
Engineered Microphysiological Systems for Testing Effectiveness of Cell-Based Cancer Immunotherapies


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
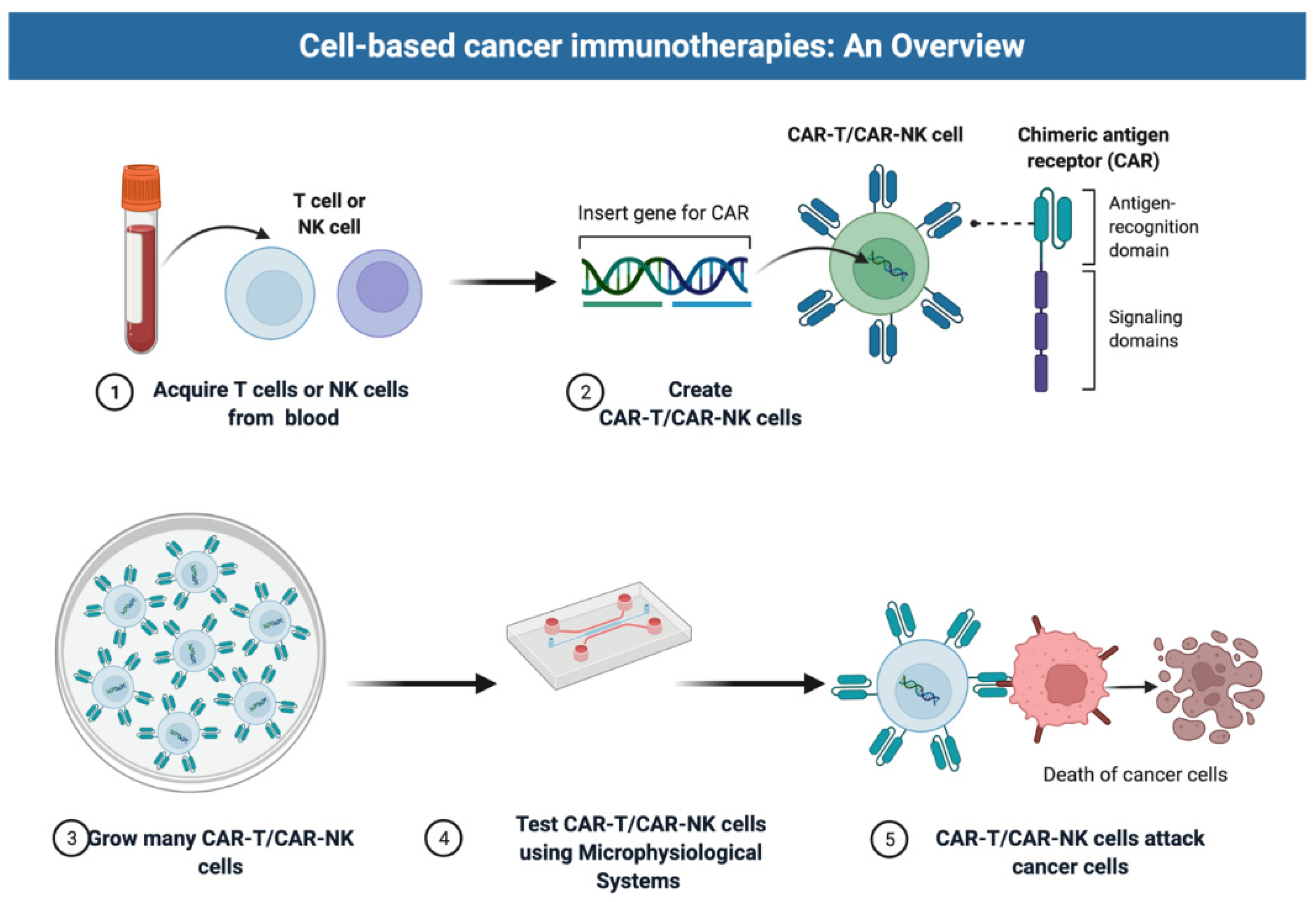
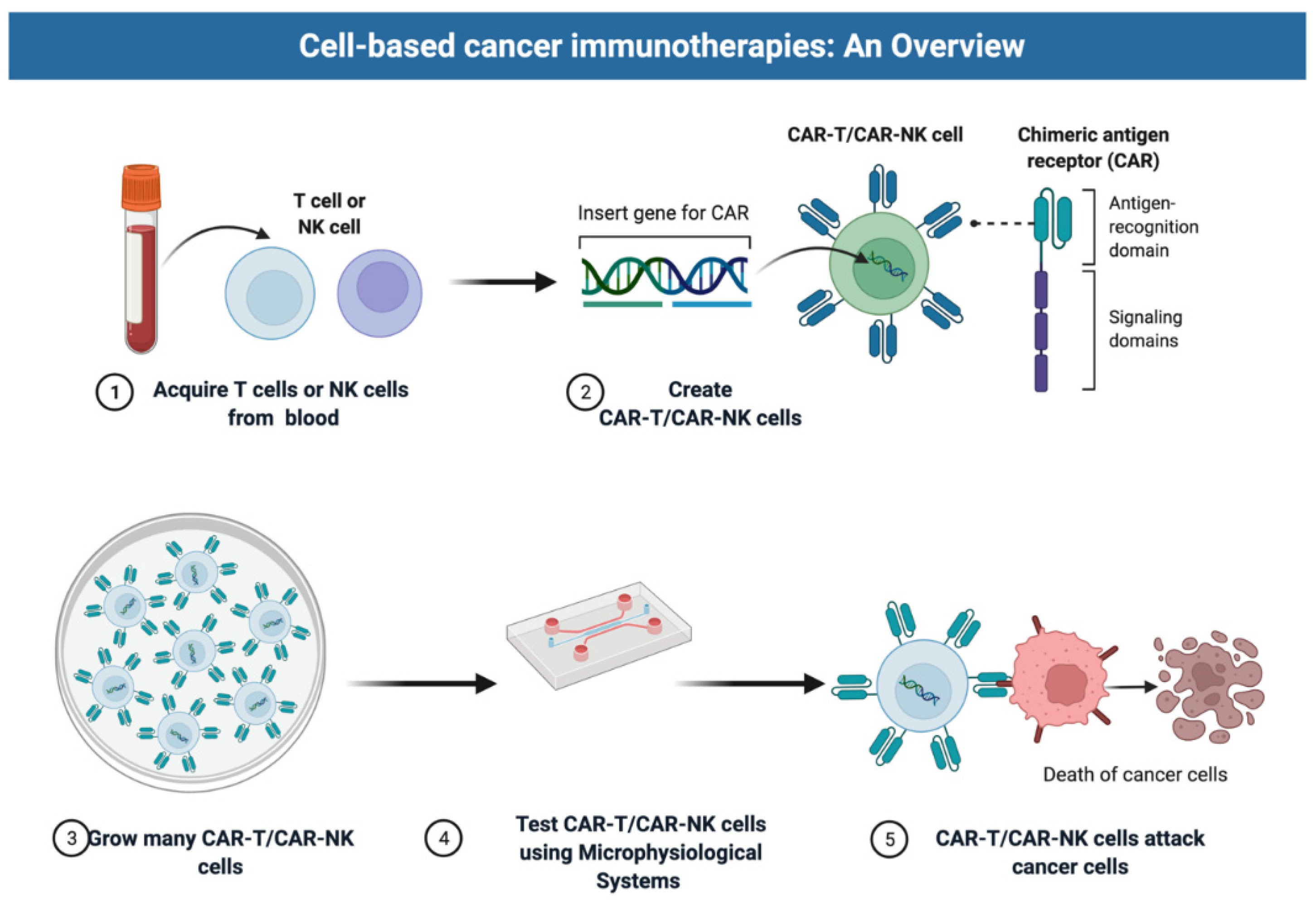
1. Introduction: Promise and Challenges of Cell Therapies
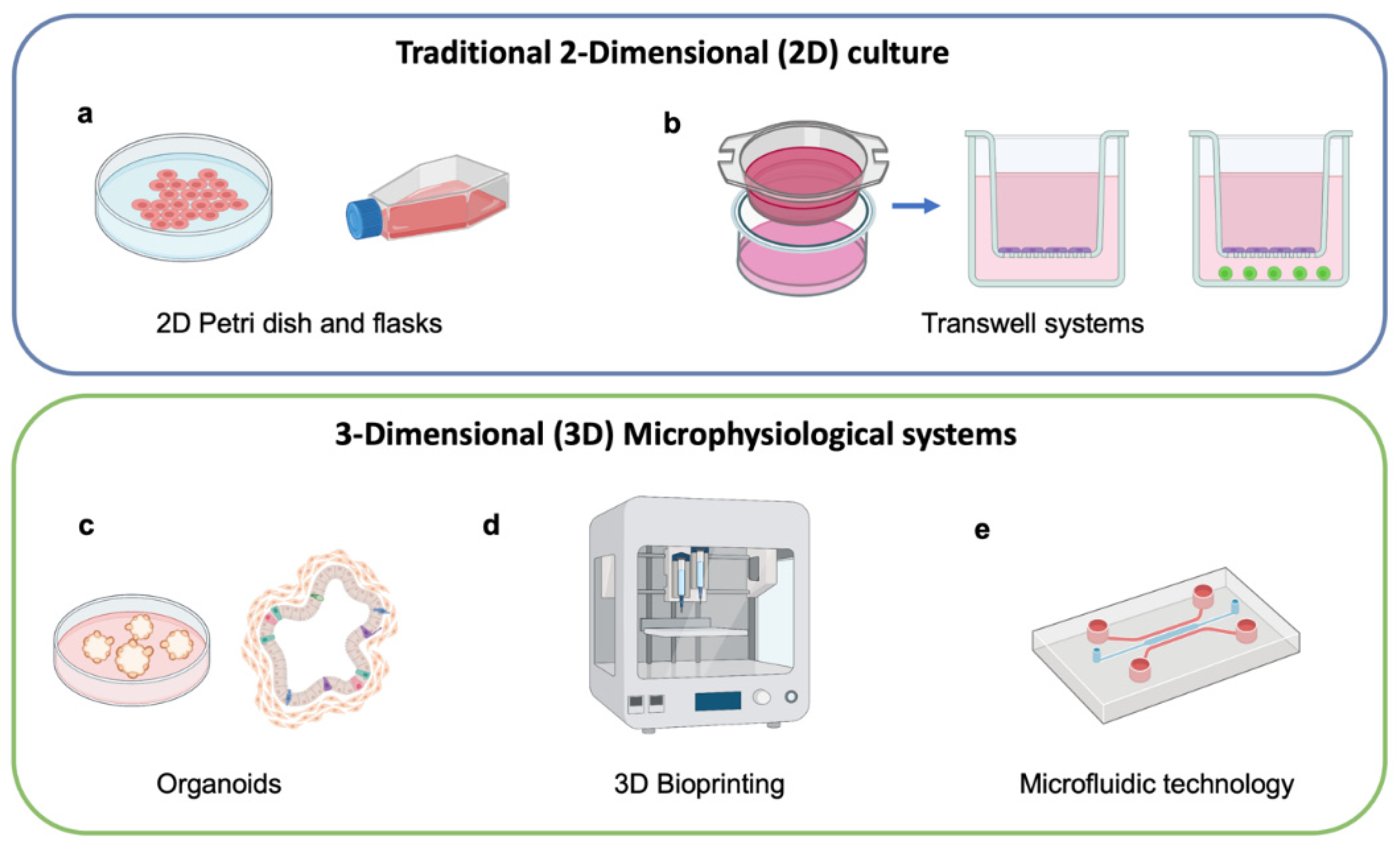
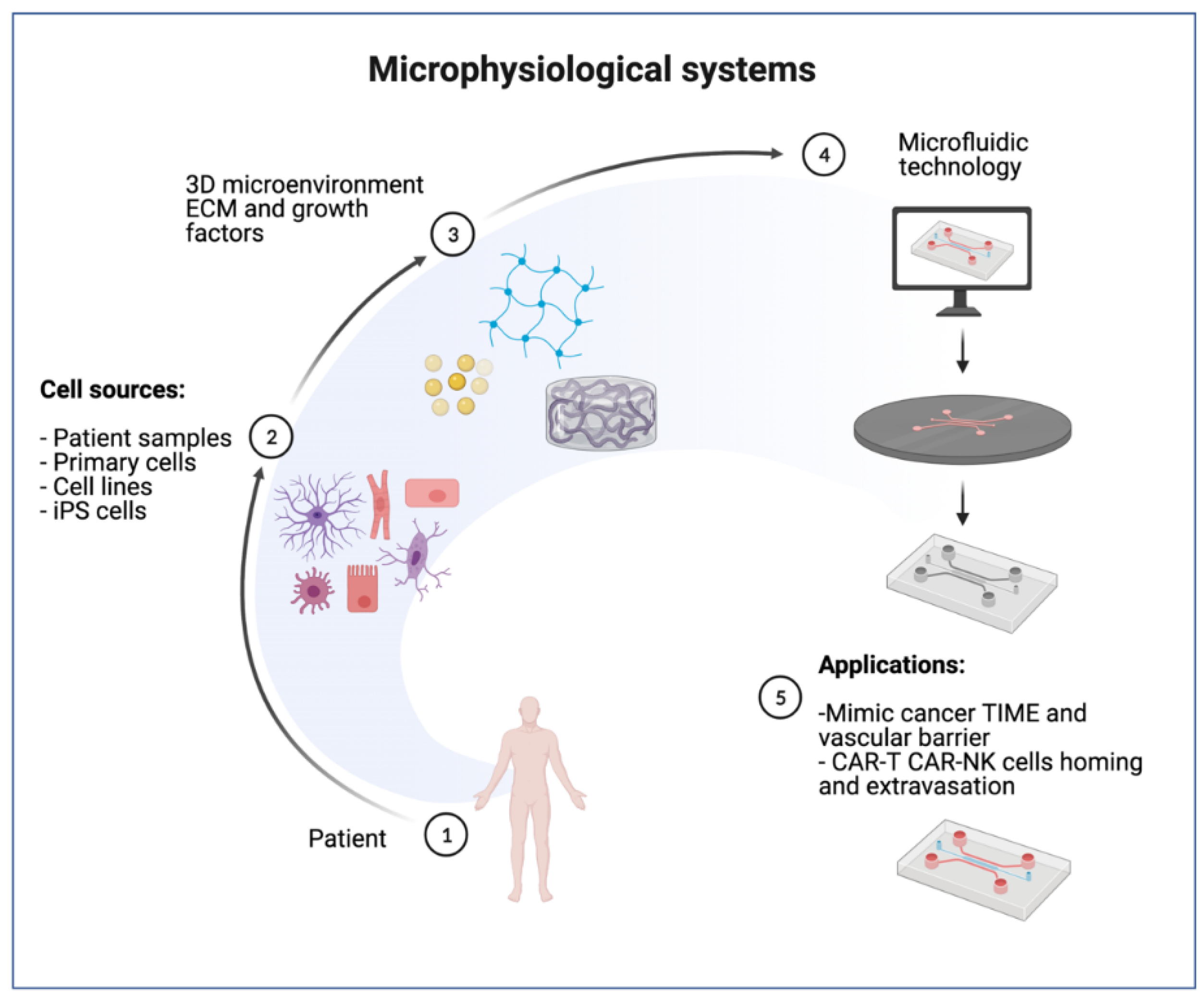
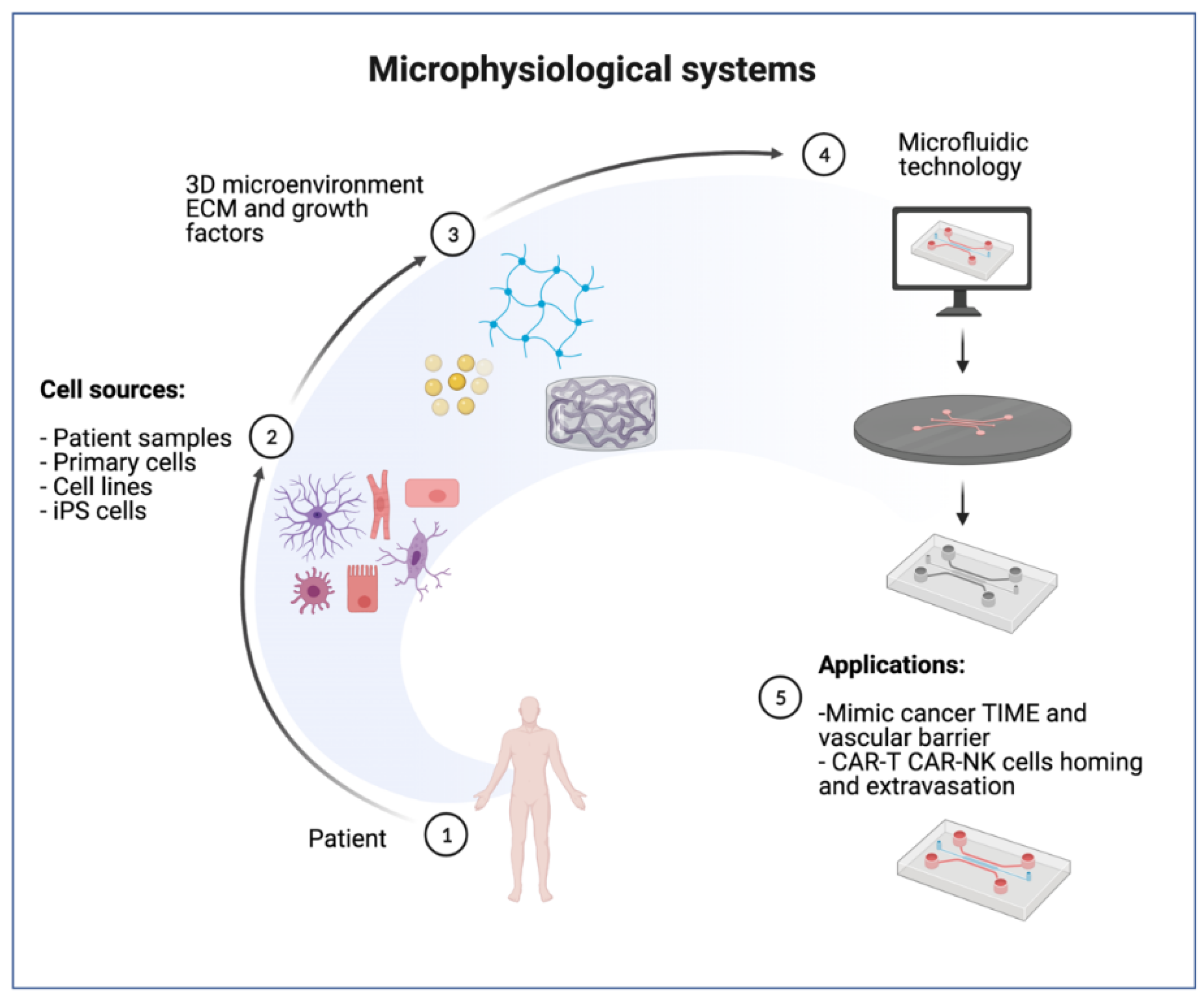
2. Microphysiological Models for Cell Therapy Testing
2.1. Selection of Cell Types
2.2. MPS Engineering with ECM
3. Advantages of Microphysiological Systems (MPS)
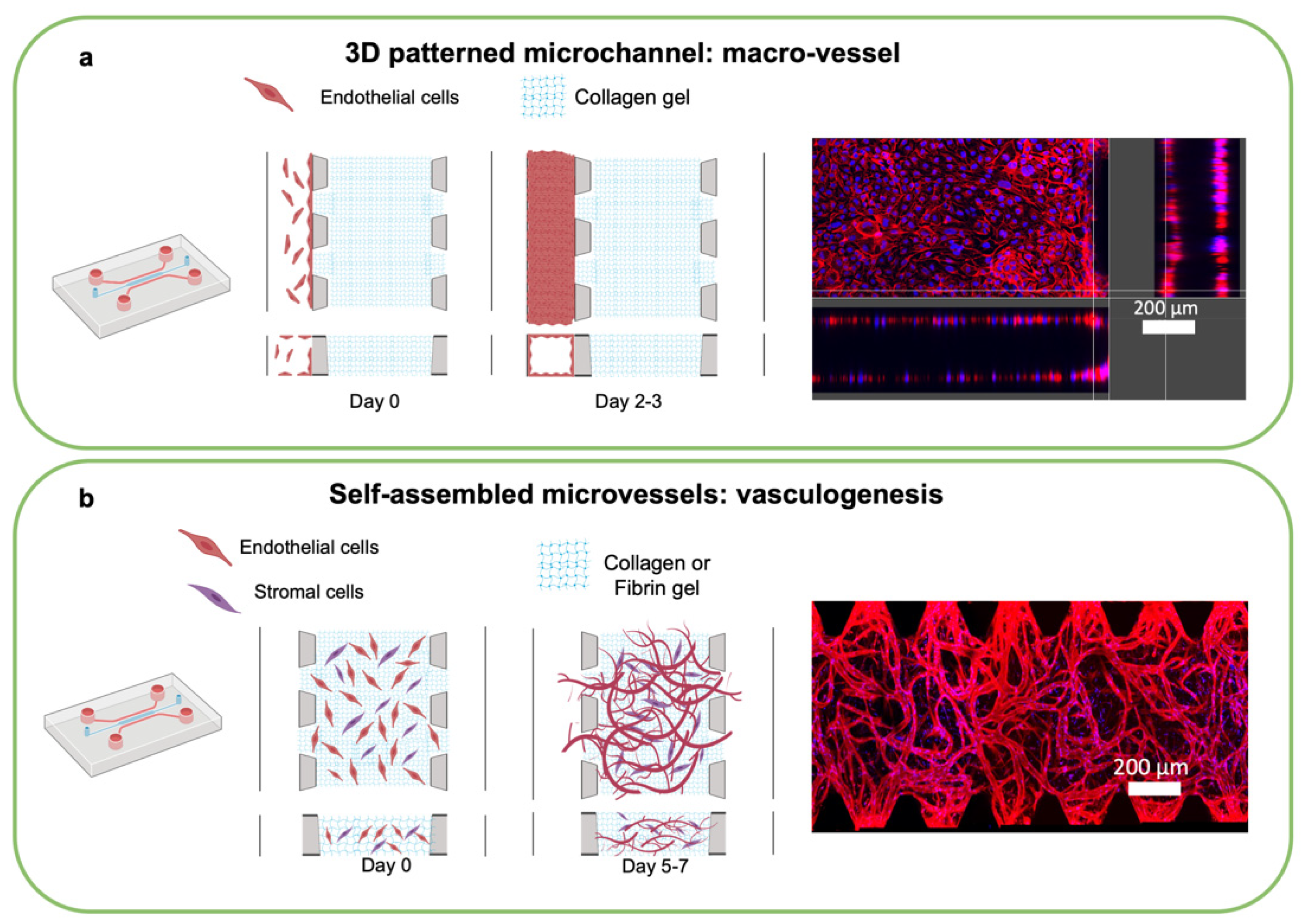
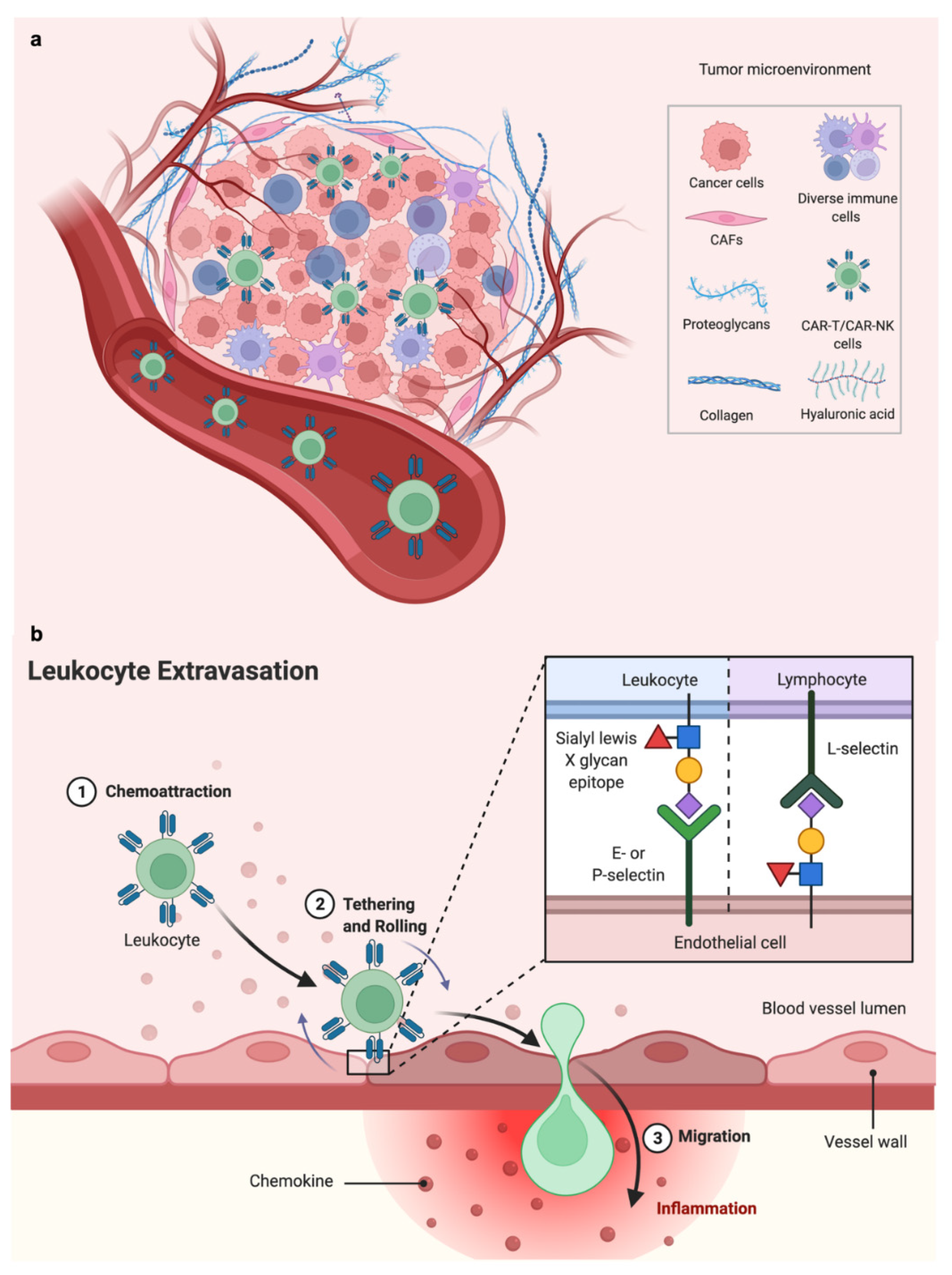
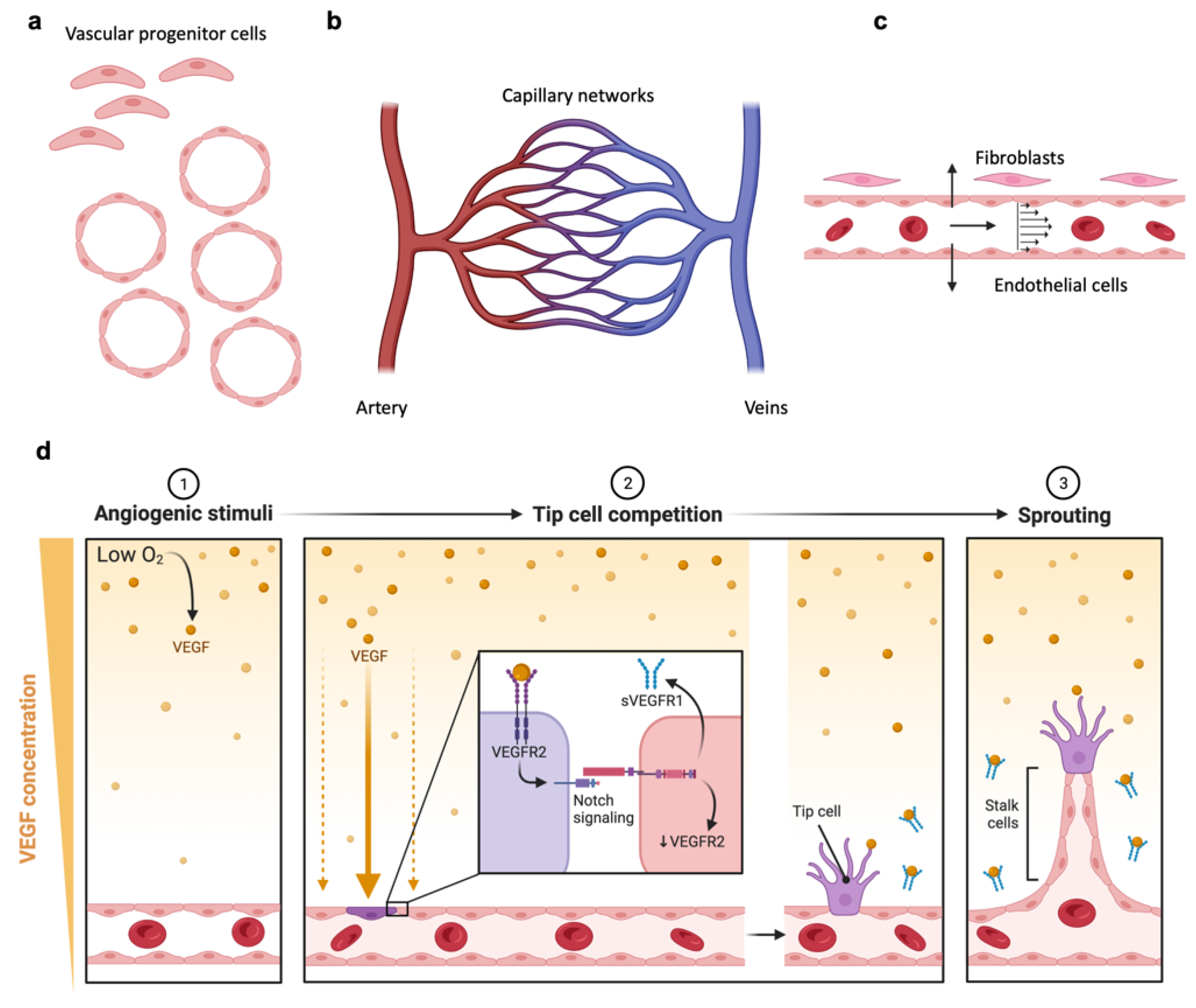
4. Microphysiological Systems including Microvasculature
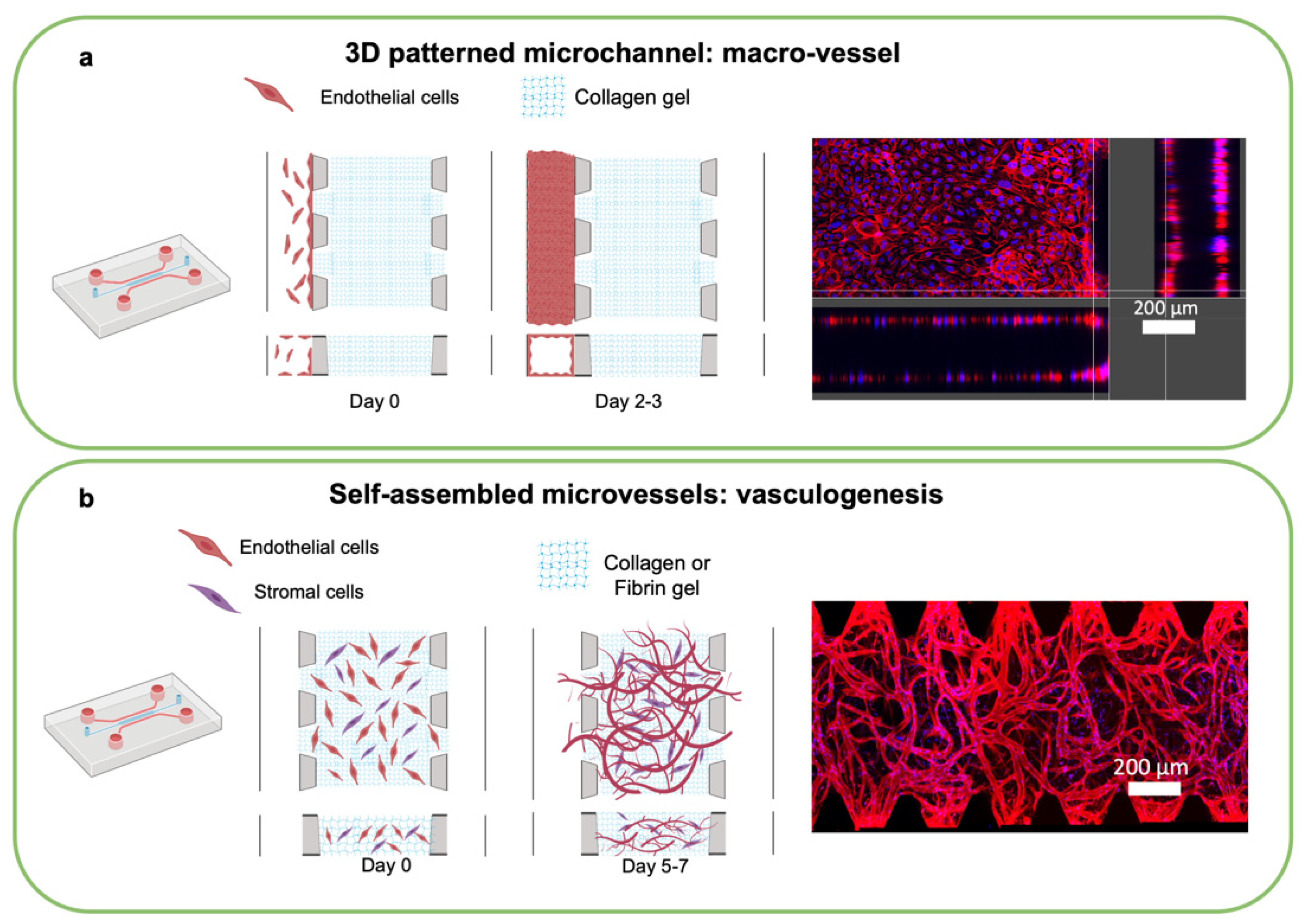
4.1. Engineering Vascularized MPS
4.2. Perfusing Vascularized MPS
5. Design of 3D MPS Using Microfluidic Technology
6. Translational Applications of MPS
7. Predicting Therapeutic Vulnerabilities and Biologic Barriers Using MPS
8. Remaining Challenges in Modeling Response to Cell Therapies
9. Microphysiological Systems in Pharmaceutical Development
