
Selective Targeting of Protein Kinase C (PKC)-θ Nuclear Translocation Reduces Mesenchymal Gene Signatures and Reinvigorates Dysfunctional CD8+ T Cells in Immunotherapy-Resistant and Metastatic Cancers

 , ,
, ,  ,
,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MCF-7 Inducible Model (MCF-7-IM)
2.3. Reconstitution of Nuclear Import
2.4. AlphaScreen® Binding Assay
2.5. CD8+ and CTC Enrichment from Blood
2.6. Cell and Tissue Processing and Immunofluorescence Microscopy
2.7. Applied Spectral Imaging (ASI) Digital Pathology
2.8. Proximity Ligation Assay (PLA)
2.9. Nuclear to Cytoplasmic Fluorescence Ratio (Fn/c) Analysis
2.10. PKC-θ Activity Assay
2.11. WST-1 Cell Viability Assay
2.12. IncuCyte® Scratch Wound Assay
2.13. In Vivo Mouse Xenograft Model
2.14. Flow Cytometry
2.15. RNA Sequencing
2.16. RNA-Seq Bioinformatics
2.17. Synthesis of Inhibitor Peptides
2.18. PBMC RNA Extraction and Quantitative Reverse Transcription PCR (qRT-PCR)
2.19. NanoString nCounter Assay
2.20. Peptide Microarrays
2.21. Quantification and Statistical Analysis
3. Results
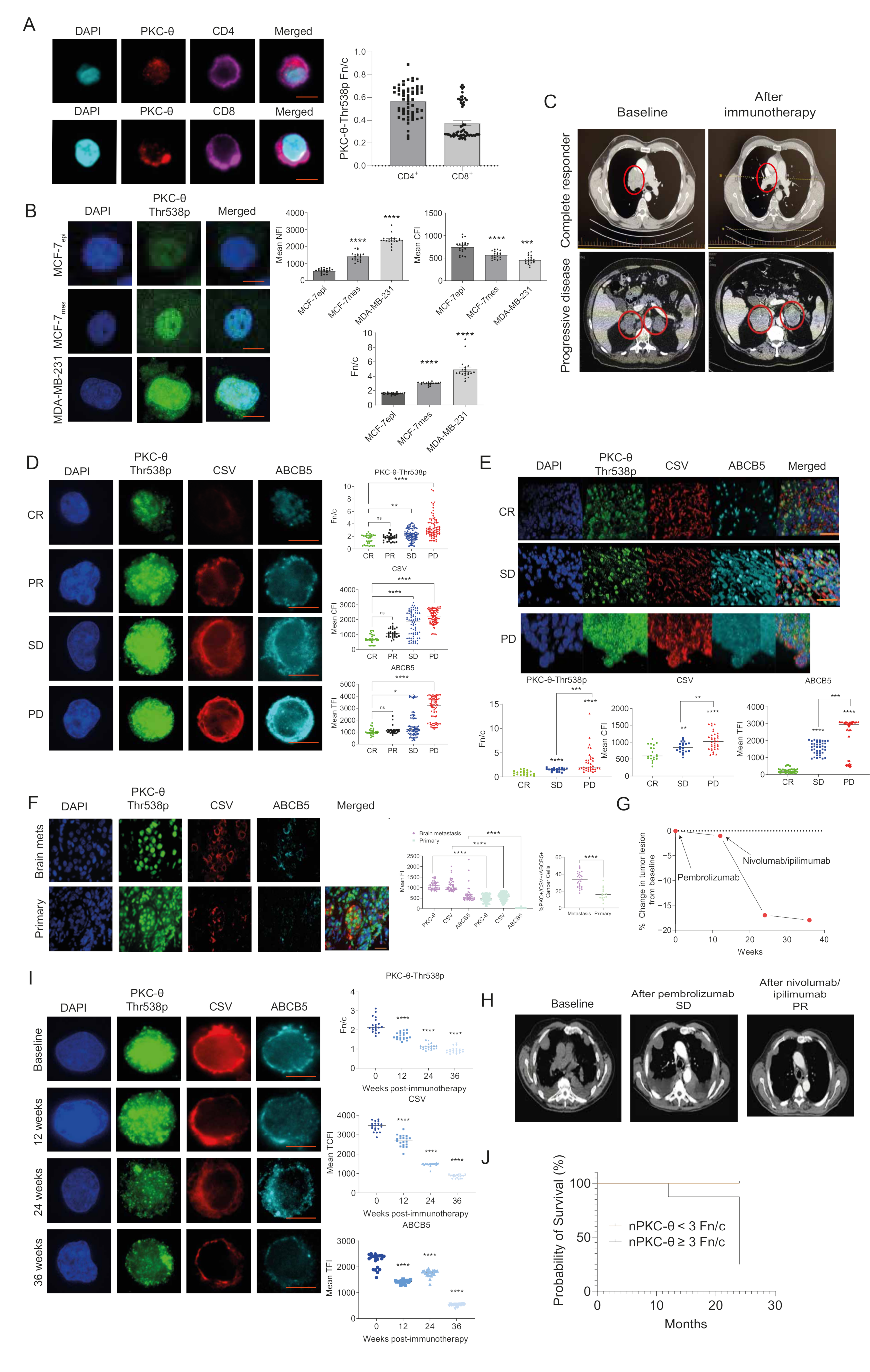
3.1. nPKC-θ Is Enriched in Immunotherapy-Resistant CTCs and Is Associated with Poor Patient Survival in Immunotherapy-Resistant Metastatic Disease
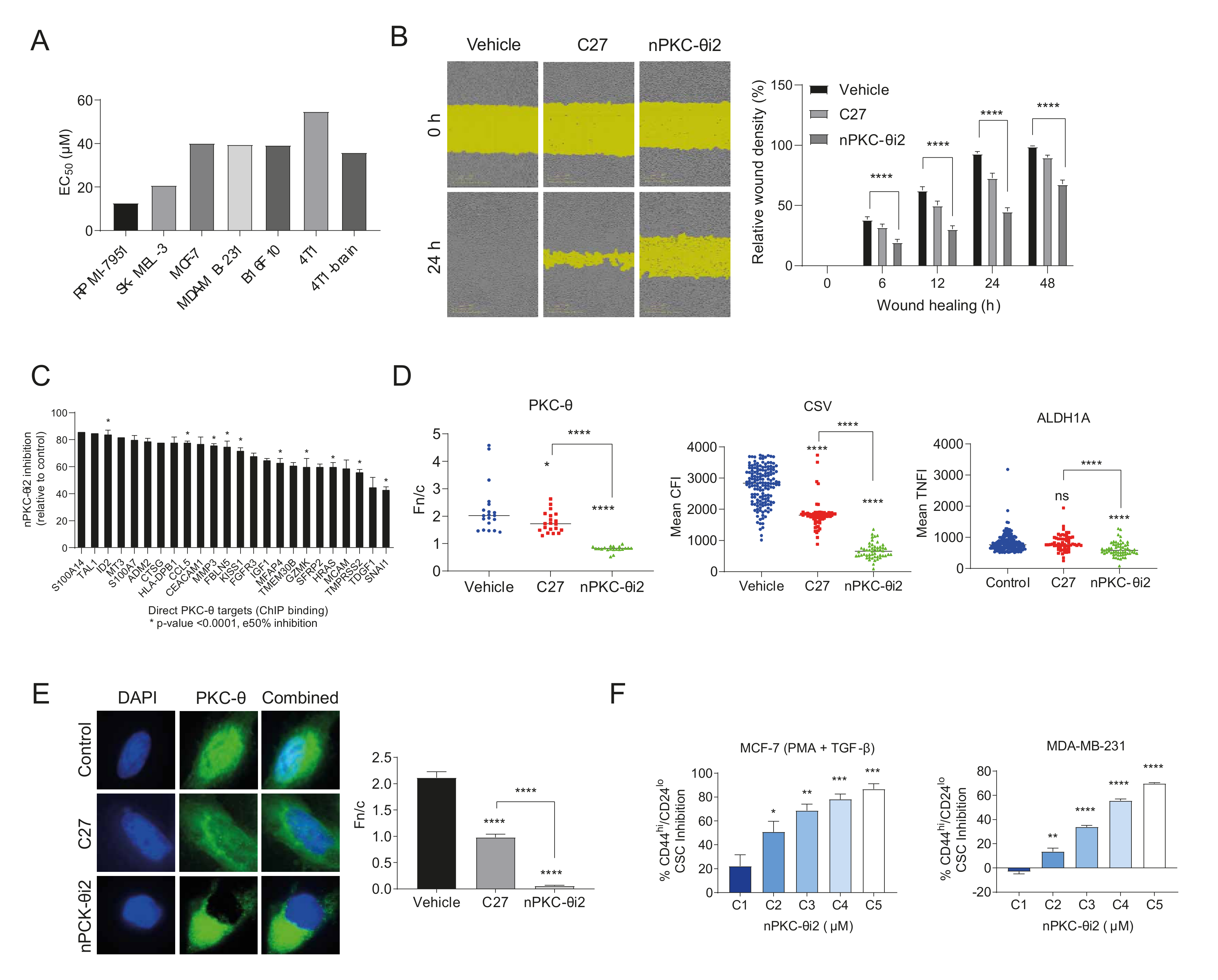
3.2. ATP Competitive Catalytic PKC-θ Inhibitors Do Not Target Its Nuclear Import
3.3. A Novel PKC-θ Peptide Inhibitor Specifically Inhibits Nuclear Translocation of PKC-θ while Preserving PKC-θ Catalytic Activity
3.4. nPKC-θ Induces CSC Signatures and Mesenchymal Pathways in Metastatic and Resistant Cancer Cell Lines
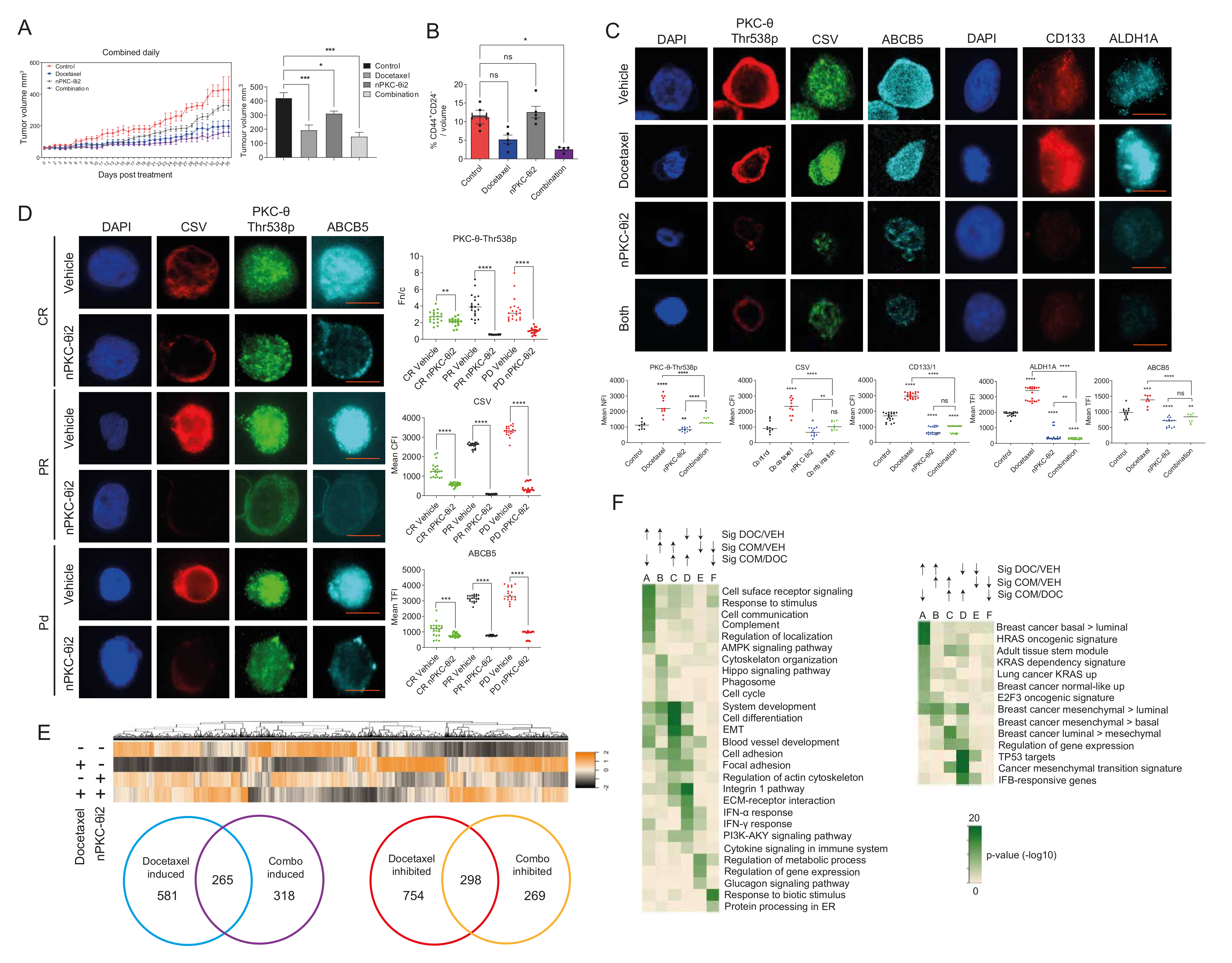
3.5. Inhibition of nPKC-θ Reduces Mesenchymal Signatures in Primary Tumors and CTCs In Vivo
3.6. Targeting nPKC-θ Reduces the Chemotherapy-Induced Mesenchymal Signature on Tumor Cell Transcriptome
3.7. PKC-θ Is Enriched in the Nuclei of CD8+ T Cells Isolated from Stage IV Metastatic Cancers
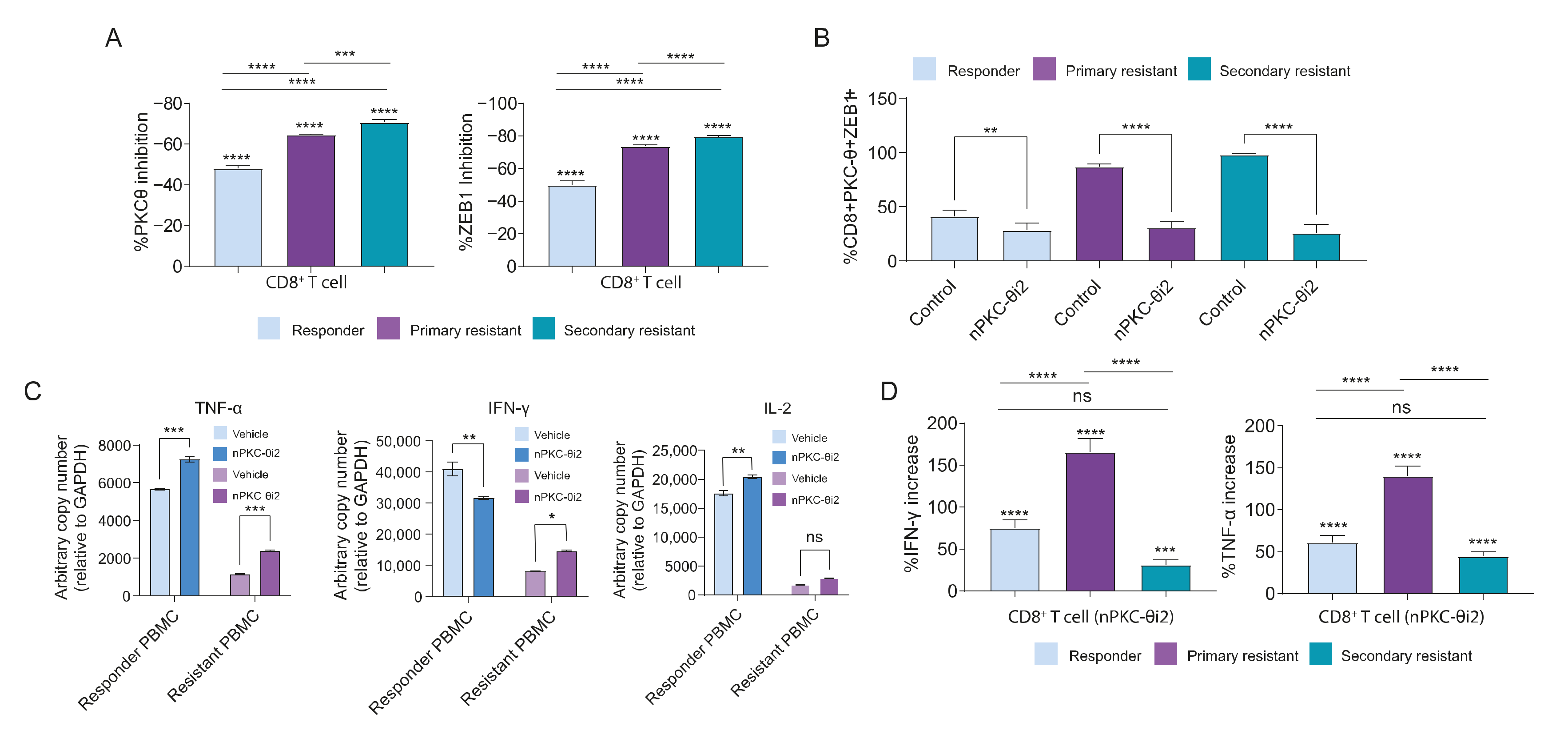
3.8. PKC-θ Forms a Repressive Complex with ZEB1 in the Nuclei of CD8+ T Cells Isolated from Metastatic Cancer Patients
3.9. Inhibition of nPKC-θ Inhibits Dysfunctional ZEB1/PKC-θ Nuclear Complex and Induces Cytokine Expression in CD8+ T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhanasekaran, N.; Premkumar Reddy, E. Signaling by Dual Specificity Kinases. Oncogene 1998, 17, 1447–1455. [Google Scholar] [CrossRef]
