
SMYD3 Promotes Cell Cycle Progression by Inducing Cyclin D3 Transcription and Stabilizing the Cyclin D1 Protein in Medulloblastoma

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Transfection
2.2. Plasmids, Antibodies, Chemicals, and Inhibitors
2.3. In Silico Analysis
2.4. Immunocytochemistry (ICC), Immunohistochemistry (IHC), Nuclear Extraction, Immunoprecipitation (IP), and Mass-Spectrometry
2.5. Ubiquitination Assay
2.6. Chromatin Immunoprecipitation (ChIP), Real-Time PCR, and RNA-Seq
2.7. Cloning and Luciferase Reporter Gene Assay
2.8. Cell Cycle, Annexin V Staining, TUNEL, Colony-Forming, and Cell Viability Assays
2.9. Slice Culture and Lactate Dehydrogenase (LDH) Detection
2.10. Statistical Analysis
3. Results
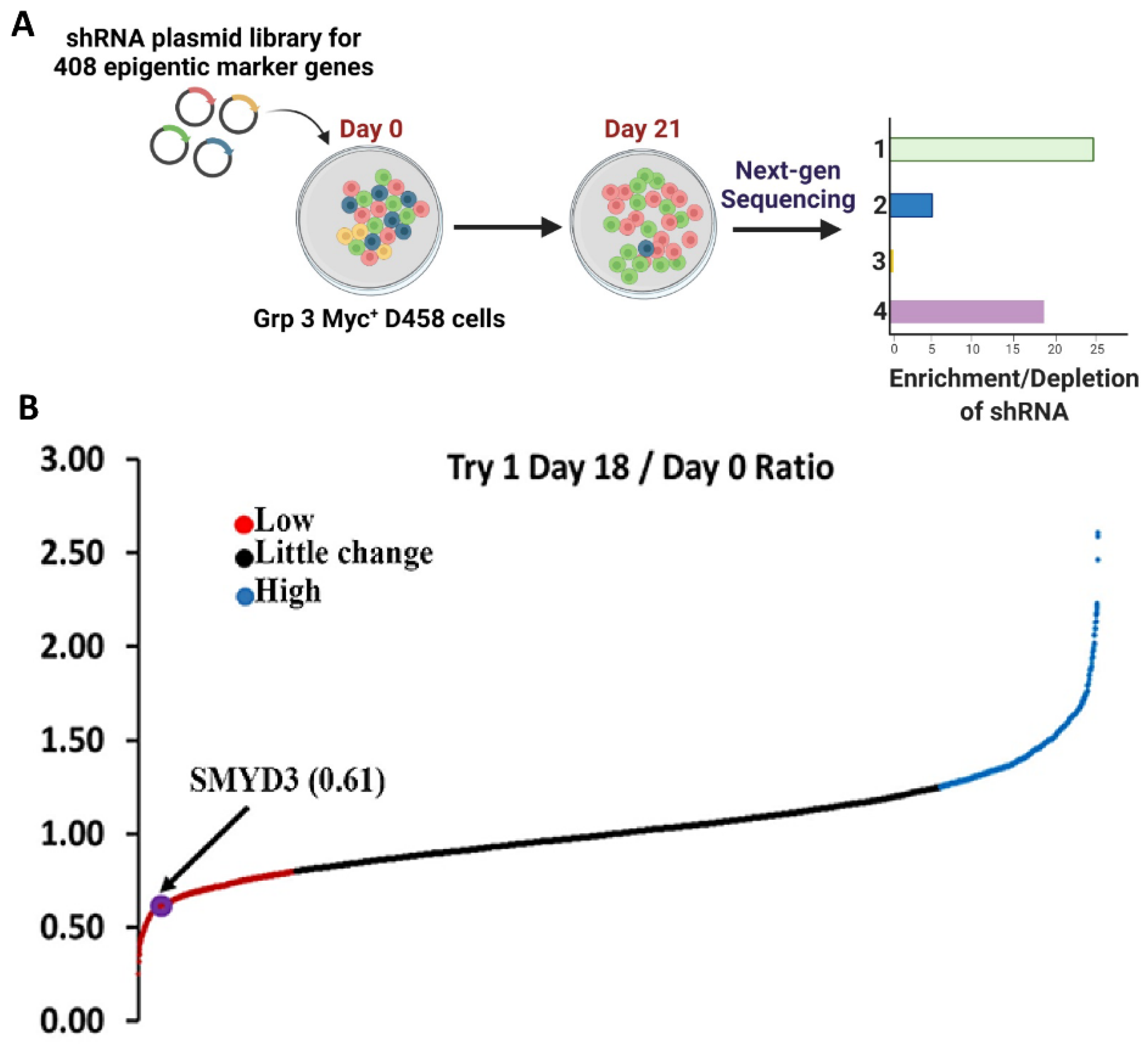
3.1. SMYD3 Is Crucial for MB Cell Survival
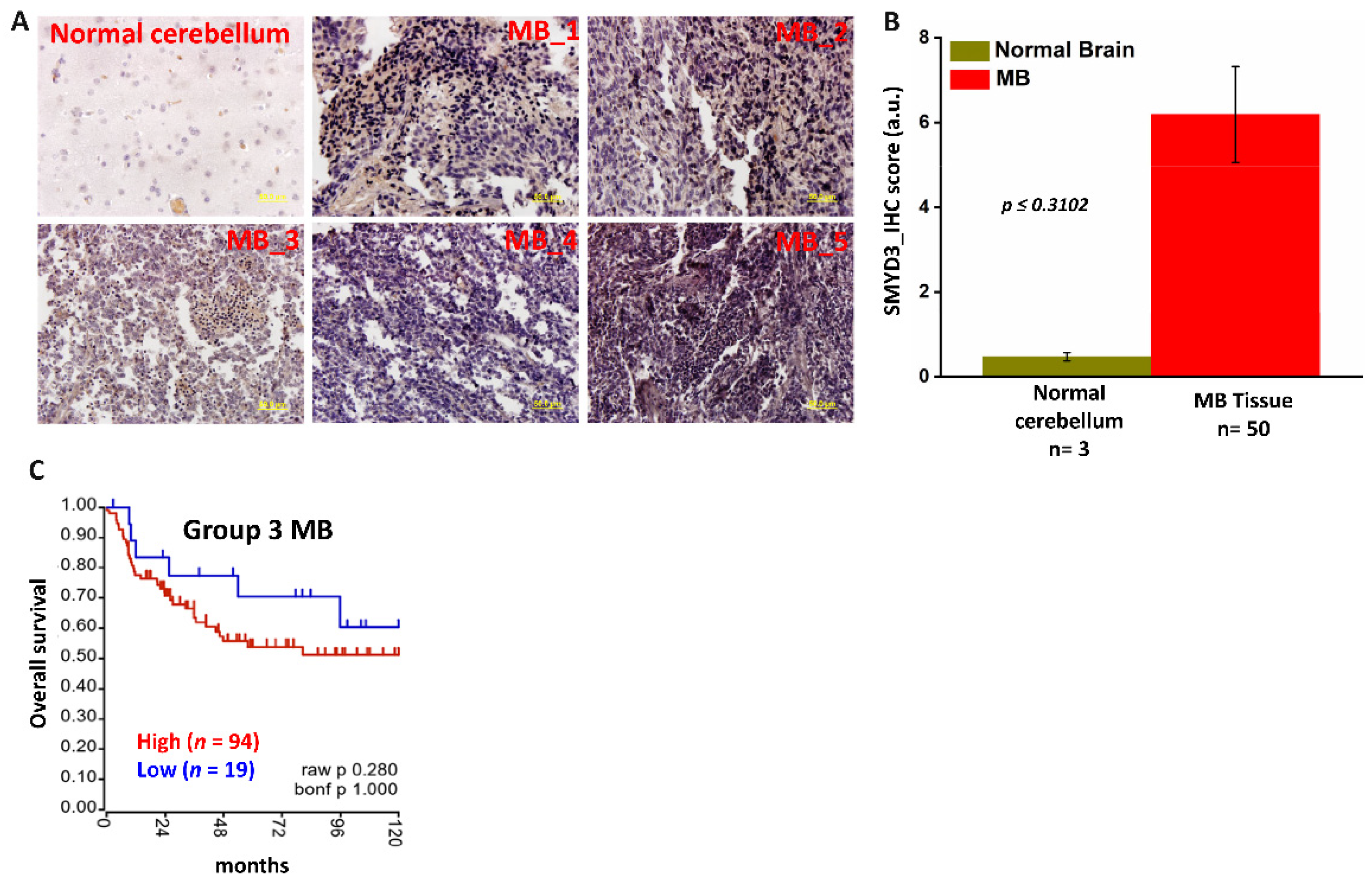
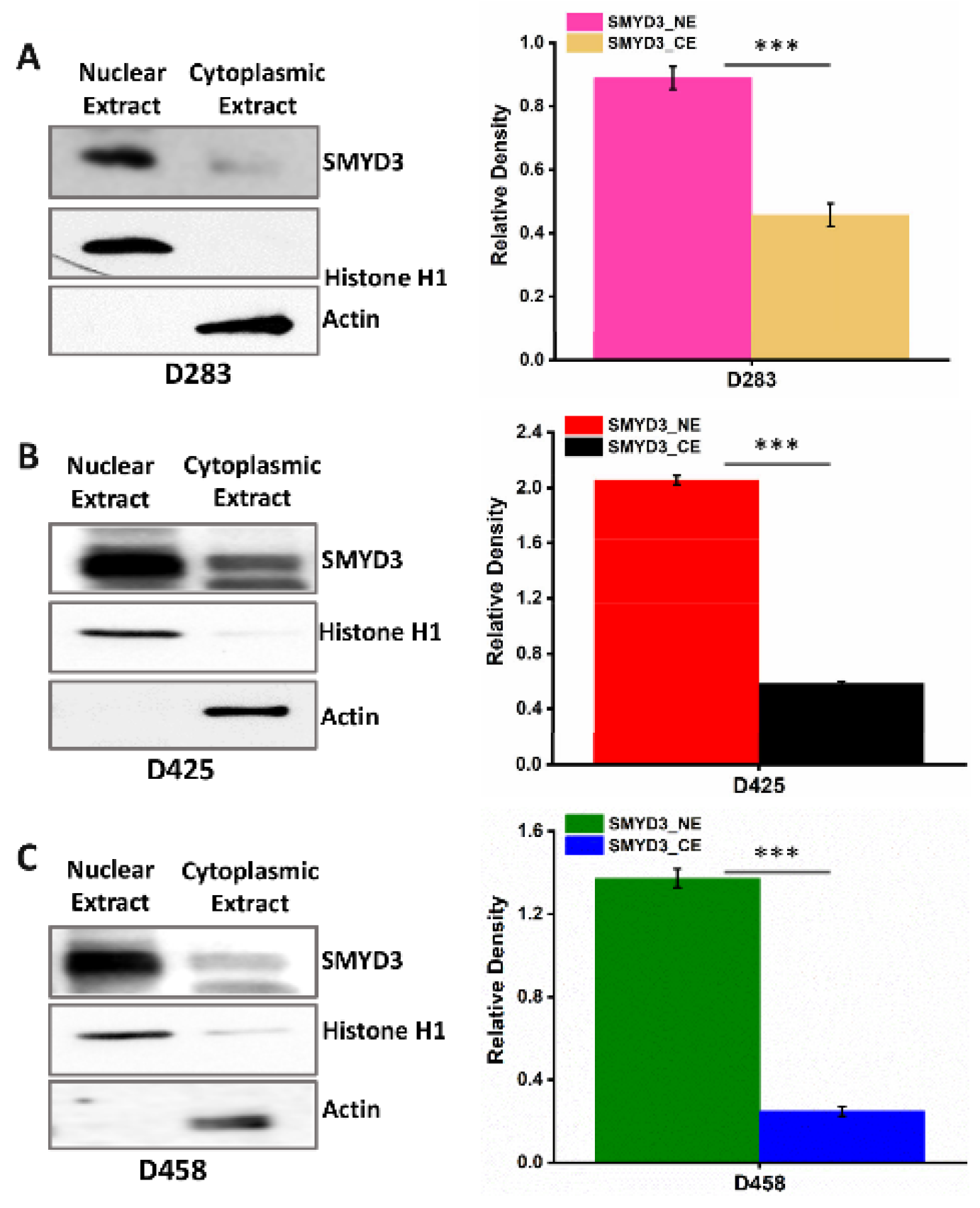
3.2. SMYD3 Is Primarily Expressed in the Nuclei of MB Cells and Targets Cell Cycle Regulators
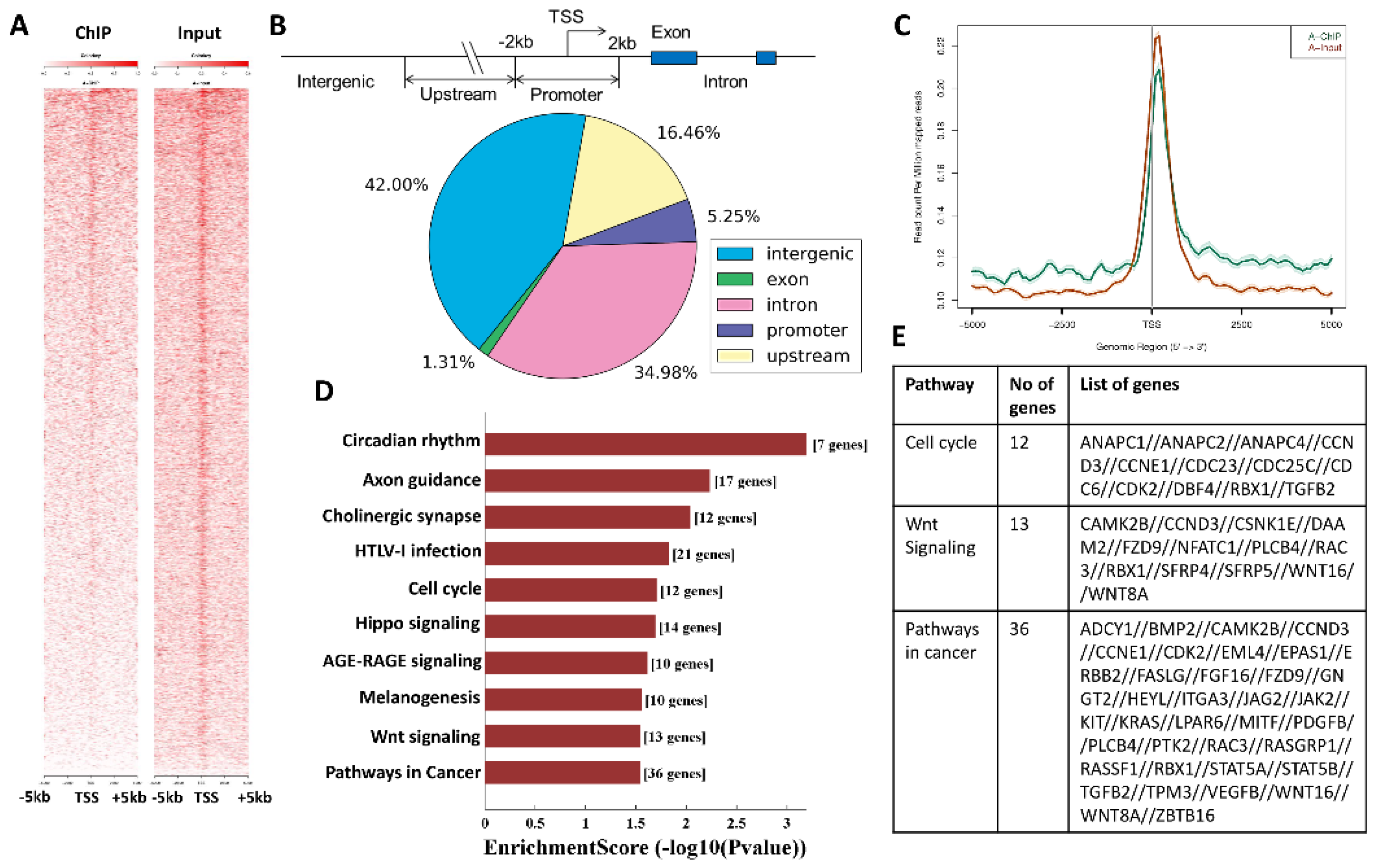
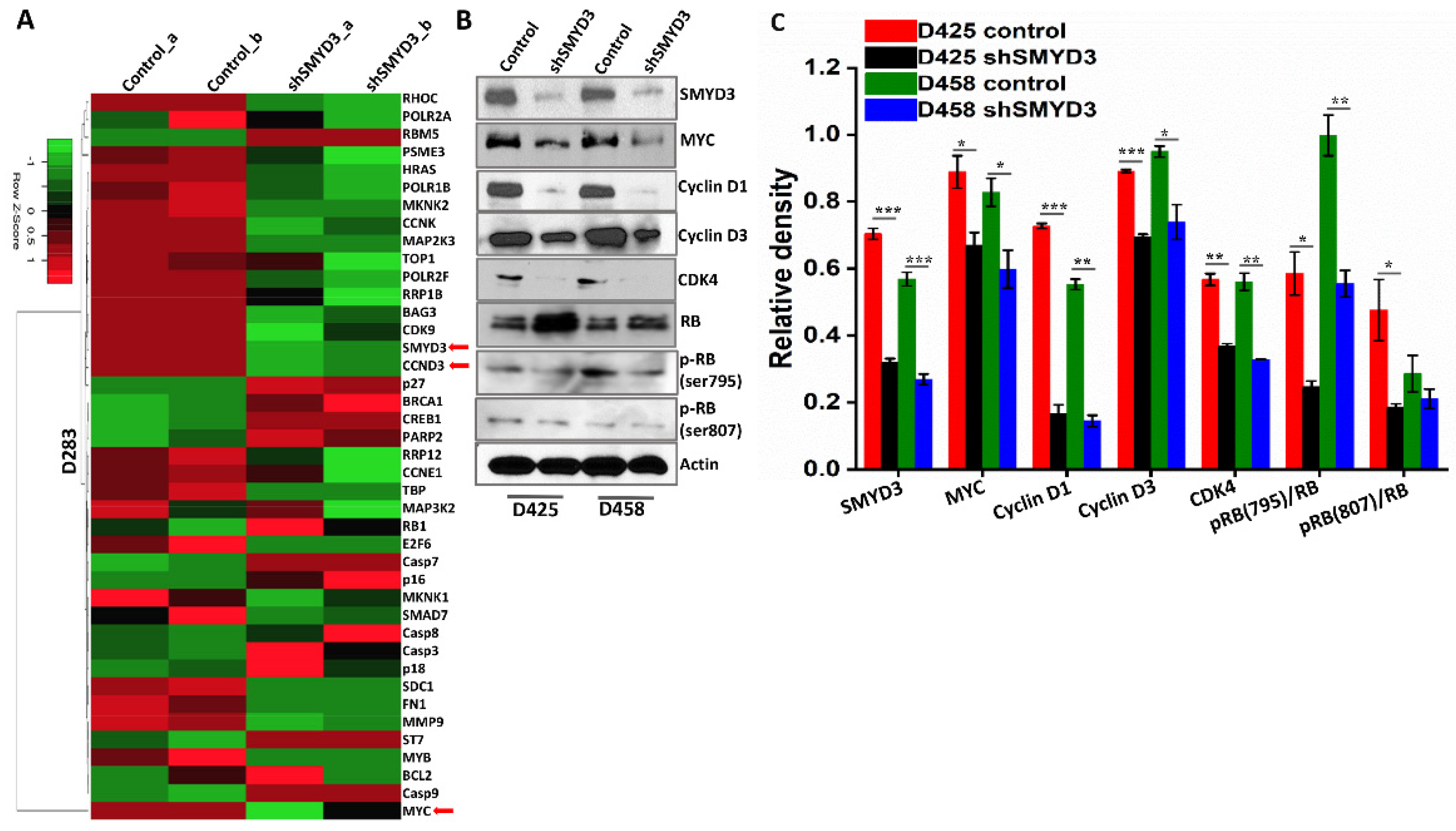
3.3. SMYD3 Transcriptionally Regulates Cell Cycle Effectors
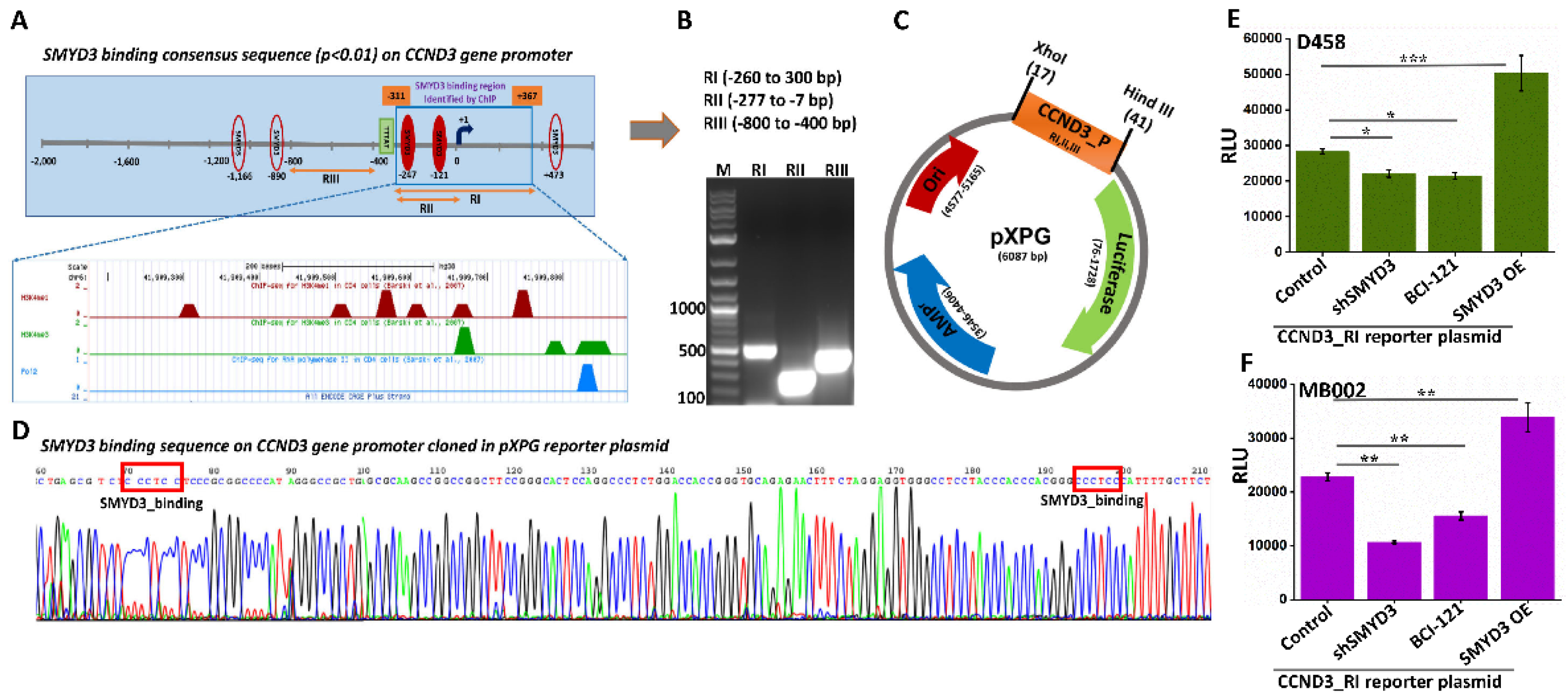
3.4. SMYD3 Induces Cyclin D3 Transcription
3.5. RNA Sequencing Supports the Role of SMYD3 in Regulating the Cell Cycle
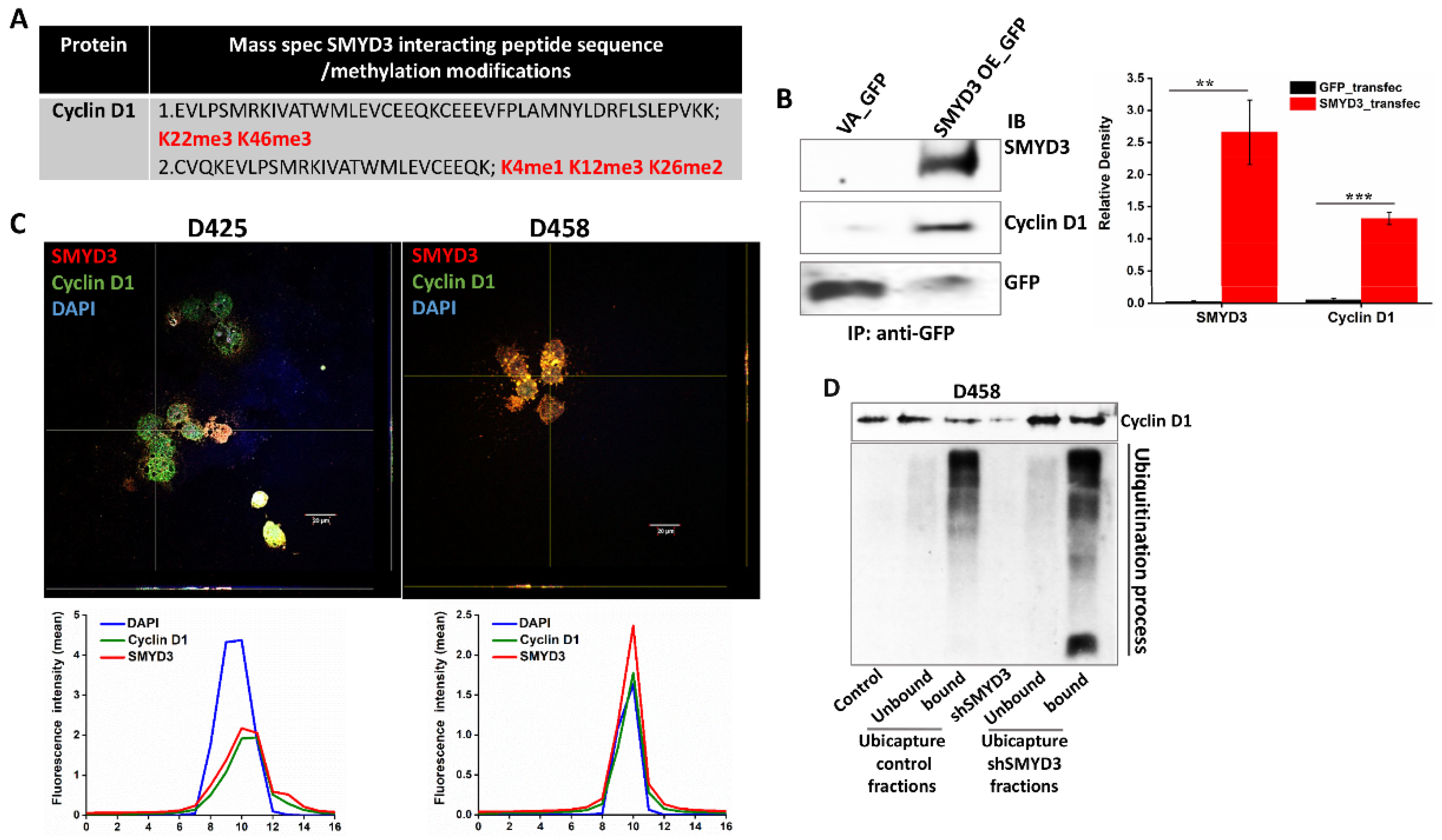
3.6. SMYD3 Knockdown Is Associated with Increased Ubiquitination of Cyclin D1
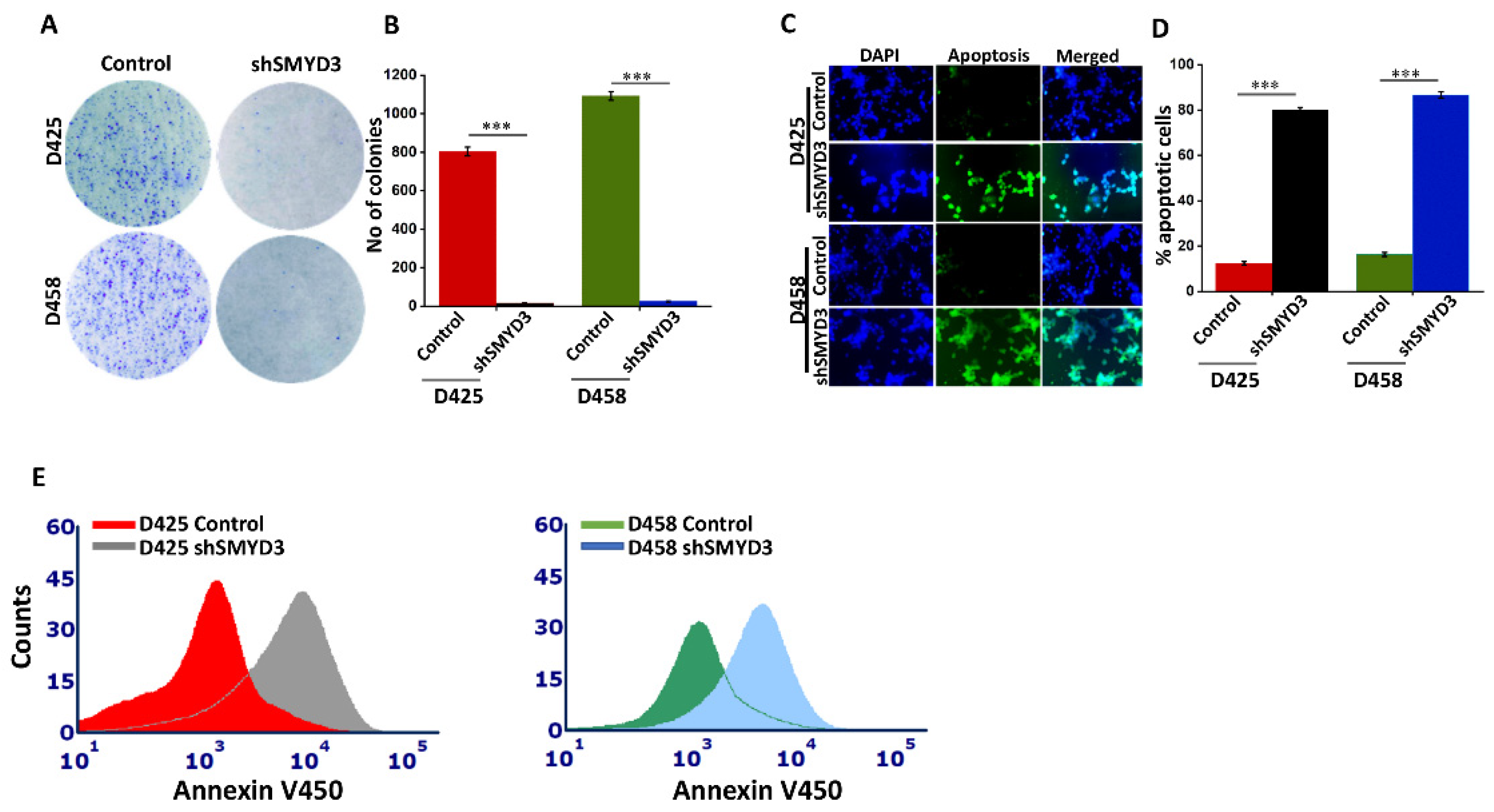
3.7. SMYD3 Knockdown Impairs Group 3 MB Cell Proliferation and Augments Apoptosis
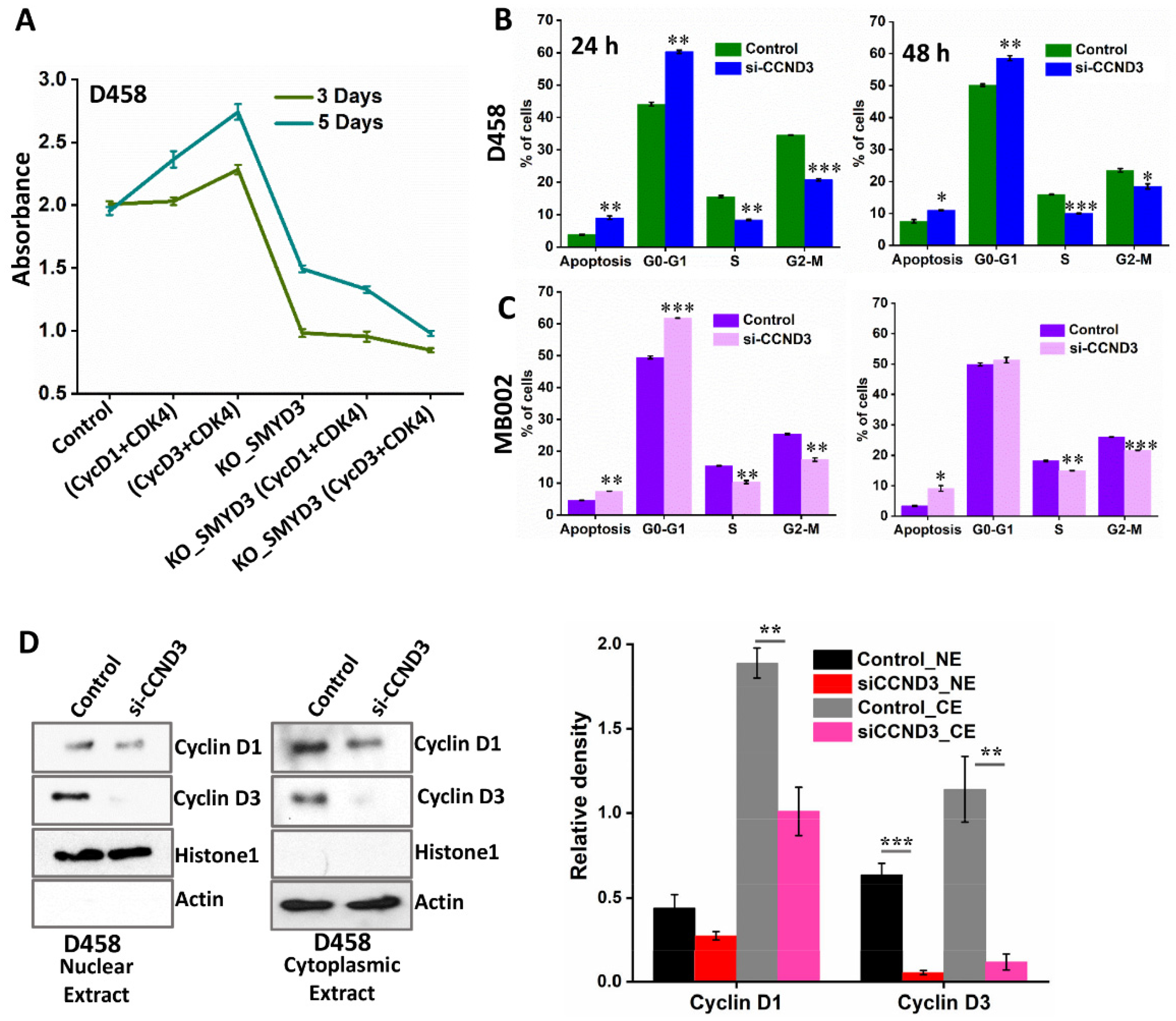
3.8. SMYD3 Regulates Downstream Effectors, Cyclin D3 and Cyclin D1, to Enhance MB Cell Viability
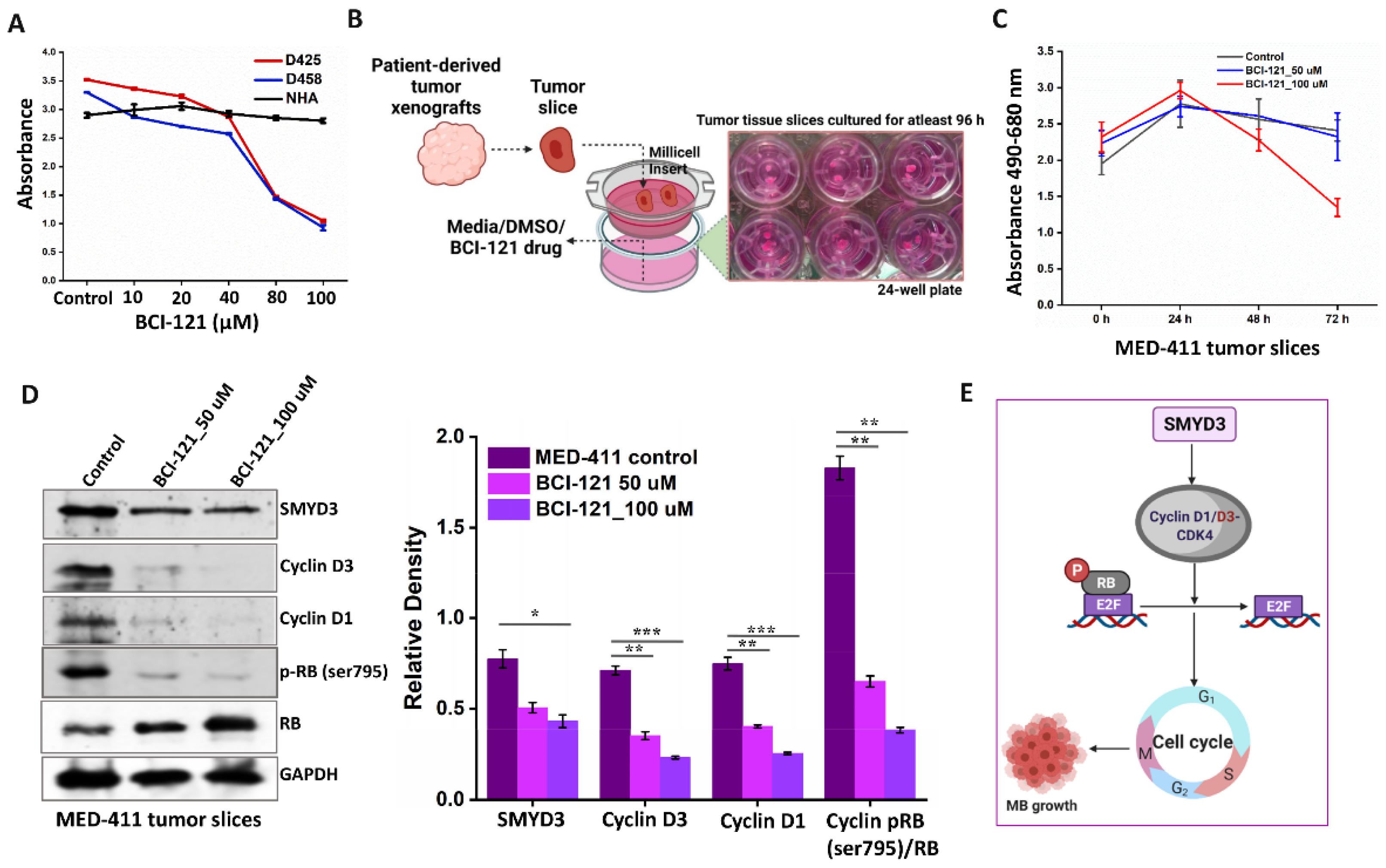