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Longo, D.L.; Wilson, W.H. CAR T-Cell Therapy for Large B-Cell Lymphoma—Who, When, and How? N. Engl. J. Med. 2021, 386, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N. The landscape of car-t cell clinical trials against solid tumors—a comprehensive overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Woll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, srep39833. [Google Scholar] [CrossRef] [Green Version]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Joseph Espat, N.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Munger, M.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Levine, B.L.; et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010, 116, 2484–2493. [Google Scholar] [CrossRef]
- Tarannum, M.; Romee, R.; Shapiro, R.M. Innovative Strategies to Improve the Clinical Application of NK Cell-Based Immunotherapy. Front. Immunol. 2022, 13, 859177. [Google Scholar] [CrossRef] [PubMed]
- Ruppel, K.E.; Fricke, S.; Köhl, U.; Schmiedel, D. Taking Lessons from CAR-T Cells and Going Beyond: Tailoring Design and Signaling for CAR-NK Cells in Cancer Therapy. Front. Immunol. 2022, 13, 822298. [Google Scholar] [CrossRef] [PubMed]
- Abou-El-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable Manufacturing of CAR T Cells for Cancer Immunotherapy. Blood Cancer Discov. 2021, 2, 408–422. [Google Scholar] [CrossRef]
- Kumari, R.; Ouyang, X.; Wang, J.; Xu, X.; Zheng, M.; An, X.; Li, Q.-X. Preclinical pharmacology modeling of chimeric antigen receptor T therapies. Curr. Opin. Pharmacol. 2021, 61, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Man, K.; Liu, J.; Liu, Y.; Chen, Q.; Zhou, Y.; Yang, Y. Microphysiological Systems: Design, Fabrication, and Applications. ACS Biomater. Sci. Eng. 2020, 6, 3231–3257. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A.; Kim, B.Y.S.; Trounson, A. How to design preclinical studies in nanomedicine and cell therapy to maximize the prospects of clinical translation. Nat. Biomed. Eng. 2018, 2, 797–809. [Google Scholar] [CrossRef]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2020, 3, 040401. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Hickman, J.J.; Huh, D.; Kamm, R.D. Microphysiological systems. APL Bioeng. 2019, 3, 040401. [Google Scholar] [CrossRef] [Green Version]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Laurent, J.; Blin, G.; Chatelain, F.; Vanneaux, V.; Fuchs, A.; Larghero, J.; Théry, M. Convergence of microengineering and cellular self-organization towards functional tissue manufacturing. Nat. Biomed. Eng. 2017, 1, 939–956. [Google Scholar] [CrossRef] [PubMed]