- Sutcliffe, E.L.; Li, J.; Zafar, A.; Hardy, K.; Ghildyal, R.; McCuaig, R.; Norris, N.C.; Lim, P.S.; Milburn, P.J.; Casarotto, M.G.; et al. Chromatinized Protein Kinase CΘ: Can It Escape the Clutches of NF-ΚB? Front. Immunol. 2012, 3, 1–13. [Google Scholar] [CrossRef]
- Misheva, M.; Kaur, G.; Ngoei, K.R.W.; Yeap, Y.Y.; Ng, I.H.W.; Wagstaff, K.M.; Ng, D.C.H.; Jans, D.A.; Bogoyevitch, M.A. Intracellular Mobility and Nuclear Trafficking of the Stress-Activated Kinase JNK1 Are Impeded by Hyperosmotic Stress. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, E.L.; Bunting, K.L.; He, Y.Q.; Li, J.; Phetsouphanh, C.; Seddiki, N.; Zafar, A.; Hindmarsh, E.J.; Parish, C.R.; Kelleher, A.D.; et al. Chromatin-Associated Protein Kinase C-θ Regulates an Inducible Gene Expression Program and MicroRNAs in Human T Lymphocytes. Mol. Cell 2011, 41, 704–719. [Google Scholar] [CrossRef]
- Li, J.; Hardy, K.; Phetsouphanh, C.; Tu, W.J.; Sutcliffe, E.L.; McCuaig, R.; Sutton, C.R.; Zafar, A.; Mee Ling Munier, C.; Zaunders, J.J.; et al. Nuclear PKC-θ Facilitates Rapid Transcriptional Responses in Human Memory CD4+T Cells through P65 and H2B Phosphorylation. J. Cell Sci. 2016, 129, 2448–2461. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Wu, F.; Hardy, K.; Li, J.; Tu, W.J.; McCuaig, R.; Harris, J.; Khanna, K.K.; Attema, J.; Gregory, P.A.; et al. Chromatinized Protein Kinase C-θ Directly Regulates Inducible Genes in Epithelial to Mesenchymal Transition and Breast Cancer Stem Cells. Mol. Cell. Biol. 2014, 34, 2961–2980. [Google Scholar] [CrossRef] [PubMed]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.K.; Bert, A.G.; et al. LSD1 Activation Promotes Inducible EMT Programs and Modulates the Tumour Microenvironment in Breast Cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; Tanaka, Y.; Coudronniere, N.; Sugie, K.; Hong, S.; Stipdonk, M.J. van; Altman, A. Antigen-Induced Translocation of PKC-Theta to Membrane Rafts Is Required for T Cell Activation. Nat. Immunol. 2001, 2, 556–563. [Google Scholar] [CrossRef]