3.9. Pharmacological Targeting of SMYD3 Attenuates MB Proliferation Ex Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.-J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Badodi, S.; Pomella, N.; Zhang, X.; Rosser, G.; Whittingham, J.; Niklison-Chirou, M.V.; Lim, Y.M.; Brandner, S.; Morrison, G.; Pollard, S.M.; et al. Inositol treatment inhibits medulloblastoma through suppression of epigenetic-driven metabolic adaptation. Nat. Commun. 2021, 12, 2148. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.W.; Jaeger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2011, 81, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, R.; Gupta, K.; Gupta, S. Cancer epigenetics: An introduction. Methods Mol. Biol. 2015, 1238, 3–25. [Google Scholar] [CrossRef]
- Roussel, M.F.; Stripay, J.L. Epigenetic Drivers in Pediatric Medulloblastoma. Cerebellum 2017, 17, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Solberg, M.; Rossen, J.L. Center for Advanced Food Technology: A Successful Example of University-Industry Cooperation in Food Technology Development: Gearing for the Year 2000. Bibl. Nutr. Dieta 1990, 47, 38–50. [Google Scholar] [CrossRef]
- Veo, B.; Danis, E.; Pierce, A.; Sola, I.; Wang, D.; Foreman, N.K.; Jin, J.; Ma, A.; Serkova, N.; Venkataraman, S.; et al. Combined functional genomic and chemical screens identify SETD8 as a therapeutic target in MYC-driven medulloblastoma. JCI Insight 2019, 4, e122933. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Purvis, I.J.; Avilala, J.; Guda, M.R.; Venkataraman, S.; Vibhakar, R.; Tsung, A.J.; Velpula, K.K.; Asuthkar, S. Role of MYC-miR-29-B7-H3 in Medulloblastoma Growth and Angiogenesis. J. Clin. Med. 2019, 8, 