- Deglincerti, A.; Croft, G.F.; Pietila, L.N.; Zernicka-Goetz, M.; Siggia, E.D.; Brivanlou, A.H. Self-organization of the in vitro attached human embryo. Nature 2016, 533, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Jacob, F.; Ming, G.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef]
- Ando, Y.; Mariano, C.; Shen, K. Engineered in vitro tumor models for cell-based immunotherapy. Acta Biomater. 2021, 132, 345–359. [Google Scholar] [CrossRef]
- Veninga, V.; Voest, E.E. Tumor organoids: Opportunities and challenges to guide precision medicine. Cancer Cell 2021, 39, 1190–1201. [Google Scholar] [CrossRef]
- YBar-Ephraim, E.; Kretzschmar, K.; Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 2020, 20, 279–293. [Google Scholar] [CrossRef]
- Knelson, E.H.; Ivanova, E.V.; Tarannum, M.; Campisi, M.; Lizotte, P.H.; Booker, M.A.; Ozgenc, I.; Noureddine, M.; Meisenheimer, B.; Chen, M.; et al. Activation of tumor-cell STING primes NK-cell therapy. Cancer Immunol. Res. 2022, 2022, OF1–OF15. [Google Scholar] [CrossRef] [PubMed]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [Green Version]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W.; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.