- Monks, C.R.; Kupfer, H.; Tamir, I.; Barlow, A.; Kupfer, A. Selective Modulation of Protein Kinase C-Theta during T-Cell Activation. Nature 1997, 385, 83–86. [Google Scholar] [CrossRef]
- Brezar, V.; Tu, W.J.; Seddiki, N. PKC-Theta in Regulatory and Effector T-Cell Functions. Front. Immunol. 2015, 6, 530. [Google Scholar] [CrossRef]
- Isakov, N.; Altman, A. PKC-Theta-Mediated Signal Delivery from the TCR/CD28 Surface Receptors. Front. Immunol. 2012, 3, 2643–2649. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Dustin, M.L.; Blazar, B.R. PKC-θ Function at the Immunological Synapse: Prospects for Therapeutic Targeting. Trends Immunol. 2011, 32, 358–363. [Google Scholar] [CrossRef]
- Coudronniere, N.; Villalba, M.; Englund, N.; Altman, A. NF-ΚB Activation Induced by T Cell Receptor/CD28 Costimulation Is Mediated by Protein Kinase C-θ. Proc. Natl. Acad. Sci. USA 2000, 97, 3394–3399. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Arendt, C.W.; Ellmeier, W.; Schaeffer, E.M.; Sunshine, M.J.; Gandhl, L.; Annes, J.; Petrzilka, D.; Kupfer, A.; Schwartzberg, P.L.; et al. PKC-θ Is Required for TCR-Induced NF-ΚB Activation in Mature but Not Immature T Lymphocytes. Nature 2000, 404, 402–407. [Google Scholar] [CrossRef]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The Protein Kinase Cβ-Selective Inhibitor, Enzastaurin (LY317615.HCl), Suppresses Signaling through the AKT Pathway, Induces Apoptosis, and Suppresses Growth of Human Colon Cancer and Glioblastoma Xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef]
- Haarberg, K.M.K.; Li, J.; Heinrichs, J.; Wang, D.; Liu, C.; Bronk, C.C.; Kaosaard, K.; Owyang, A.M.; Holland, S.; Masuda, E.; et al. Pharmacologic Inhibition of PKCa & PKCu Prevents GVHD While Preserving GVL Activity in Mice. Blood 2013, 122, 2500–2511. [Google Scholar] [CrossRef]
- Jourdan, E.; Leblond, V.; Maisonneuve, H.; Benhadji, K.A.; Hossain, A.M.; Nguyen, T.S.; Wooldridge, J.E.; Moreau, P. A Multicenter Phase II Study of Single-Agent Enzastaurin in Previously Treated Multiple Myeloma. Leuk. Lymphoma 2014, 55, 2013–2017. [Google Scholar] [CrossRef]
- Kovarik, J.M.; Stitah, S.; Slade, A.; Vitaliti, A.; Straube, F.; Grenet, O.; Winter, S.; Sfikas, N.; Seiberling, M. Sotrastaurin and Cyclosporine Drug Interaction Study in Healthy Subjects. Biopharm. Drug Dispos. 2010, 31, 331–339. [Google Scholar] [CrossRef]