1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veo, B.; Danis, E.; Pierce, A.; Wang, D.; Fosmire, S.; Sullivan, K.D.; Joshi, M.; Khanal, S.; Dahl, N.; Karam, S.; et al. Transcriptional control of DNA repair networks by CDK7 regulates sensitivity to radiation in MYC-driven medulloblastoma. Cell Rep. 2021, 35, 109013. [Google Scholar] [CrossRef] [PubMed]
- Asuthkar, S.; Velpula, K.K.; Elustondo, P.A.; Demirkhanyan, L.; Zakharian, E. TRPM8 channel as a novel molecular target in androgen-regulated prostate cancer cells. Oncotarget 2015, 6, 17221–17236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, T.-H.; Romanova, E.; Roberts-Galbraith, R.H.; Yang, N.; Zimmerman, T.; Collins, J.J.; Lee, J.E.; Kelleher, N.L.; Newmark, P.; Sweedler, J.V. Mass Spectrometry Imaging and Identification of Peptides Associated with Cephalic Ganglia Regeneration in Schmidtea mediterranea. J. Biol. Chem. 2016, 291, 8109–8120. [Google Scholar] [CrossRef] [Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Asuthkar, S.; Gogineni, V.R.; Rao, J.S.; Velpula, K.K. Nuclear Translocation of Hand-1 Acts as a Molecular Switch to Regulate Vascular Radiosensitivity in Medulloblastoma Tumors: The Protein uPAR Is a Cytoplasmic Sequestration Factor for Hand-1. Mol. Cancer Ther. 2014, 13, 1309–1322. [Google Scholar] [CrossRef] [Green Version]
- Asuthkar, S.; Gondi, C.S.; Nalla, A.K.; Velpula, K.K.; Gorantla, B.; Rao, J.S. Urokinase-type Plasminogen Activator Receptor (uPAR)-mediated Regulation of WNT/β-Catenin Signaling Is Enhanced in Irradiated Medulloblastoma Cells. J. Biol. Chem. 2012, 287, 20576–20589. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, S.; Birks, D.K.; Balakrishnan, I.; Alimova, I.; Harris, P.S.; Patel, P.R.; Handler, M.H.; Dubuc, A.; Taylor, M.; Foreman, N.; et al. MicroRNA 218 Acts as a Tumor Suppressor by Targeting Multiple Cancer Phenotype-associated Genes in Medulloblastoma. J. Biol. Chem. 2013, 288, 1918–1928. [Google Scholar] [CrossRef] [Green Version]
