
High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand

 ,
,  ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
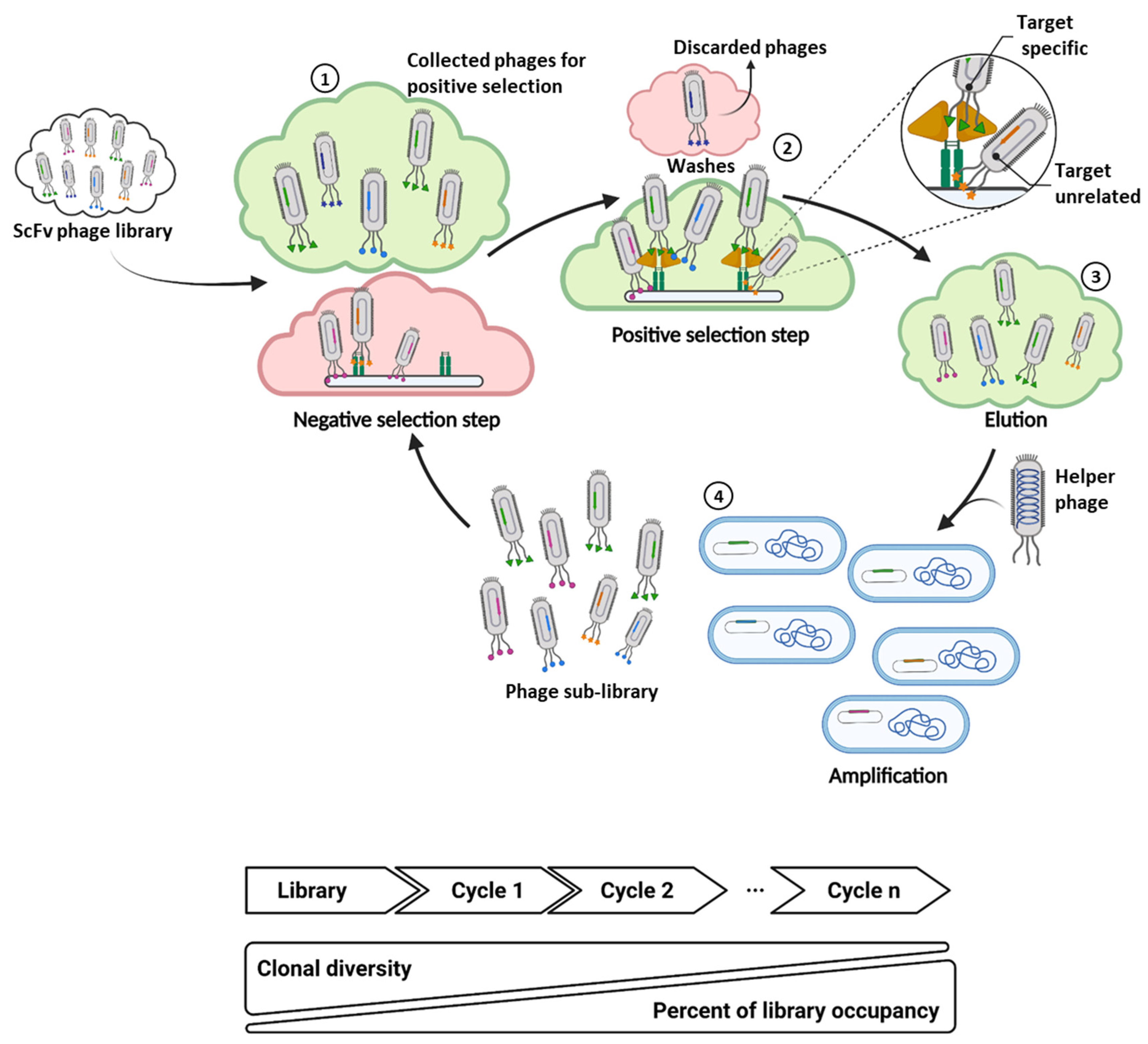
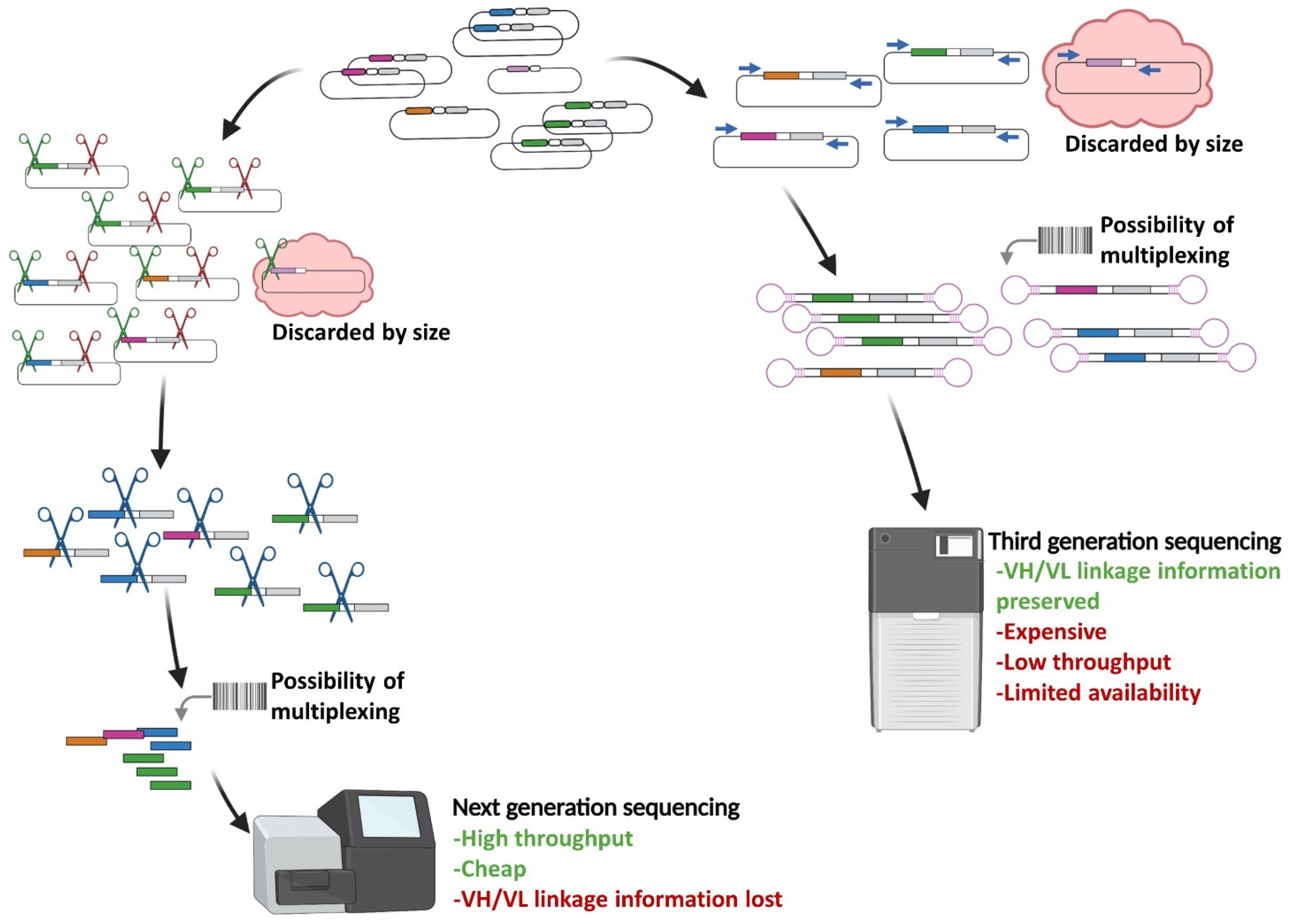
2. Implementation of Next Generation Sequencing to Phage Display scFv Library Screening
2.1. Sequencing-Guided Antibody Discovery: State-of-the-Art and Beyond
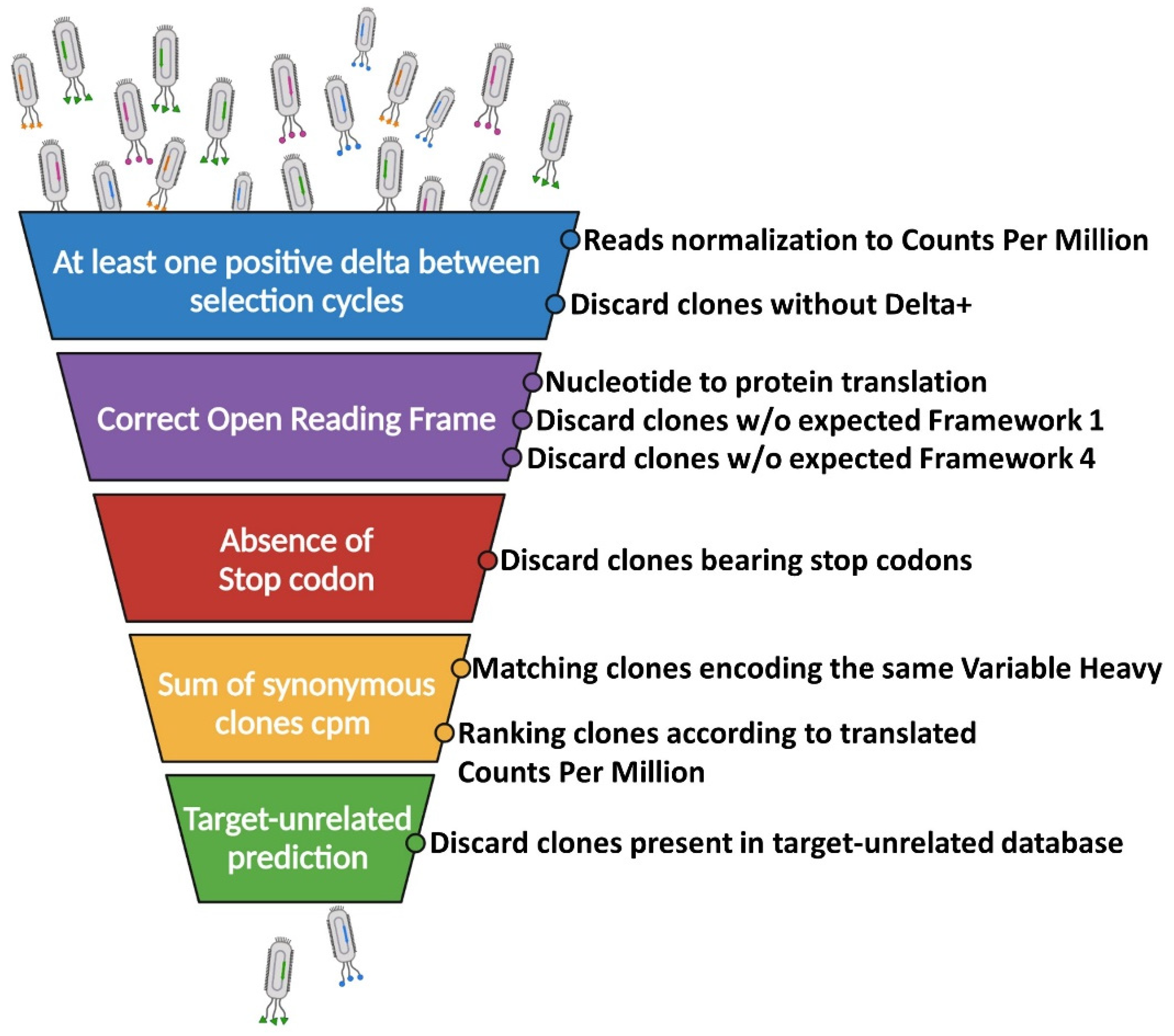
2.2. Filtering of Potential Binders: The Funnel Approach
2.2.1. Identification of Break Point Selection Cycle
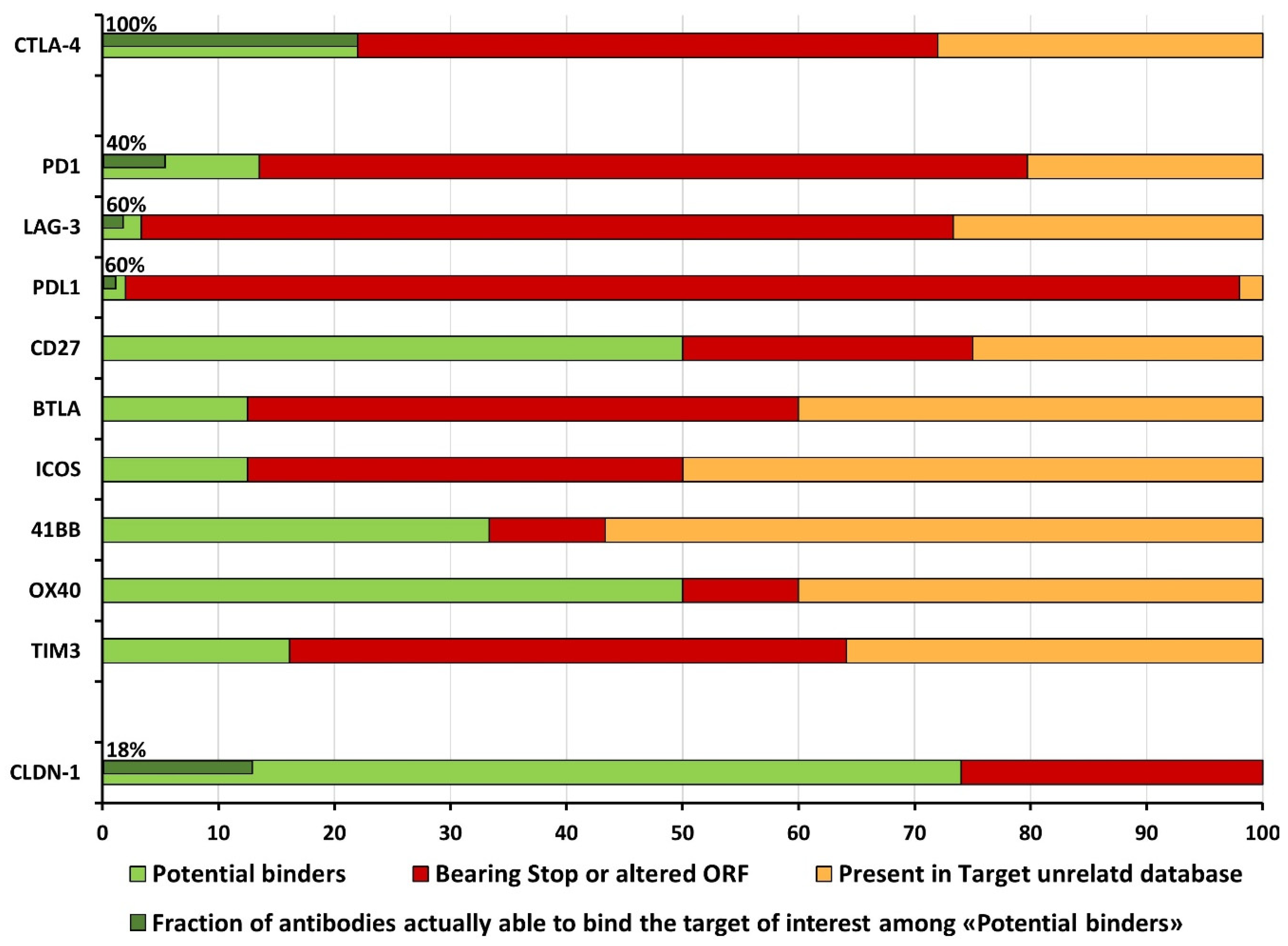
2.2.2. The Issue of False-Positive Clones: Target-Unrelated and Non-Valid Insert Identification
2.2.3. A Computational Strategy for Rapid Discovery of Potential scFv Binders
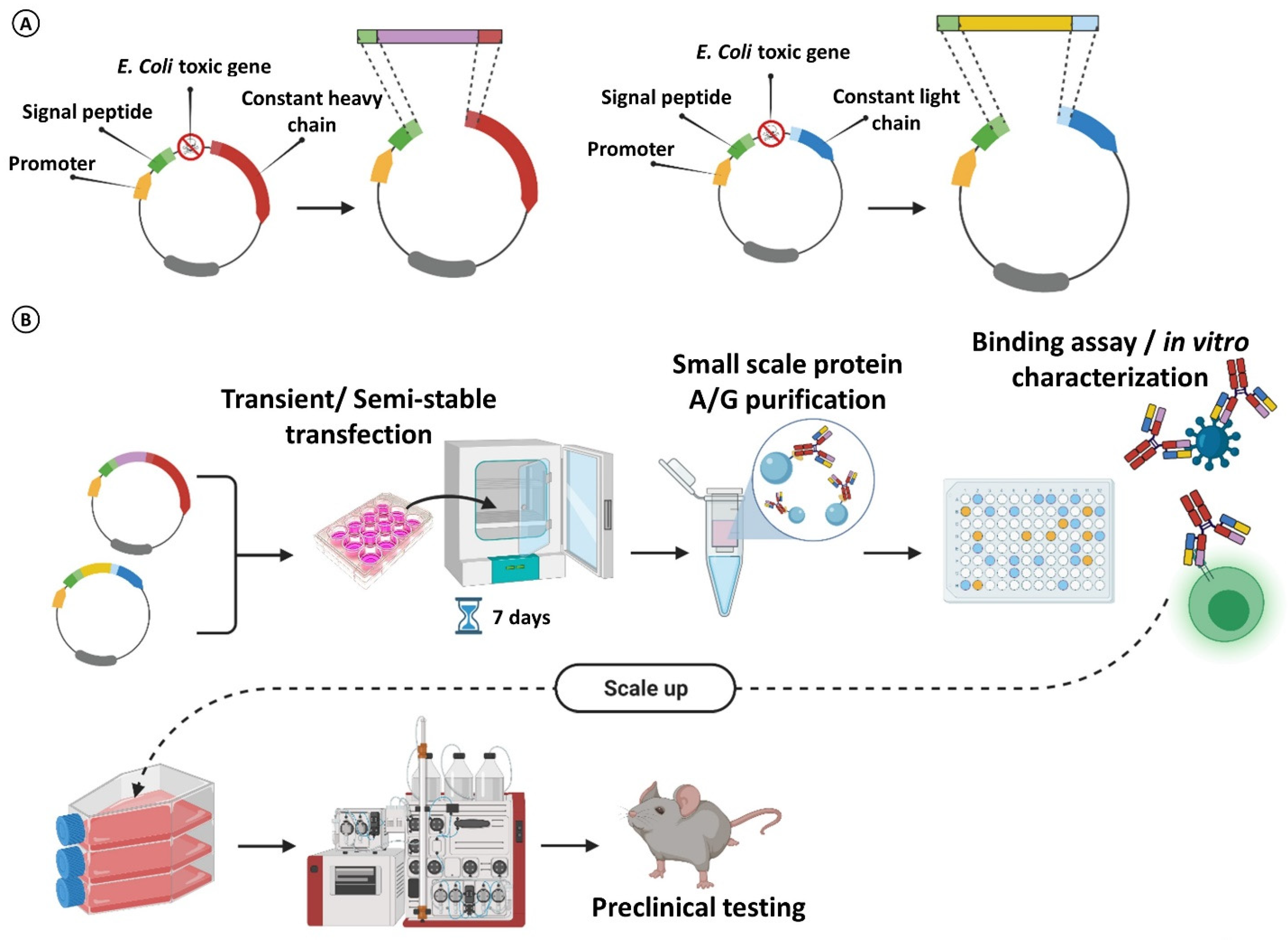
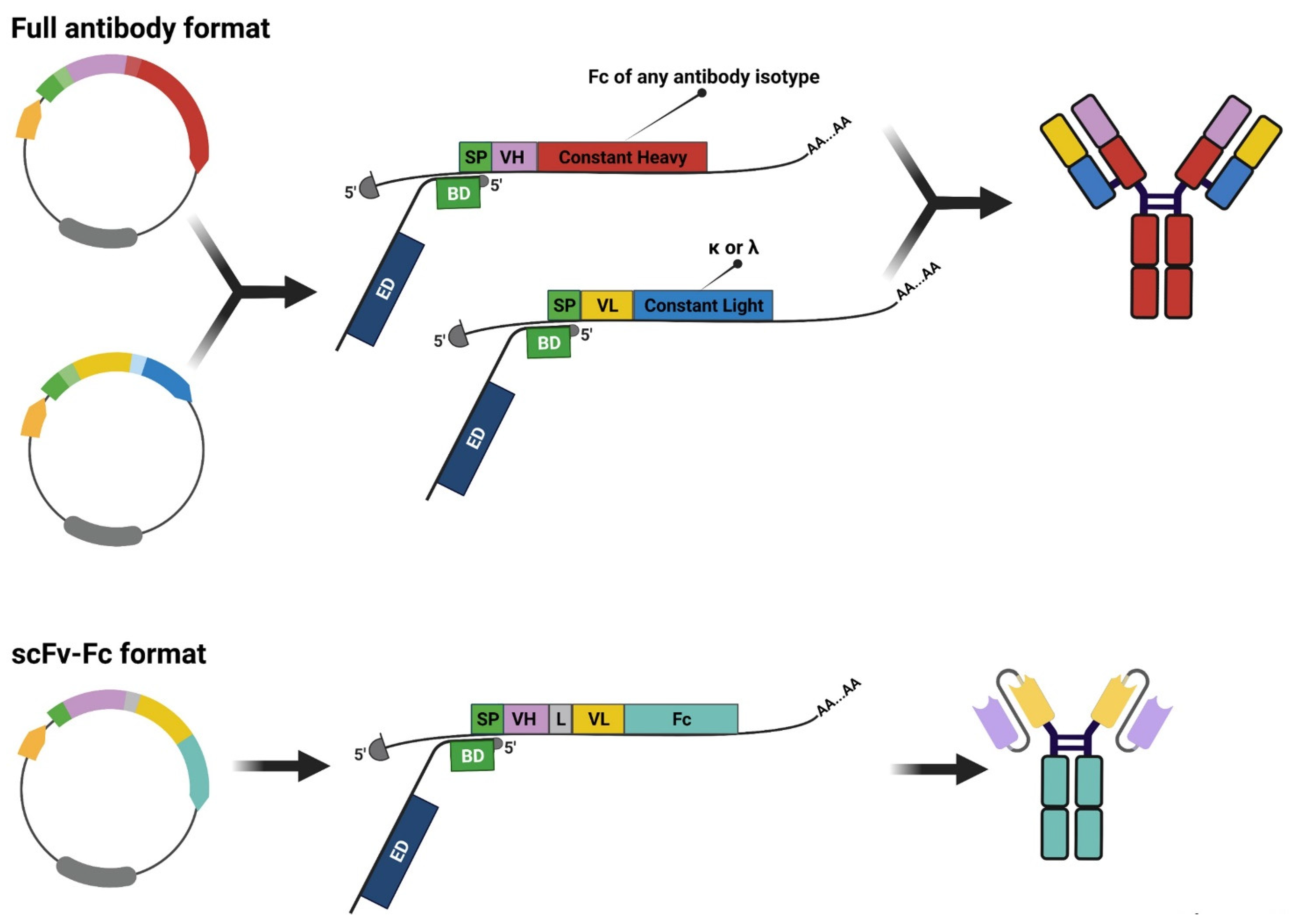
3. Rescuing and Cloning Strategies
4. Maximizing mAb Production for Massive Parallel Characterization
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. mAbs 2019, 12, 1703531. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Moorkens, E.; Vulto, A.G.; Huys, I. An overview of patents on therapeutic monoclonal antibodies in Europe: Are they a hurdle to biosimilar market entry? mAbs 2020, 12, 1743517. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.S.; Shereef, F.; Shareef, F. The influence of domestic manufacturing capabilities on biologic pricing in emerging economies. Nat. Biotechnol. 2019, 37, 498–501, Erratum in Nat. Biotechnol. 2019, 37, 1521. [Google Scholar] [CrossRef]
- Andreano, E.; Nicastri, E.; Paciello, I.; Pileri, P.; Manganaro, N.; Piccini, G.; Manenti, A.; Pantano, E.; Kabanova, A.; Troisi, M.; et al. Extremely potent human monoclonal antibodies from COVID-19 convalescent patients. Cell 2021, 184, 1821–1835.e16. [Google Scholar] [CrossRef]
- Jaworski, J.P. Neutralizing monoclonal antibodies for COVID-19 treatment and prevention. Biomed. J. 2021, 44, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Alape-Girón, A.; Moreira-Soto, A.; Arguedas, M.; Brenes, H.; Buján, W.; Corrales-Aguilar, E.; Díaz, C.; Echeverri, A.; Flores-Díaz, M.; Gómez, A.; et al. Heterologous hyperimmune polyclonal antibodies against SARS-CoV-2: A broad coverage, affordable, and scalable potential immunotherapy for COVID-19. Front. Med. 2021, 8, 743325. [Google Scholar] [CrossRef]
- Kelley, B.; Renshaw, T.; Kamarck, M. Process and operations strategies to enable global access to antibody therapies. Biotechnol. Prog. 2021, 37, e3139. [Google Scholar] [CrossRef]
- Sasso, E.; D’Alise, A.M.; Zambrano, N.; Scarselli, E.; Folgori, A.; Nicosia, A. New viral vectors for infectious diseases and cancer. Semin. Immunol. 2020, 50, 101430. [Google Scholar] [CrossRef]
- Andreano, E.; Seubert, A.; Rappuoli, R. Human monoclonal antibodies for discovery, therapy, and vaccine acceleration. Curr. Opin. Immunol. 2019, 59, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Parray, H.A.; Shukla, S.; Samal, S.; Shrivastava, T.; Ahmed, S.; Sharma, C.; Kumar, R. Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. Int. Immunopharmacol. 2020, 85, 106639. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Murawsky, C.M. Strategies for generating diverse antibody repertoires using transgenic animals expressing human antibodies. Front. Immunol. 2018, 9, 460. [Google Scholar] [CrossRef] [PubMed]
- Laffleur, B.; Pascal, V.; Sirac, C.; Cogné, M. Production of human or humanized antibodies in mice. Methods Mol. Biol. 2012, 901, 149–159. [Google Scholar] [CrossRef]
- Foster, R.H.; Wiseman, L.R. Abciximab. An updated review of its use in ischaemic heart disease. Drugs 1998, 56, 629–665, Erratum in Drugs 1999, 57, 91. [Google Scholar] [CrossRef]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Tsurushita, N.; Hinton, P.R.; Kumar, S. Design of humanized antibodies: From anti-Tac to Zenapax. Methods 2005, 36, 69–83. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef]
- Sheets, M.D.; Amersdorfer, P.; Finnern, R.; Sargent, P.; Lindqvist, E.; Schier, R.; Hemingsen, G.; Wong, C.; Gerhart, J.C.; Marks, J.D. Efficient construction of a large nonimmune phage antibody library: The production of high-affinity human single-chain antibodies to protein antigens. Proc. Natl. Acad. Sci. USA 1998, 95, 6157–6162, Erratum in Proc. Natl. Acad. Sci. USA 1999, 96, 795. [Google Scholar] [CrossRef] [PubMed]