- Matz, M.; Weber, U.; Mashreghi, M.F.; Lorkowski, C.; Ladhoff, J.; Kramer, S.; Neumayer, H.H.; Budde, K. Effects of the New Immunosuppressive Agent AEB071 on Human Immune Cells. Nephrol. Dial. Transplant. 2010, 25, 2159–2167. [Google Scholar] [CrossRef][Green Version]
- Jimenez, J.-M.; Boyall, D.; Brenchley, G.; Collier, P.N.; Davis, J.C.; Damien, F.; Keily, S.B.; Henderson, J.; Miller, A.; Pierard, F.; et al. Design and Optimization of Selective Protein Kinase C θ (PKCθ) Inhibitors for the Treatment of Autoimmune Diseases. J. Med. Chem. 2013, 56, 1799–1810. [Google Scholar] [CrossRef]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T Cell Exhaustion in Infection and Cancer Kristen. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The Yin and Yang of Co-Inhibitory Receptors: Toward Anti-Tumor Immunity without Autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef]
- Rowe, C.L.; Wagstaff, K.M.; Oksayan, S.; Glover, D.J.; Jans, D.; Moseley, G.W. Nuclear Trafficking of the Rabies Virus Interferon Antagonist P-Protein Is Regulated by an Importin-Binding Nuclear Localization Sequence in the C-Terminal Domain. PLoS ONE 2016, 11, e0150477. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Headey, S.; Telwatte, S.; Tyssen, D.; Hearps, A.C.; Thomas, D.R.; Tachedjian, G.; Jans, D.A. Molecular Dissection of an Inhibitor Targeting the HIV Integrase Dependent Preintegration Complex Nuclear Import. Cell. Microbiol. 2018, 21, e12953. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Jans, D.A. Intramolecular Masking of Nuclear Localization Signals: Analysis of Importin Binding Using a Novel AlphaScreen-Based Method. Anal. Biochem. 2006, 348, 49–56. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Rawlinson, S.M.; Hearps, A.C.; Jans, D.A. An AlphaScreen®-Based Assay for High-Throughput Screening for Specific Inhibitors of Nuclear Import. J. Biomol. Screen. 2011, 16, 192–200. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Glover, D.J.; Tremethick, D.J.; Jans, D.A. Histone-Mediated Transduction as an Efficient Means for Gene Delivery. Mol. Ther. 2007, 15, 721–731. [Google Scholar] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.H.; Litière, S.; De Vries, E.; Ford, R.