- Asuthkar, S.; Nalla, A.K.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Mohanam, S.; Rao, J.S. Gadd45a sensitizes medulloblastoma cells to irradiation and suppresses MMP-9–mediated EMT. Neuro-Oncol. 2011, 13, 1059–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarris, M.E.; Moulos, P.; Haroniti, A.; Giakountis, A.; Talianidis, I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazur, P.; Reynoird, N.; Khatri, P.; Jansen, P.W.T.C.; Wilkinson, A.W.; Liu, S.; Barbash, O.; Van Aller, G.S.; Huddleston, M.J.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, D.A.; Fargo, D.C.; Adelman, K. Using ChIP-chip and ChIP-seq to study the regulation of gene expression: Genome-wide localization studies reveal widespread regulation of transcription elongation. Methods 2009, 48, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2018, 62, 4233–4251. [Google Scholar] [CrossRef]
- Tetsu, O.; McCormick, F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 2003, 3, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, M.; Brill, J.A.; Fink, G.R.; Weinberg, R.A. Collaboration of G1 cyclins in the functional inactivation of the retinoblastoma protein. Genes Dev. 1994, 8, 1759–1771. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Stacey, D.W.; Hitomi, M. Post-transcriptional regulation of cyclin D1 expression during G2 phase. Oncogene 2002, 21, 7545–7556. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Masclef, L.; Dehennaut, V.; Mortuaire, M.; Schulz, C.; Leturcq, M.; Lefebvre, T.; Vercoutter-Edouart, A.-S. Cyclin D1 Stability Is Partly Controlled by O-GlcNAcylation. Front. Endocrinol. 