- Valldorf, B.; Hinz, S.C.; Russo, G.; Pekar, L.; Mohr, L.; Klemm, J.; Doerner, A.; Krah, S.; Hust, M.; Zielonka, S. Antibody display technologies: Selecting the cream of the crop. Biol. Chem. 2021. [Google Scholar] [CrossRef]
- Dufner, P.; Jermutus, L.; Minter, R.R. Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 2006, 24, 523–529. [Google Scholar] [CrossRef]
- Cembrola, B.; Ruzza, V.; Troise, F.; Esposito, M.L.; Sasso, E.; Cafaro, V.; Passariello, M.; Visconte, F.; Raia, M.; Del Vecchio, L.; et al. Rapid affinity maturation of novel anti-PD-L1 antibodies by a fast drop of the antigen concentration and FACS selection of yeast libraries. Biomed. Res. Int. 2019, 2019, 6051870. [Google Scholar] [CrossRef]
- Barderas, R.; Desmet, J.; Timmerman, P.; Meloen, R.; Casal, J.I. Affinity maturation of antibodies assisted byin silicomodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 9029–9034. [Google Scholar] [CrossRef]
- Noy-Porat, T.; Makdasi, E.; Alcalay, R.; Mechaly, A.; Levy, Y.; Bercovich-Kinori, A.; Zauberman, A.; Tamir, H.; Yahalom-Ronen, Y.; Israeli, M.; et al. A panel of human neutralizing mAbs targeting SARS-CoV-2 spike at multiple epitopes. Nat. Commun. 2020, 11, 4303. [Google Scholar] [CrossRef]
- Hust, M.; Meyer, T.; Voedisch, B.; Rülker, T.; Thie, H.; El-Ghezal, A.; Kirsch, M.I.; Schütte, M.; Helmsing, S.; Meier, D.; et al. A human scFv antibody generation pipeline for proteome research. J. Biotechnol. 2011, 152, 159–170. [Google Scholar] [CrossRef]
- de Haard, H.J.W.; van Neer, N.; Reurs, A.; Hufton, S.E.; Roovers, R.C.; Henderikx, P.; de Bruïne, A.P.; Arends, J.-W.; Hoogenboom, H.R. A large non-immunized human fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J. Biol. Chem. 1999, 274, 18218–18230. [Google Scholar] [CrossRef]
- Gonzalez-Sapienza, G.; Rossotti, M.A.; Rosa, S.T.-D. Single-domain antibodies as versatile affinity reagents for analytical and diagnostic applications. Front. Immunol. 2017, 8, 977. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Pekar, L.; Bauer, C.; Ciesielski, E.; Krah, S.; Becker, S.; Toleikis, L.; Kügler, J.; Frenzel, A.; Valldorf, B.; et al. A one-step process for the construction of phage display scFv and VHH libraries. Mol. Biotechnol. 2020, 62, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Sasso, E.; D’Avino, C.; Passariello, M.; D’Alise, A.M.; Siciliano, D.; Esposito, M.L.; Froechlich, G.; Cortese, R.; Scarselli, E.; Zambrano, N.; et al. Massive parallel screening of phage libraries for the generation of repertoires of human immunomodulatory monoclonal antibodies. mAbs 2018, 10, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Johns, M.; George, A.J.; Ritter, M.A. In vivo selection of sFv from phage display libraries. J. Immunol. Methods 2000, 239, 137–151. [Google Scholar] [CrossRef]
- Newton-Northup, J.R.; Dickerson, M.T.; Kumar, S.R.; Smith, G.P.; Quinn, T.P.; Deutscher, S.L. In vivo bacteriophage peptide display to tailor pharmacokinetics of biological nanoparticles. Mol. Imaging Biol. 2014, 16, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage display derived monoclonal antibodies: From bench to bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef]
- Pecetta, S.; Finco, O.; Seubert, A. Quantum leap of monoclonal antibody (mAb) discovery and development in the COVID-19 era. Semin. Immunol. 2020, 50, 101427. [Google Scholar] [CrossRef]
- Passariello, M.; Gentile, C.; Ferrucci, V.; Sasso, E.; Vetrei, C.; Fusco, G.; Viscardi, M.; Brandi, S.; Cerino, P.; Zambrano, N.; et al. Novel human neutralizing mAbs specific for Spike-RBD of SARS-CoV-2. Sci. Rep. 2021, 11, 11046. [Google Scholar] [CrossRef]
- Paciello, R.; Urbanowicz, R.; Riccio, G.; Sasso, E.; McClure, P.; Zambrano, N.; Ball, J.; Cortese, R.; Nicosia, A.; De Lorenzo, C. Novel human anti-claudin 1 mAbs inhibit hepatitis C virus infection and may synergize with anti-SRB1 mAb. J. Gen. Virol. 2016, 97, 82–94. [Google Scholar] [CrossRef]
- Goydel, R.S.; Rader, C. Antibody-based cancer therapy. Oncogene 2021, 40, 3655–3664. [Google Scholar] [CrossRef]
- Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; De Lucia, M.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the antiviral STING-mediated DNA sensing in tumor cells is required to sustain the immunotherapeutic efficacy of herpes simplex oncolytic virus. Cancers 2020, 12, 3407. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Funchain, P.; Song, J.M.; Rayman, P.; Tannenbaum, C.; Ko, J.; McNamara, M.; Diaz-Montero, C.M.; Gastman, B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: A case series. J. Immunother. Cancer 2018, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Sasso, E.; Froechlich, G.; Cotugno, G.; D’Alise, A.M.; Gentile, C.; Bignone, V.; De Lucia, M.; Petrovic, B.; Campadelli-Fiume, G.; Scarselli, E.; et al. Replicative conditioning of herpes simplex type 1 virus by survivin promoter, combined to ERBB2 retargeting, improves tumour cell-restricted oncolysis. Sci. Rep. 2020, 10, 4307. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and multi-cytokine-armed herpes virus is a potent cancer endovaccine for local and systemic anti-tumor treatment. Mol. Ther. Oncolytics 2020, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Chicas-Sett, R.; Zafra-Martin, J.; Morales-Orue, I.; Castilla-Martinez, J.; Berenguer-Frances, M.A.; Gonzalez-Rodriguez, E.; Rodriguez-Abreu, D.; Couñago, F. Immunoradiotherapy as an effective therapeutic strategy in lung cancer: From palliative care to curative intent. Cancers 2020, 12, 2178. [Google Scholar] [CrossRef]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition—Novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 2021, 18, 199–214. [Google Scholar] [CrossRef]
- Passariello, M.; Vetrei, C.; Sasso, E.; Froechlich, G.; Gentile, C.; D’Alise, A.; Zambrano, N.; Scarselli, E.; Nicosia, A.; De Lorenzo, C. Isolation of two novel human anti-CTLA-4 mAbs with intriguing biological properties on tumor and NK cells. Cancers 2020, 12, 2204. [Google Scholar] [CrossRef]
- Fenwick, C.; Loredo-Varela, J.-L.; Joo, V.; Pellaton, C.; Farina, A.; Rajah, N.; Esteves-Leuenberger, L.; Decaillon, T.; Suffiotti, M.; Noto, A.; et al. Tumor suppression of novel anti–PD-1 antibodies mediated through CD28 costimulatory pathway. J. Exp. Med. 2019, 216, 1525–1541. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef]
- Blind, M.; Blank, M. Aptamer selection technology and recent advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef] [PubMed]
- Bakhshinejad, B.; Zade, H.M.; Shekarabi, H.S.Z.; Neman, S. Phage display biopanning and isolation of target-unrelated peptides: In search of nonspecific binders hidden in a combinatorial library. Amino Acids 2016, 48, 2699–2716. [Google Scholar] [CrossRef] [PubMed]
- Vodnik, M.; Zager, U.; Strukelj, B.; Lunder, M. Phage display: Selecting straws instead of a needle from a haystack. Molecules 2011, 16, 790–817. [Google Scholar] [CrossRef] [PubMed]
- Hoen, P.A.C.T.; Jirka, S.M.G.; Broeke, B.R.T.; Schultes, E.A.; Aguilera, B.; Pang, K.H.; Heemskerk, H.; Aartsma-Rus, A.; van Ommen, G.J.; Dunnen, J.T.D. Phage display screening without repetitious selection rounds. Anal. Biochem. 2012, 421, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yoon, A.; Lee, S.; Kim, S.; Han, J.; Chung, J. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp. Mol. Med. 2017, 49, e308. [Google Scholar] [CrossRef]
- Ljungars, A.; Svensson, C.; Carlsson, A.; Birgersson, E.; Tornberg, U.-C.; Frendéus, B.; Ohlin, M.; Mattsson, M. Deep mining of complex antibody phage pools generated by cell panning enables discovery of rare antibodies binding new targets and epitopes. Front. Pharmacol. 2019, 10, 847. [Google Scholar] [CrossRef]
- Rouet, R.; Jackson, K.J.L.; Langley, D.B.; Christ, D. Next-Generation Sequencing of Antibody Display Repertoires. Front. Immunol. 2018, 9, 118. [Google Scholar] [CrossRef]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; De Marco, A.; Ligat, L.; Rain, J.-C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife 2016, 5, e16228. [Google Scholar] [CrossRef]
- Ravn, U.; Gueneau, F.; Baerlocher, L.; Osteras, M.; Desmurs, M.; Malinge, P.; Magistrelli, G.; Farinelli, L.; Kosco-Vilbois, M.; Fischer, N. By-passing in vitro screening—Next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010, 38, e193. [Google Scholar] [CrossRef]
- He, L.; Lin, X.; De Val, N.; Saye-Francisco, K.L.; Mann, C.J.; Augst, R.; Morris, C.D.; Azadnia, P.; Zhou, B.; Sok, D.; et al. Hidden lineage complexity of glycan-dependent HIV-1 broadly neutralizing antibodies uncovered by digital panning and native-like gp140 trimer. Front. Immunol. 2017, 8, 1025. [Google Scholar] [CrossRef]
