; Oursler, M.J.; Rasmussen, K.; Riggs, B.L.; Spelsberg, T.C. Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995, 23, 4907–4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblin, T. Chronic lymphocytic leukaemia: One disease or two? Ann. Hematol. 2002, 81, 299–303. [Google Scholar]
- Ten Hacken, E.; Burger, J.A. Microenvironment interactions and B-cell receptor signaling in Chronic Lymphocytic Leukemia: Implications for disease pathogenesis and treatment. Biochim. Biophys. Acta 2016, 1863, 401–413. [Google Scholar] [CrossRef]
- Calissano, C.; Damle, R.N.; Marsilio, S.; Yan, X.J.; Yancopoulos, S.; Hayes, G.; Emson, C.; Murphy, E.J.; Hellerstein, M.K.; Sison, C.; et al. Intraclonal complexity in chronic lymphocytic leukemia: Fractions enriched in recently born/divided and older/quiescent cells. Mol. Med. 2011, 17, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Prieto, D.; Sotelo, N.; Seija, N.; Sernbo, S.; Abreu, C.; Duran, R.; Gil, M.; Sicco, E.; Irigoin, V.; Oliver, C.; et al. S100-A9 protein in exosomes from chronic lymphocytic leukemia cells promotes NF-kappaB activity during disease progression. Blood 2017, 130, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Meier-Abt, F.; Lu, J.; Cannizzaro, E.; Pohly, M.F.; Kummer, S.; Pfammatter, S.; Kunz, L.; Collins, B.C.; Nadeu, F.; Lee, K.S.; et al. The protein landscape of chronic lymphocytic leukemia. Blood 2021, 138, 2514–2525. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.B.; Taylor, K.H.; Arthur, G.L.; Rahmatpanah, F.B.; Hooshmand, S.I.; Caldwell, C.W. Epigenetic regulation of WNT signaling in chronic lymphocytic leukemia. Epigenomics 2010, 2, 53–70. [Google Scholar] [PubMed] [Green Version]
- Arruga, F.; Gizdic, B.; Serra, S.; Vaisitti, T.; Ciardullo, C.; Coscia, M.; Laurenti, L.; D’Arena, G.; Jaksic, O.; Inghirami, G.; et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia 2014, 28, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Oppezzo, P.; Dighiero, G. Role of the B-cell receptor and the microenvironment in chronic lymphocytic leukemia. Blood Cancer J. 2013, 3, e149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anelli, L.; Zagaria, A.; Specchia, G.; Musto, P.; Albano, F. Dysregulation of miRNA in Leukemia: Exploiting miRNA Expression Profiles as Biomarkers. Int. J. Mol. Sci. 2021, 22, 7156. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Ruhela, V.; Rani, L.; Gupta, A.; Sriram, K.; Gogia, A.; Sharma, A.; Kumar, L.; Gupta, R. RNA-Seq profiling of deregulated miRs in CLL and their impact on clinical outcome. Blood Cancer J. 2020, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrajoli, A.; Shanafelt, T.D.; Ivan, C.; Shimizu, M.; Rabe, K.G.; Nouraee, N.; Ikuo, M.; Ghosh, A.K.; Lerner, S.; Rassenti, L.Z.; et al. Prognostic value of miR-155 in individuals with monoclonal B-cell lymphocytosis and patients with B chronic lymphocytic leukemia. Blood 2013, 122, 1891–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulci, V.; Chiaretti, S.; Goldoni, M.; Azzalin, G.; Carucci, N.; Tavolaro, S.; Castellano, L.; Magrelli, A.; Citarella, F.; Messina, M.; et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 2007, 109, 4944–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Chun, H.J.; Azuma, J.; Maegdefessel, L.; Kundu, R.K.; Quertermous, T.; Tsao, P.S.; Spin, J.M. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J. Cell. Physiol. 2011, 226, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Lagneaux, L.; Delforge, A.; Bron, D.; Massy, M.; Bernier, M.; Stryckmans, P. Heterogenous response of B lymphocytes to transforming growth factor-beta in B-cell chronic lymphocytic leukaemia: Correlation with the expression of TGF-beta receptors. Br. J. Haematol. 1997, 97, 612–620. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rational Study | CLL # | Age | Sex | Stage Binet | L.C. (×103/µL) | FISH | AID | LPL | IgHV Status Homology (%) | IgHV Rearrangement | Clinical Disease Status | T.F.T. (Years) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study cohort (progressive patients) | 1 | 58 | M | C | 62.000 | del (17p) | (+) | (+) | 100 | VH1-02 | Progressive | <1 |
| 2 | 52 | M | B | 26.200 | del (13q) | (+) | (+) | 98.0 | VH4b*02 | Progressive | 2.4 | |
| 3 | 66 | F | B | 59.250 | Normal | (+) | (+) | 99.6 | VH2-5*10 | Progressive | 3 | |
| 4 | 58 | M | C | 72.100 | del (13q)/del (11) | (+) | (+) | 100 | VH3-48*03 | Progressive | <1 | |
| 5 | 72 | M | B | 56.200 | Normal | (+) | (+) | 100 | VH3-11*01 | Progressive | <1 | |
| 6 | 53 | M | B | 116.000 | Normal | (+) | (+) | 99.0 | VH1-69 | Progressive | <1 | |
| Study cohort (stable patients) | 15 | 66 | F | A | 7520 | Normal | (−) | (+) | 100 | VH3-9*01 | Stable | N/T |
| 16 | 67 | M | A | 9520 | Normal | (−) | (+) | 100 | VH3-13*01 | Stable | N/T | |
| 17 | 72 | M | A | 10.830 | del (13q) | (−) | (+) | 99.7 | VH3-23*01 | Stable | N/T | |
| 18 | 64 | M | B | 15.960 | Normal | (−) | (+) | 100 | VH1-69*01 | Stable | N/T | |
| 19 | 63 | F | A | Normal | (−) | (+) | 100 | VH2-5*01 | Stable | N/T | ||
| Validation cohort (progressive patients) | 7 | 59 | M | B | 26.200 | Normal | (+) | (+) | 100 | VH3-7*01 | Progressive | 1 |
| 8 | 77 | M | B | 34.600 | Normal | (+) | (+) | 99.1 | VH1-69*12 | Progressive | 1.7 | |
| 9 | 55 | M | 300.250 | del (17p) | (+) | (+) | 99.2 | VH3-15*01 | Progressive | 2.8 | ||
| 10 | 53 | F | C | 55.360 | del (17p) | (+) | (+) | 100 | VH1-69 | Progressive | <1 | |
| 11 | 49 | M | B | 154.830 | del (13p) | (+) | (+) | 100 | VH1-69*01 | Progressive | 1.5 | |
| 12 | 59 | M | C | 94.000 | Normal | (+) | (+) | 100 | VH4-34*01 | Progressive | 1.2 | |
| 13 | 64 | M | B | 64.740 | Normal | (+) | (+) | 100 | VH1-2*02 | Progressive | 2.5 | |
| 14 | 52 | M | C | 421.000 | Tris12 | (+) | (+) | 100 | VH1-69*01 | Progressive | <1 | |
| Validation cohort (stable patients) | 20 | 67 | M | A | 9.179 | Normal | (−) | (+) | 100 | VH1-69*13 | Stable | N/T |
| 21 | 61 | M | A | 6.230 | Normal | (−) | (+) | 98.6 | VH3-30 | Stable | N/T | |