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Hayes, W.; Hodi, F.S.; et al. RECIST 1.1—Update and Clarification: From the RECIST Committee. Eur. J. Cancer 2016, 62, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.J.; McCuaig, R.D.; Melino, M.; Rawle, D.J.; Le, T.T.; Yan, K.; Suhrbier, A.; Johnston, R.L.; Koufariotis, L.T.; Waddell, N.; et al. Targeting Novel LSD1-Dependent ACE2 Demethylation Domains Inhibits SARS-CoV-2 Replication. Cell Discov. 2021, 7, 37. [Google Scholar] [CrossRef]
- Tu, W.J.; McCuaig, R.D.; Tan, A.H.Y.; Hardy, K.; Seddiki, N.; Ali, S.; Dahlstrom, J.E.; Bean, E.G.; Dunn, J.; Forwood, J.; et al. Targeting Nuclear LSD1 to Reprogram Cancer Cells and Reinvigorate Exhausted T Cells via a Novel LSD1-EOMES Switch. Front. Immunol. 2020, 11, 1228. [Google Scholar] [CrossRef]
- Wu, F.; McCuaig, R.D.; Sutton, C.R.; Tan, A.H.Y.; Jeelall, Y.; Bean, E.G.; Dai, J.; Prasanna, T.; Batham, J.; Malik, L.; et al. Nuclear-Biased DUSP6 Expression Is Associated with Cancer Spreading Including Brain Metastasis in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2019, 20, 3080. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Lassmann, T.; Hayashizaki, Y.; Daub, C.O. TagDust—A Program to Eliminate Artifacts from next Generation Sequencing Data. Bioinformatics 2009, 25, 2839–2840. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg1, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements Daehwan HHS Public Access. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown Bridges the Gap between Transcriptome Assembly and Expression Analysis. Nat. Biotechnol. 2015, 33, 243–246. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Enseñat-Waser, R.; Martin, F.; Barahona, F.; Vazquez, J.; Soria, B.; Reig, J.A. Direct Visualization by Confocal Fluorescent Microscopy of the Permeation of Myristoylated Peptides through the Cell Membrane. IUBMB Life 2002, 54, 33–36. [Google Scholar] [CrossRef]
- Liu, Y.; Graham, C.; Li, A.; Fisher, R.J.; Shaw, S. Phosphorylation of the Protein Kinase C-Theta Activation Loop and Hydrophobic Motif Regulates Its Kinase Activity, but Only Activation Loop Phosphorylation Is Critical to in Vivo Nuclear-Factor-ΚB Induction. Biochem. J. 2002, 361, 255–265. [Google Scholar] [CrossRef]