- Sasso, E.; Paciello, R.; D’Auria, F.; Riccio, G.; Froechlich, G.; Cortese, R.; Nicosia, A.; De Lorenzo, C.; Zambrano, N. One-step recovery of scFv clones from high-throughput sequencing-based screening of phage display libraries challenged to cells expressing native claudin-1. Biomed. Res. Int. 2015, 2015, 703213. [Google Scholar] [CrossRef]
- Hu, D.; Hu, S.; Wan, W.; Xu, M.; Du, R.; Zhao, W.; Gao, X.; Liu, J.; Liu, H.; Hong, J. Effective optimization of antibody affinity by phage display integrated with high-throughput DNA synthesis and sequencing technologies. PLoS ONE 2015, 10, e0129125. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.; Pandolfini, L.; Lisi, S.; Chirichella, M.; Arisi, I.; Terrigno, M.; Goracci, M.; Cremisi, F.; Cattaneo, A. Assessment of antibody library diversity through next generation sequencing and technical error compensation. PLoS ONE 2017, 12, e0177574. [Google Scholar] [CrossRef]
- Barreto, K.; Maruthachalam, B.V.; Hill, W.; Hogan, D.; Sutherland, A.R.; Kusalik, A.; Fonge, H.; DeCoteau, J.F.; Geyer, C.R. Next-generation sequencing-guided identification and reconstruction of antibody CDR combinations from phage selection outputs. Nucleic Acids Res. 2019, 47, e50. [Google Scholar] [CrossRef] [PubMed]
- Gavrielatos, M.; Kyriakidis, K.; Spandidos, D.A.; Michalopoulos, I. Benchmarking of next and third generation sequencing technologies and their associated algorithms for de novo genome assembly. Mol. Med. Rep. 2021, 23, 251. [Google Scholar] [CrossRef]
- Hemadou, A.; Giudicelli, V.; Smith, M.L.; Lefranc, M.-P.; Duroux, P.; Kossida, S.; Heiner, C.; Hepler, N.L.; Kuijpers, J.; Groppi, A.; et al. Pacific biosciences sequencing and IMGT/HighV-QUEST analysis of full-length single chain fragment variable from an in vivo selected phage-display combinatorial library. Front. Immunol. 2017, 8, 1796. [Google Scholar] [CrossRef] [PubMed]
- Han, S.Y.; Antoine, A.; Howard, D.; Chang, B.; Chang, W.S.; Slein, M.; Deikus, G.; Kossida, S.; Duroux, P.; Lefranc, M.-P.; et al. Coupling of single molecule, long read sequencing with IMGT/HighV-QUEST analysis expedites identification of SIV gp140-specific antibodies from scFv phage display libraries. Front. Immunol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Nannini, F.; Senicar, L.; Parekh, F.; Kong, K.J.; Kinna, A.; Bughda, R.; Sillibourne, J.; Hu, X.; Ma, B.; Bai, Y.; et al. Combining phage display with SMRTbell next-generation sequencing for the rapid discovery of functional scFv fragments. mAbs 2021, 13, 1864084. [Google Scholar] [CrossRef]
- Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A Highly functional synthetic phage display library containing over 40 billion human antibody clones. PLoS ONE 2014, 9, e100000. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Adamkiewicz, M.A.; Hebert, L.E.; Zygiel, E.M.; Boyle, H.R.; Martone, C.M.; Meléndez-Ríos, C.B.; Noren, K.A.; Noren, C.J.; Hall, M.F. Identification and characterization of mutant clones with enhanced propagation rates from phage-displayed peptide libraries. Anal. Biochem. 2014, 462, 35–43. [Google Scholar] [CrossRef]
- Stellwagen, S.D.; Sarkes, D.A.; Adams, B.L.; Hunt, M.A.; Renberg, R.L.; Hurley, M.M.; Stratis-Cullum, D.N. The next generation of biopanning: Next gen sequencing improves analysis of bacterial display libraries. BMC Biotechnol. 2019, 19, 100. [Google Scholar] [CrossRef] [PubMed]
- Cárcamo, J.; Ravera, M.W.; Brissette, R.; Dedova, O.; Beasley, J.R.; Alam-Moghé, A.; Wan, C.; Blume, A.; Mandecki, W. Unexpected frameshifts from gene to expressed protein in a phage-displayed peptide library. Proc. Natl. Acad. Sci. USA 1998, 95, 11146–11151. [Google Scholar] [CrossRef] [PubMed]
- Høydahl, L.S.; Nilssen, N.R.; Gunnarsen, K.S.; Du Pré, M.F.; Iversen, R.; Roos, N.; Chen, X.; Michaelsen, T.E.; Sollid, L.M.; Sandlie, I.; et al. Multivalent pIX phage display selects for distinct and improved antibody properties. Sci. Rep. 2016, 6, 39066. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.A.; Cox, F.; Van Der Horst, M.; Oudenrijn, S.V.D.; Res, P.C.M.; Bia, J.; Logtenberg, T.; De Kruif, J. A novel helper phage that improves phage display selection efficiency by preventing the amplification of phages without recombinant protein. Nucleic Acids Res. 2003, 31, e59. [Google Scholar] [CrossRef]