-S.; et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017, 8, 66747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid modeling of the tumor immune microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van De Haar, J.; Fanchi, L.F.; Slagter, M.; Van Der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Han, S.; Sanny, A.; Chan, D.L.-K.; van Noort, D.; Lim, W.; Tan, A.H.-M.; Park, S. 3D hanging spheroid plate for high-throughput CAR T cell cytotoxicity assay. J. Nanobiotechnol. 2022, 20, 30. [Google Scholar] [CrossRef]
- Barisam, M.; Niavol, F.R.; Kinj, M.A.; Saidi, M.S.; Ghanbarian, H.; Kashaninejad, N. Enrichment of cancer stem-like cells by controlling oxygen, glucose and fluid shear stress in a microfluidic spheroid culture device. J. Sci. Mater. Devices 2022, 7, 100439. [Google Scholar] [CrossRef]
- Madrigal, J.L.; Schoepp, N.G.; Xu, L.; Powell, C.S.; Delley, C.L.; Siltanen, C.A.; Danao, J.; Srinivasan, M.; Cole, R.H.; Abate, A.R. Characterizing cell interactions at scale with made-to-order droplet ensembles (MODEs). Proc. Natl. Acad. Sci. USA 2022, 119, e2110867119. [Google Scholar] [CrossRef]