- Fillmore, C.M.; Kuperwasser, C. Human Breast Cancer Cell Lines Contain Stem-like Cells That Self-Renew, Give Rise to Phenotypically Diverse Progeny and Survive Chemotherapy. Breast Cancer Res. 2008, 10, 1–13. [Google Scholar] [CrossRef]
- Yang, S.N.; Atkinson, S.C.; Wang, C.; Lee, A.; Bogoyevitch, M.A.; Borg, N.A.; Jans, D.A. The Broad Spectrum Antiviral Ivermectin Targets the Host Nuclear Transport Importin α/Β1 Heterodimer. Antivir. Res. 2020, 177, 104760. [Google Scholar] [CrossRef]
- Martin, A.J.; Jans, D.A. Antivirals That Target the Host IMPα/Β1-Virus Interface. Biochem. Soc. Trans. 2021, 49, 281–295. [Google Scholar] [CrossRef]
- DeVries, T.A.; Neville, M.C.; Reyland, M.E. Nuclear Import of PKCδ Is Required for Apoptosis: Identification of a Novel Nuclear Import Sequence. EMBO J. 2002, 21, 6050–6060. [Google Scholar] [CrossRef]
- Smith, K.; Himiari, Z.; Tsimbalyuk, S.; Forwood, J. Structural Basis for Importin-α Binding of the Human Immunodeficiency Virus Tat. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Entani, T.; Takayama, S.; Tomita, M.; Yanagawa, H. Design of Peptide Inhibitors for the Importin α/β Nuclear Import Pathway by Activity-Based Profiling. Chem. Biol. 2008, 15, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Faenza, I.; Billi, A.M.; Fala, F.; Coco, L.; Manzoli, L. Nuclear Protein Kinase C Isoforms: Key Players in Multiple Cell Functions? Histol. Histopathol. 2003, 18, 1301–1312. [Google Scholar] [PubMed]
- Belguise, K.; Milord, S.; Galtier, F.; Moquet-Torcy, G.; Piechaczyk, M.; Chalbos, D. The PKCθ Pathway Participates in the Aberrant Accumulation of Fra-1 Protein in Invasive ER-Negative Breast Cancer Cells. Oncogene 2012, 31, 4889–4897. [Google Scholar] [CrossRef]
- Byerly, J.; Halstead-Nussloch, G.; Ito, K.; Katsyv, I.; Irie, H.Y. PRKCQ Promotes Oncogenic Growth and Anoikis Resistance of a Subset of Triple-Negative Breast Cancer Cells. Breast Cancer Res. 2016, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.B.; Harpole, D.; Lancaster, J.M.; Berchuck, A.; et al. Oncogenic Pathway Signatures in Human Cancers as a Guide to Targeted Therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adélaïde, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene Expression Profiling of Breast Cell Lines Identifies Potential New Basal Markers. Oncogene 2006, 25, 2273–2284. [Google Scholar] [CrossRef]