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.C.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240. [Google Scholar] [CrossRef]
- Camacho, F.; Sarmiento, M.E.; Reyes, F.; Kim, L.; Huggett, J.; Lepore, M.; Otero, O.; Gilleron, M.; Puzo, G.; Norazmi, M.N.; et al. Selection of phage-displayed human antibody fragments specific for CD1b presenting the Mycobacterium tuberculosis glycolipid Ac2SGL. Int. J. Mycobacteriol. 2016, 5, 120–127. [Google Scholar] [CrossRef]
- Rusciano, G.; Sasso, E.; Capaccio, A.; Zambrano, N.; Sasso, A. Revealing membrane alteration in cells overexpressing CA IX and EGFR by Surface-Enhanced Raman Scattering. Sci. Rep. 2019, 9, 1832, Erratum in Sci. Rep. 2019, 9, 9001. [Google Scholar] [CrossRef]
- Weber, T.; Pscherer, S.; Gamerdinger, U.; Teigler-Schlegel, A.; Rutz, N.; Blau, W.; Rummel, M.; Gattenlöhner, S.; Tur, M.K. Parallel evaluation of cell-based phage display panning strategies: Optimized selection and depletion steps result in AML blast-binding consensus antibodies. Mol. Med. Rep. 2021, 24, 767. [Google Scholar] [CrossRef]
- Lim, C.C.; Woo, P.C.Y.; Lim, T.S. Development of a phage display panning strategy utilizing crude antigens: Isolation of MERS-CoV nucleoprotein human antibodies. Sci. Rep. 2019, 9, 6088. [Google Scholar] [CrossRef]
- Even-Desrumeaux, K.; Nevoltris, D.; Lavaut, M.N.; Alim, K.; Borg, J.-P.; Audebert, S.; Kerfelec, B.; Baty, D.; Chames, P. Masked Selection: A Straightforward and Flexible Approach for the Selection of Binders Against Specific Epitopes and Differentially Expressed Proteins by Phage Display. Mol. Cell. Proteom. 2014, 13, 653–665. [Google Scholar] [CrossRef]
- He, B.; Yang, S.; Long, J.; Chen, X.; Zhang, Q.; Gao, H.; Chen, H.; Huang, J. TUPDB: Target-Unrelated Peptide Data Bank. Interdiscip. Sci. Comput. Life Sci. 2021, 13, 426–432. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Dzisoo, A.M.; Derda, R.; Huang, J. Development and Application of Computational Methods in Phage Display Technology. Curr. Med. Chem. 2020, 26, 7672–7693. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ru, B.; Li, S.; Lin, H.; Guo, F.-B. SAROTUP: Scanner and Reporter of Target-Unrelated Peptides. J. Biomed. Biotechnol. 2010, 2010, 101932. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chai, G.; Duan, Y.; Yan, Z.; Qiu, L.; Zhang, H.; Liu, Z.; He, Q.; Han, K.; Ru, B.; et al. BDB: Biopanning data bank. Nucleic Acids Res. 2016, 44, D1127–D1132. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.D.; Golomb, M.; Smith, G.P. Corruption of phage display libraries by target-unrelated clones: Diagnosis and countermeasures. Anal. Biochem. 2010, 407, 237–240. [Google Scholar] [CrossRef]
- He, B.; Jiang, L.; Duan, Y.; Chai, G.; Fang, Y.; Kang, J.; Yu, M.; Li, N.; Tang, Z.; Yao, P.; et al. Biopanning data bank 2018: Hugging next generation phage display. Database 2018, 2018, bay032. [Google Scholar] [CrossRef]
- Spiliotopoulos, A.; Owen, J.; Maddison, B.; Dreveny, I.; Rees, H.; Gough, K. Sensitive recovery of recombinant antibody clones after their in silico identification within NGS datasets. J. Immunol. Methods 2015, 420, 50–55. [Google Scholar] [CrossRef]
- Li, F.; Liu, S.-L.; Mullins, J.I. Site-directed mutagenesis using uracil-containing double-stranded DNA templates and DpnI digestion. Biotechniques 1999, 27, 734–738. [Google Scholar] [CrossRef]
- Ferrara, F.; Teixeira, A.A.; Naranjo, L.; Erasmus, M.F.; D’Angelo, S.; Bradbury, A.R. Exploiting next-generation sequencing in antibody selections—A simple PCR method to recover binders. mAbs 2020, 12, 1701792. [Google Scholar] [CrossRef]
- Zhang, H.; Torkamani, A.; Jones, T.M.; Ruiz, D.I.; Pons, J.; Lerner, R.A. Phenotype-information-phenotype cycle for deconvolution of combinatorial antibody libraries selected against complex systems. Proc. Natl. Acad. Sci. USA 2011, 108, 13456–13461. [Google Scholar] [CrossRef]
- Noh, J.; Kim, O.; Jung, Y.; Han, H.; Kim, J.-E.; Kim, S.; Lee, S.; Park, J.; Jung, R.H.; Kim, S.I.; et al. High-throughput retrieval of physical DNA for NGS-identifiable clones in phage display library. mAbs 2019, 11, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Vaisman-Mentesh, A.; Wine, Y. Monitoring phage biopanning by next-generation sequencing. Methods Mol. Biol. 2018, 1701, 463–473. [Google Scholar] [CrossRef]