- Sarkar, S.; Sabhachandani, P.; Stroopinsky, D.; Palmer, K.; Cohen, N.; Rosenblatt, J.; Avigan, D.; Konry, T. Dynamic analysis of immune and cancer cell interactions at single cell level in microfluidic droplets. Biomicrofluidics 2016, 10, 54115. [Google Scholar] [CrossRef] [Green Version]
- Segaliny, A.I.; Li, G.; Kong, L.; Ren, C.; Chen, X.; Wang, J.K.; Baltimore, D.; Wu, G.; Zhao, W. Functional TCR T cell screening using single-cell droplet microfluidics. Lab Chip 2018, 18, 3733–3749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wong, J.K.-U.; Shi, J.; Jia, Y.; Deng, C.; Jiang, L.; Li, P.; Wong, A.H.-H. Assessment of chimeric antigen receptor T (CAR-T) cell cytotoxicity using droplet microflluidics. In Proceedings of the 23rd International Conference on Miniaturized Systems for Chemistry and Life Sciences, MicroTAS 2019, Basel, Switzerland, 27–31 October 2019. [Google Scholar]
- Wong, K.U.; Shi, J.; Li, P.; Wang, H.; Jia, Y.; Deng, C.; Jiang, L.; Wong, A.H.-H. Assessment of chimeric antigen receptor T cytotoxicity by droplet microfluidics in vitro. Antib. Ther. 2022, 5, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Langer, E.M.; Allen-Petersen, B.L.; King, S.M.; Kendsersky, N.D.; Turnidge, M.A.; Kuziel, G.M.; Riggers, R.; Samatham, R.; Amery, T.S.; Jacques, S.L.; et al. Modeling Tumor Phenotypes In Vitro with Three-Dimensional Bioprinting. Cell Rep. 2019, 26, 608–623.