- Moserle, L.; Indraccolo, S.; Ghisi, M.; Frasson, C.; Fortunato, E.; Canevari, S.; Miotti, S.; Tosello, V.; Zamarchi, R.; Corradin, A.; et al. The Side Population of Ovarian Cancer Cells Is a Primary Target of IFN-α Antitumor Effects. Cancer Res. 2008, 68, 5658–5668. [Google Scholar] [CrossRef]
- Anastassiou, D. Universality of a Mesenchymal Transition Signature in Invasive Solid Cancers. Nat. Preced. 2012, 2–6. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-Cell Invigoration to Tumor Burden Ration Associated with Anti-PD-1 Response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis Is Regulated via MicroRNA-200/ZEB1 Axis Control of Tumour Cell PD-L1 Expression and Intratumoral Immunosuppression. Nat. Commun. 2014, 5, 611–629. [Google Scholar] [CrossRef]
- Guo, Y.; Lu, X.; Chen, Y.; Rendon, B.; Mitchell, R.A.; Cuatrecasas, M.; Cortés, M.; Postigo, A.; Liu, Y.; Dean, D.C. Zeb1 Induces Immune Checkpoints to Form an Immunosuppressive Envelope around Invading Cancer Cells. Sci. Adv. 2021, 7, eabd7455. [Google Scholar] [CrossRef]
- Guan, T.; Dominguez, C.X.; Amezquita, R.A.; Laidlaw, B.J.; Cheng, J.; Henao-Mejia, J.; Williams, A.; Flavell, R.A.; Lu, J.; Kaech, S.M. ZEB1, ZEB2, and the MiR-200 Family Form a Counterregulatory Network to Regulate CD8+ T Cell Fates. J. Exp. 2018, 215, 1153–1168. [Google Scholar] [CrossRef]
- Drápela, S.; Bouchal, J.; Jolly, M.K.; Culig, Z.; Souček, K. ZEB1: A Critical Regulator of Cell Plasticity, DNA Damage Response, and Therapy Resistance. Front. Mol. Biosci. 2020, 7, 36. [Google Scholar] [CrossRef]
- Belguise, K.; Sonenshein, G.E. PKCθ Promotes C-Rel-Driven Mammary Tumorigenesis in Mice and Humans by Repressing Estrogen Receptor α Synthesis. J. Clin. Investig. 2007, 117, 4009–4021. [Google Scholar] [CrossRef][Green Version]
- Chu, P.-Y.; Chung-Heng Hsu, N.; Tai, H.-C.; Yeh, C.-M.; Lin, S.-H.; Hou, M.-F.; Yeh, K.-T. High Nuclear Protein Kinase C Theta Expression May Correlate with Disease Recurrence and Poor Survival in Oral Squamous Cell Carcinoma. Hum. Pathol. 2012, 43, 276–281. [Google Scholar] [CrossRef]