- Prabakaran, P.; Glanville, J.; Ippolito, G.C. Editorial: Next-generation sequencing of human antibody repertoires for exploring B-cell landscape, antibody discovery and vaccine development. Front. Immunol. 2020, 11, 1344. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef]
- Lanzavecchia, A. Dissecting human antibody responses: Useful, basic and surprising findings. EMBO Mol. Med. 2018, 10, e8879. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, C.; Tedesco, A.; Terrazzano, G.; Cozzolino, R.; Laccetti, P.; Piccoli, R.; D’Alessio, G. A human, compact, fully functional anti-ErbB2 antibody as a novel antitumour agent. Br. J. Cancer 2004, 91, 1200–1204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Lorenzo, C.; D’Alessio, G. Human anti-ErbB2 immunoagents-immunoRNases and compact antibodies. FEBS J. 2009, 276, 1527–1535. [Google Scholar] [CrossRef]
- Riccio, G.; Ricardo, A.R.; Passariello, M.; Saraiva, K.; Rubino, V.; Cunnah, P.; Mertens, N.; De Lorenzo, C. T-cell activating tribodies as a novel approach for efficient killing of ErbB2-positive cancer cells. J. Immunother. 2019, 42, 1–10. [Google Scholar] [CrossRef]
- Spidel, J.L.; Vaessen, B.; Chan, Y.Y.; Grasso, L.; Kline, J.B. Rapid high-throughput cloning and stable expression of antibodies in HEK293 cells. J. Immunol. Methods 2016, 439, 50–58. [Google Scholar] [CrossRef]
- Lund, B.A.; Leiros, H.-K.S.; Bjerga, G.K. A high-throughput, restriction-free cloning and screening strategy based on ccd B-gene replacement. Microb. Cell Fact. 2014, 13, 38. [Google Scholar] [CrossRef]
- Carrara, S.C.; Ulitzka, M.; Grzeschik, J.; Kornmann, H.; Hock, B.; Kolmar, H. From cell line development to the formulated drug product: The art of manufacturing therapeutic monoclonal antibodies. Int. J. Pharm. 2021, 594, 120164. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.G.; White, R.N.; Barnard, G.C. Development of a high cell density transient CHO platform yielding mAb titers greater than 2 g/L in only 7 days. Biotechnol. Prog. 2020, 36, e3047. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Lombardi, R.; Nevoltris, D.; Luthra, A.; Schofield, P.; Zimmermann, C.; Christ, D. Transient expression of human antibodies in mammalian cells. Nat. Protoc. 2018, 13, 99–117. [Google Scholar] [CrossRef]
- Jäger, V.; Büssow, K.; Wagner, A.; Weber, S.; Hust, M.; Frenzel, A.; Schirrmann, T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol. 2013, 13, 52. [Google Scholar] [CrossRef]
- Knight, T.J.; Turner, S.; Jaques, C.M.; Smales, C.M. Selection of CHO host and recombinant cell pools by inhibition of the proteasome results in enhanced product yields and cell specific productivity. J. Biotechnol. 2021, 337, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, J.F.; Arnall, C.L.; Patel, Y.D.; Barber, N.O.; Lovelady, C.S.; Rosignoli, G.; Harris, C.L.; Dunn, S.; Field, R.P.; Dean, G.; et al. A platform for context-specific genetic engineering of recombinant protein production by CHO cells. J. Biotechnol. 2020, 312, 11–22. [Google Scholar] [CrossRef]
- Vink, T.; Oudshoorn-Dickmann, M.; Roza, M.; Reitsma, J.-J.; de Jong, R.N. A simple, robust and highly efficient transient expression system for producing antibodies. Methods 2014, 65, 5–10. [Google Scholar] [CrossRef]
- Dumont, A.J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R.R. Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef]
- Gieselmann, L.; Kreer, C.; Ercanoglu, M.S.; Lehnen, N.; Zehner, M.; Schommers, P.; Potthoff, J.; Gruell, H.; Klein, F. Effective high-throughput isolation of fully human antibodies targeting infectious pathogens. Nat. Protoc. 2021, 16, 3639–3671. [Google Scholar] [CrossRef]
- Backliwal, G.; Hildinger, M.; Chenuet, S.; Wulhfard, S.; De Jesus, M.; Wurm, F.M. Rational vector design and multi-pathway modulation of HEK 293E cells yield recombinant antibody titers exceeding 1 g/l by transient transfection under serum-free conditions. Nucleic Acids Res. 2008, 36, e96. [Google Scholar] [CrossRef]
- Toki, N.; Takahashi, H.; Sharma, H.; Valentine, M.N.Z.; Rahman, F.-U.M.; Zucchelli, S.; Gustincich, S.; Carninci, P. SINEUP long non-coding RNA acts via PTBP1 and HNRNPK to promote translational initiation assemblies. Nucleic Acids Res. 2020, 48, 11626–11644. [Google Scholar] [CrossRef]