2019, 10, 106. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Suzuki, T.; Matsuo, Y.; Nakakido, M.; Tsurita, G.; Simone, C.; Watanabe, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. SMYD3-mediated lysine methylation in the PH domain is critical for activation of AKT1. Oncotarget 2016, 7, 75023–75037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sakamoto, K.; Liu, C.; Triplett, A.A.; Lin, W.-C.; Rui, H.; Wagner, K.-U. Cyclin D3 Compensates for the Loss of Cyclin D1 during ErbB2-Induced Mammary Tumor Initiation and Progression. Cancer Res. 2011, 71, 7513–7524. [Google Scholar] [CrossRef] [Green Version]
- Radulovich, N.; Pham, N.-A.; Strumpf, D.; Leung, L.; Xie, W.; Jurisica, I.; Tsao, M.-S. Differential roles of cyclin D1 and D3 in pancreatic ductal adenocarcinoma. Mol. Cancer 2010, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; da Silva Santos, R.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lyu, T.; Che, X.; Jia, N.; Li, Q.; Feng, W. Overexpression of SMYD3 in Ovarian Cancer is Associated with Ovarian Cancer Proliferation and Apoptosis via Methylating H3K4 and H4K20. J. Cancer 2019, 10, 4072–4084. [Google Scholar] [CrossRef]
- Peserico, A.; Germani, A.; Sanese, P.; Barbosa, A.M.; Di Virgilio, V.; Fittipaldi, R.; Fabini, E.; Bertucci, C.; Varchi, G.; Moyer, M.P.; et al. A SMYD3 Small-Molecule Inhibitor Impairing Cancer Cell Growth. J. Cell. Physiol. 2015, 230, 2447–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xie, B.-H.; Lin, W.-H.; Huang, Y.-H.; Ni, J.-Y.; Hu, J.; Cui, W.; Zhou, J.; Shen, L.; Xu, L.-F.; et al. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene 2019, 38, 4948–4961. [Google Scholar] [CrossRef]