- Ou, W.B.; Zhu, M.J.; Demetri, G.D.; Fletcher, C.D.M.; Fletcher, J.A. Protein Kinase C-θ Regulates KIT Expression and Proliferation in Gastrointestinal Stromal Tumors. Oncogene 2008, 27, 5624–5634. [Google Scholar] [CrossRef]
- Byerly, J.H.; Port, E.R.; Irie, H.Y. PRKCQ Inhibition Enhances Chemosensitivity of Triple-Negative Breast Cancer by Regulating Bim. Breast Cancer Res. 2020, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Rao, S. Epigenetics and Immunotherapy: The Current State of Play. Mol. Immunol. 2017, 87, 227–239. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunn, J.; McCuaig, R.D.; Tan, A.H.Y.; Tu, W.J.; Wu, F.; Wagstaff, K.M.; Zafar, A.; Ali, S.; Diwakar, H.; Dahlstrom, J.E.; et al. Selective Targeting of Protein Kinase C (PKC)-θ Nuclear Translocation Reduces Mesenchymal Gene Signatures and Reinvigorates Dysfunctional CD8+ T Cells in Immunotherapy-Resistant and Metastatic Cancers. Cancers 2022, 14, 1596. https://doi.org/10.3390/cancers14061596
Dunn J, McCuaig RD, Tan AHY, Tu WJ, Wu F, Wagstaff KM, Zafar A, Ali S, Diwakar H, Dahlstrom JE, et al. Selective Targeting of Protein Kinase C (PKC)-θ Nuclear Translocation Reduces Mesenchymal Gene Signatures and Reinvigorates Dysfunctional CD8+ T Cells in Immunotherapy-Resistant and Metastatic Cancers. Cancers. 2022; 14(6):1596. https://doi.org/10.3390/cancers14061596
Chicago/Turabian StyleDunn, Jenny, Robert D. McCuaig, Abel H. Y. Tan, Wen Juan Tu, Fan Wu, Kylie M. Wagstaff, Anjum Zafar, Sayed Ali, Himanshu Diwakar, Jane E. Dahlstrom, and et al. 2022. "Selective Targeting of Protein Kinase C (PKC)-θ Nuclear Translocation Reduces Mesenchymal Gene Signatures and Reinvigorates Dysfunctional CD8+ T Cells in Immunotherapy-Resistant and Metastatic Cancers" Cancers 14, no. 6: 1596. https://doi.org/10.3390/cancers14061596
APA StyleDunn, J., McCuaig, R. D., Tan, A. H. Y., Tu, W. J., Wu, F., Wagstaff, K. M., Zafar, A., Ali, S., Diwakar, H., Dahlstrom, J. E., Bean, E. G., Forwood, J. K., Tsimbalyuk, S., Cross, E. M., Hardy, K., Bain, A. L., Ahern, E., Dolcetti, R., Mazzieri, R., ... Rao, S. (2022). Selective Targeting of Protein Kinase C (PKC)-θ Nuclear Translocation Reduces Mesenchymal Gene Signatures and Reinvigorates Dysfunctional CD8+ T Cells in Immunotherapy-Resistant and Metastatic Cancers. Cancers, 14(6), 1596. https://doi.org/10.3390/cancers14061596