- Ohyama, T.; Takahashi, H.; Sharma, H.; Yamazaki, T.; Gustincich, S.; Ishii, Y.; Carninci, P. An NMR-based approach reveals the core structure of the functional domain of SINEUP lncRNAs. Nucleic Acids Res. 2020, 48, 9346–9360. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Sharma, H.; Carninci, P. Cell based assays of SINEUP non-coding RNAs that can specifically enhance mRNA translation. J. Vis. Exp. 2019, 144, e58627. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Patrucco, L.; Persichetti, F.; Gustincich, S.; Cotella, D. Engineering translation in mammalian cell factories to increase protein yield: The unexpected use of long non-coding SINEUP RNAs. Comput. Struct. Biotechnol. J. 2016, 14, 404–410. [Google Scholar] [CrossRef]
- Zucchelli, S.; Fasolo, F.; Russo, R.; Cimatti, L.; Patrucco, L.; Takahashi, H.; Jones, M.; Santoro, C.; Sblattero, D.; Cotella, D.; et al. SINEUPs are modular antisense long non-coding RNAs that increase synthesis of target proteins in cells. Front. Cell. Neurosci. 2015, 9, 174. [Google Scholar] [CrossRef]
- Espinoza, S.; Bon, C.; Valentini, P.; Pierattini, B.; Matey, A.T.; Damiani, D.; Pulcrano, S.; Sanges, R.; Persichetti, F.; Takahashi, H.; et al. SINEUPs: A novel toolbox for RNA therapeutics. Essays Biochem. 2021, 65, 775–789. [Google Scholar] [CrossRef]
- Espinoza, S.; Scarpato, M.; Damiani, D.; Managò, F.; Mereu, M.; Contestabile, A.; Peruzzo, O.; Carninci, P.; Santoro, C.; Papaleo, F.; et al. SINEUP non-coding RNA targeting GDNF rescues motor deficits and neurodegeneration in a mouse model of Parkinson’s disease. Mol. Ther. 2020, 28, 642–652. [Google Scholar] [CrossRef]
- Bon, C.; Luffarelli, R.; Russo, R.; Fortuni, S.; Pierattini, B.; Santulli, C.; Fimiani, C.; Persichetti, F.; Cotella, D.; Mallamaci, A.; et al. SINEUP non-coding RNAs rescue defective frataxin expression and activity in a cellular model of Friedreich’s Ataxia. Nucleic Acids Res. 2019, 47, 10728–10743. [Google Scholar] [CrossRef]
- Patrucco, L.; Chiesa, A.; Soluri, M.F.; Fasolo, F.; Takahashi, H.; Carninci, P.; Zucchelli, S.; Santoro, C.; Gustincich, S.; Sblattero, D.; et al. Engineering mammalian cell factories with SINEUP noncoding RNAs to improve translation of secreted proteins. Gene 2015, 569, 287–293. [Google Scholar] [CrossRef]
- Yao, Y.; Jin, S.; Long, H.; Yu, Y.; Zhang, Z.; Cheng, G.; Xu, C.; Ding, Y.; Guang-Jun, H.; Li, N.; et al. RNAe: An effective method for targeted protein translation enhancement by artificial non-coding RNA with SINEB2 repeat. Nucleic Acids Res. 2015, 43, e58. [Google Scholar] [CrossRef] [PubMed]
- Sasso, E.; Latino, D.; Froechlich, G.; Succoio, M.; Passariello, M.; De Lorenzo, C.; Nicosia, A.; Zambrano, N. A long non-coding SINEUP RNA boosts semi-stable production of fully human monoclonal antibodies in HEK293E cells. mAbs 2018, 10, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Hoseinpoor, R.; Kazemi, B.; Rajabibazl, M.; Rahimpour, A. Improving the expression of anti-IL-2Rα monoclonal antibody in the CHO cells through optimization of the expression vector and translation efficiency. J. Biotechnol. 2020, 324, 112–120, Erratum in J. Biotechnol. 2021, 325, 397. [Google Scholar] [CrossRef] [PubMed]
- Pasche, N.; Wulhfard, S.; Pretto, F.; Carugati, E.; Neri, D. The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clin. Cancer Res. 2012, 18, 4092–4103. [Google Scholar] [CrossRef]
- Neri, D. Antibody-cytokine fusions: Versatile products for the modulation of anticancer immunity. Cancer Immunol. Res. 2019, 7, 348–354. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-drug conjugates: A comprehensive review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Mitra, S.; Tomar, P.C. Hybridoma technology; advancements, clinical significance, and future aspects. J. Genet. Eng. Biotechnol. 2021, 19, 159. [Google Scholar] [CrossRef]