- Roelants, C.; Pillet, C.; Franquet, Q.; Sarrazin, C.; Peilleron, N.; Giacosa, S.; Guyon, L.; Fontanell, A.; Fiard, G.; Long, J.-A.; et al. Ex-Vivo Treatment of Tumor Tissue Slices as a Predictive Preclinical Method to Evaluate Targeted Therapies for Patients with Renal Carcinoma. Cancers 2020, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, Y.; Liu, K.-W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC- Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET Bromodomain Inhibition of MYC-Amplified Medulloblastoma. Clin. Cancer Res. 2013, 20, 912–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhshinyan, D.; Venugopal, C.; Adile, A.A.; Garg, N.; Manoranjan, B.; Hallett, R.; Wang, X.; Mahendram, S.; Vora, P.; Vijayakumar, T.; et al. BMI1 is a therapeutic target in recurrent medulloblastoma. Oncogene 2018, 38, 1702–1716. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef]
- Dong, S.-W.; Zhang, H.; Wang, B.-L.; Sun, P.; Wang, Y.-G.; Zhang, P.; Sun, P. Effect of the downregulation of SMYD3 expression by RNAi on RIZ1 expression and proliferation of esophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-H.; Chen, Y.-J.; Yu, C.-J.; Tzeng, S.-R.; Wu, I.-C.; Kuo, W.-H.; Lin, M.-C.; Chan, N.-L.; Wu, K.-J.; Teng, S.-C. SMYD3-Mediated H2A.Z.1 Methylation Promotes Cell Cycle and Cancer Proliferation. Cancer Res. 2016, 76, 6043–6053. [Google Scholar] [CrossRef] [Green Version]
- Toyoshima, H.; Hunter, T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef]
- Giakountis, A.; Moulos, P.; Sarris, M.E.; Hatzis, P.; Talianidis, I. Smyd3-associated regulatory pathways in cancer. Semin. Cancer Biol. 2017, 42, 70–80. [Google Scholar] [CrossRef]