| 22 | 79 | M | A | 8.900 | Normal | (−) | (+) | 100 | VH1-69*01 | Stable | N/T | |
| 23 | 73 | M | A | 18.800 | Normal | (−) | (−) | 100 | VH4-30-2*01 | Stable | N/T | |
| 24 | 67 | F | A | 19.660 | del (13q) | (−) | (+) | 100 | VH2-5*01 | Stable | N/T | |
| 25 | 73 | F | A | N/D | N/D | (−) | (+) | 98.2 | VH1-18*01 | Stable | N/T | |
| 26 | 72 | M | A | 10.830 | del (13q) | (−) | (−) | 99.7 | VH3-23*01 | Stable | N/T |
| miRNAs | Fold Change | p Value | Previously Reported in CLL | References |
|---|---|---|---|---|
| Increased relative expression in progressive U-CLL | ||||
| Let-7g-5p | 1.70 | 0.005 | YES | [19] |
| Let-7i-5p | 1.79 | 0.007 | YES | [36] |
| miR-7-5p | 2.49 | 0.005 | YES | [13,14] |
| miR-19a-3p | 3.22 | <0.0001 | YES | [11,37] |
| miR-19b-3p | 2.74 | <0.0001 | YES | [11] |
| miR-142-3p | 2.54 | <0.0001 | YES | [17] |
| miR-26a-2-3p | 7.64 | 0.01 | YES | [14] |
| miR-29b-3p | 2.30 | 0.01 | YES | [38] |
| miR-29c-5p | 5.68 | 0.002 | YES | [38] |
| miR-106b-5p | 2.55 | 0.005 | YES | [14] |
| miR-142-5p | 1.76 | 0.03 | YES | [22] |
| miR-374b-5p | 1.60 | 0.004 | YES | [39] |
| miR-7-1-3p | 3.31 | 0.008 | NO | -- |
| miR-26b-5p | 3.33 | 0.02 | NO | -- |
| miR-140-5p | 2.51 | 0.02 | NO | -- |
| miR-340-5p | 1.63 | 0.01 | NO | -- |
| miR-374a-3p | 2.60 | 0.02 | NO | -- |
| miR-3613-5p | 3.63 | 0.006 | NO | -- |
| Reduced relative expression in progressive U-CLL | ||||
| miR-146a-5p | 0.56 | 0.01 | YES | [12,17] |
| miR-130b-5p | 0.40 | 0.01 | NO | -- |
| miR-505-5p | 0.42 | 0.008 | NO | -- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquez, M.E.; Sernbo, S.; Payque, E.; Uria, R.; Tosar, J.P.; Querol, J.; Berca, C.; Uriepero, A.; Prieto, D.; Alvarez-Saravia, D.; et al. TGF-β/SMAD Pathway Is Modulated by miR-26b-5p: Another Piece in the Puzzle of Chronic Lymphocytic Leukemia Progression. Cancers 2022, 14, 1676. https://doi.org/10.3390/cancers14071676
Marquez ME, Sernbo S, Payque E, Uria R, Tosar JP, Querol J, Berca C, Uriepero A, Prieto D, Alvarez-Saravia D, et al. TGF-β/SMAD Pathway Is Modulated by miR-26b-5p: Another Piece in the Puzzle of Chronic Lymphocytic Leukemia Progression. Cancers. 2022; 14(7):1676. https://doi.org/10.3390/cancers14071676
Chicago/Turabian StyleMarquez, Maria Elena, Sandra Sernbo, Eugenia Payque, Rita Uria, Juan Pablo Tosar, Juliana Querol, Catalina Berca, Angimar Uriepero, Daniel Prieto, Diego Alvarez-Saravia, and et al. 2022. "TGF-β/SMAD Pathway Is Modulated by miR-26b-5p: Another Piece in the Puzzle of Chronic Lymphocytic Leukemia Progression" Cancers 14, no. 7: 1676. https://doi.org/10.3390/cancers14071676
APA StyleMarquez, M. E., Sernbo, S., Payque, E., Uria, R., Tosar, J. P., Querol, J., Berca, C., Uriepero, A., Prieto, D., Alvarez-Saravia, D., Oliver, C., Irigoin, V., Dos Santos, G., Guillermo, C., Landoni, A. I., Navarrete, M., Palacios, F., & Oppezzo, P. (2022). TGF-β/SMAD Pathway Is Modulated by miR-26b-5p: Another Piece in the Puzzle of Chronic Lymphocytic Leukemia Progression. Cancers, 14(7), 1676. https://doi.org/10.3390/cancers14071676