- Smith, E.G.P. Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14428–14437. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Steinwand, M.; Droste, P.; Frenzel, A.; Hust, M.; Dübel, S.; Schirrmann, T. The influence of antibody fragment format on phage display based affinity maturation of IgG. mAbs 2014, 6, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Bujak, E.; Matasci, M.; Neri, D.; Wulhfard, S. Reformatting of scFv Antibodies into the scFv-Fc format and their downstream purification. Methods Mol. Biol. 2014, 1131, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Hammers, C.M.; Stanley, J.R. Antibody Phage Display: Technique and Applications. J. Investig. Dermatol. 2014, 134, e17. [Google Scholar] [CrossRef] [PubMed]
- Song, L.-F.; Deng, Z.-H.; Gong, Z.-Y.; Li, L.-L.; Li, B.-Z. Large-scale de novo oligonucleotide synthesis for whole-genome synthesis and data storage: Challenges and opportunities. Front. Bioeng. Biotechnol. 2021, 9, 689797. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, M.; Wu, Y.; Wang, W.; Wang, R.; Du, M.; Ma, P.; Zhou, X.; Wang, Y.; Cao, Y.; et al. High-throughput reformatting of phage-displayed antibody fragments to IgGs by one-step emulsion PCR. Protein Eng. Des. Sel. 2018, 31, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Reader, R.H.; Workman, R.G.; Maddison, B.C.; Gough, K.C. Advances in the production and batch reformatting of phage antibody libraries. Mol. Biotechnol. 2019, 61, 801–815. [Google Scholar] [CrossRef]
- Dinnis, D.; James, D.C. Engineering mammalian cell factories for improved recombinant monoclonal antibody production: Lessons from nature? Biotechnol. Bioeng. 2005, 91, 180–189. [Google Scholar] [CrossRef]
- Saka, K.; Kakuzaki, T.; Metsugi, S.; Kashiwagi, D.; Yoshida, K.; Wada, M.; Tsunoda, H.; Teramoto, R. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci. Rep. 2021, 11, 5852. [Google Scholar] [CrossRef]
- Liu, G.; Zeng, H.; Mueller, J.; Carter, B.; Wang, Z.; Schilz, J.; Horny, G.; Birnbaum, M.E.; Ewert, S.; Gifford, D.K. Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics 2020, 36, 2126–2133. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, G.; Liu, X.; Qian, Q. Monoclonal antibodies as therapeutic agents in oncology and antibody gene therapy. Cell Res. 2007, 17, 89–99. [Google Scholar] [CrossRef]
- Patel, A.; Bah, M.A.; Weiner, D.B. In vivo delivery of nucleic acid-encoded monoclonal antibodies. BioDrugs 2020, 34, 273–293, Erratum in BioDrugs 2020, 34, 295. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, H.; Wirth, T.; Schenkwein, D.; Räty, J.K.; Ylä-Herttuala, S. Challenges in monoclonal antibody-based therapies. Ann. Med. 2009, 41, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Hollevoet, K.; Declerck, P.J. State of play and clinical prospects of antibody gene transfer. J. Transl. Med. 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NGS-Guided | Classical (ELISA) | |
|---|---|---|
| Yield of positive hits (Number of different target-specific scFvs) | Hundreds (Top enriched and rare clones) | Few up to tens (Only top enriched) |
| Specificity confidence (Fraction of scFvs confirmed as specific by biochemical assay) | High | Low |
| Handling | Rescue step needed to physically isolate clones | Repetitive ELISA assay |
| Sequence information; VH to VL matching | - Short reads limits sequence information to VH; VH/VL matching lost (Illumina) | Entire scFv sequence available after picking (Sanger) |
| - Entire scFv sequence and VH/VL matching available (Third-generation) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zambrano, N.; Froechlich, G.; Lazarevic, D.; Passariello, M.; Nicosia, A.; De Lorenzo, C.; Morelli, M.J.; Sasso, E. High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand. Cancers 2022, 14, 1325. https://doi.org/10.3390/cancers14051325
Zambrano N, Froechlich G, Lazarevic D, Passariello M, Nicosia A, De Lorenzo C, Morelli MJ, Sasso E. High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand. Cancers. 2022; 14(5):1325. https://doi.org/10.3390/cancers14051325
Chicago/Turabian StyleZambrano, Nicola, Guendalina Froechlich, Dejan Lazarevic, Margherita Passariello, Alfredo Nicosia, Claudia De Lorenzo, Marco J. Morelli, and Emanuele Sasso. 2022. "High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand" Cancers 14, no. 5: 1325. https://doi.org/10.3390/cancers14051325
APA StyleZambrano, N., Froechlich, G., Lazarevic, D., Passariello, M., Nicosia, A., De Lorenzo, C., Morelli, M. J., & Sasso, E. (2022). High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand. Cancers, 14(5), 1325. https://doi.org/10.3390/cancers14051325