- Li, Y.; Sun, L.; Zhang, Y.; Wang, D.; Wang, F.; Liang, J.; Gui, B.; Shang, Y. The Histone Modifications Governing TFF1 Transcription Mediated by Estrogen Receptor. J. Biol. Chem. 2011, 286, 13925–13936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, T.D.; Thompson, R.C.; Faber, A.C. Cyclins D3 and E go hand in hand with Cdk4/6 in diffuse large B-cell lymphoma. Cell Cycle 2010, 9, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Boonen, G.J.J.C.; van Oirschot, B.A.; van Diepen, A.; Mackus, W.J.M.; Verdonck, L.F.; Rijksen, G.; Medema, R.H. Cyclin D3 Regulates Proliferation and Apoptosis of Leukemic T Cell Lines. J. Biol. Chem. 1999, 274, 34676–34682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawai, C.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genescà, E.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic Targeting of the Cyclin D3:CDK4/6 Complex in T Cell Leukemia. Cancer Cell 2012, 22, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Sa, G.; Hitomi, M.; Harwalkar, J.; Stacey, A.W.; Gc, G.C.; Stacey, D.W. Ras is active throughout the cell cycle, but is able to induce cyclin D1 only during G2 phase. Cell Cycle 2002, 1, 50–58. [Google Scholar] [CrossRef]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef] [Green Version]
- Feramisco, J.R.; Gross, M.; Kamata, T.; Rosenberg, M.; Sweet, R.W. Microinjection of the oncogene form of the human H-ras (t-24) protein results in rapid proliferation of quiescent cells. Cell 1984, 38, 109–117. [Google Scholar] [CrossRef]
- Mittnacht, S.; Paterson, H.; Olson, M.; Marshall, C.J. Ras signalling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr. Biol. 1997, 7, 219–221. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, L.S.; Smith, M.R.; Stacey, D.W. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 1985, 313, 241–243. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, W.; Zhou, Q.; Zhang, J.; Jiang, H.; Hao, D.; Wang, J.; Zhou, Z.; He, C.; Xiao, Z. A Novel lncRNA IHS Promotes Tumor Proliferation and Metastasis in HCC by Regulating the ERK- and AKT/GSK-3beta-Signaling Pathways. Mol. Ther. Nucleic Acids 2019, 16, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.C.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asuthkar, S.; Venkataraman, S.; Avilala, J.; Shishido, K.; Vibhakar, R.; Veo, B.; Purvis, I.J.; Guda, M.R.; Velpula, K.K. SMYD3 Promotes Cell Cycle Progression by Inducing Cyclin D3 Transcription and Stabilizing the Cyclin D1 Protein in Medulloblastoma. Cancers 2022, 14, 1673. https://doi.org/10.3390/cancers14071673
Asuthkar S, Venkataraman S, Avilala J, Shishido K, Vibhakar R, Veo B, Purvis IJ, Guda MR, Velpula KK. SMYD3 Promotes Cell Cycle Progression by Inducing Cyclin D3 Transcription and Stabilizing the Cyclin D1 Protein in Medulloblastoma. Cancers. 2022; 14(7):1673. https://doi.org/10.3390/cancers14071673
Chicago/Turabian StyleAsuthkar, Swapna, Sujatha Venkataraman, Janardhan Avilala, Katherine Shishido, Rajeev Vibhakar, Bethany Veo, Ian J. Purvis, Maheedhara R. Guda, and Kiran K. Velpula. 2022. "SMYD3 Promotes Cell Cycle Progression by Inducing Cyclin D3 Transcription and Stabilizing the Cyclin D1 Protein in Medulloblastoma" Cancers 14, no. 7: 1673. https://doi.org/10.3390/cancers14071673
APA StyleAsuthkar, S., Venkataraman, S., Avilala, J., Shishido, K., Vibhakar, R., Veo, B., Purvis, I. J., Guda, M. R., & Velpula, K. K. (2022). SMYD3 Promotes Cell Cycle Progression by Inducing Cyclin D3 Transcription and Stabilizing the Cyclin D1 Protein in Medulloblastoma. Cancers, 14(7), 1673. https://doi.org/10.3390/cancers14071673