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leberfinger, A.N.; Moncal, K.K.; Ravnic, D.J.; Ozbolat, I.T. 3D Printing for Cell Therapy Applications. In Cell Therapy. Molecular and Translational Medicine; Emerich, D., Orive, G., Eds.; Humana Press: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Jin, W.; Tamzalit, F.; Chaudhuri, P.K.; Black, C.T.; Huse, M.; Kam, L.C. T cell activation and immune synapse organization respond to the microscale mechanics of structured surfaces. Proc. Natl. Acad. Sci. USA 2019, 116, 19835–19840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, L.; Lam, T.; Andersch, L.; Klaus, A.; Schwiebert, S.; Winkler, A.; Gauert, A.; Heeren-Hagemann, A.I.; Astrahantseff, K.; Klironomos, F.; et al. A reproducible bioprinted 3D tumor model serves as a preselection tool for CAR T cell therapy optimization. Front. Immunol. 2021, 12, 2382. [Google Scholar] [CrossRef]
- Dey, M.; Kim, M.H.; Nagamine, M.; Dogan, M.; Kozhaya, L.; Unutmaz, D.; Ozbolat, I.T. 3D Bioprinted perfusable and vascularized breast tumor model for dynamic screening of chemotherapeutics and CAR-T cells. BioRxiv 2022. [Google Scholar] [CrossRef]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Shin, Y.; Han, S.; Jeon, J.S.; Yamamoto, K.; Zervantonakis, I.K.; Sudo, R.; Kamm, R.D.; Chung, S. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat. Protoc. 2012, 7, 1247–1259. [Google Scholar] [CrossRef] [Green Version]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.A.; Whitesides, G.M. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef]
- Qin, D.; Xia, Y.; Whitesides, G.M. Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 2010, 5, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Giannattasio, A.; Weil, S.; Kloess, S.; Ansari, N.; Stelzer, E.H.K.; Cerwenka, A.; Steinle, A.; Koehl, U.; Koch, J. Cytotoxicity and infiltration of human NK cells in in vivo-like tumor spheroids. BMC Cancer 2015, 15, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, K.; Paterson, S.; Mulholland, T.; Coffelt, S.B.; Zagnoni, M. Assessment of CAR-T mediated and targeted cytotoxicity in 3D microfluidic TBNC co-culture models for combination therapy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tu, H.; Wu, Z.; Xia, Y.; Chen, H.; Hu, H.; Ding, Z.; Zhou, F.; Guo, S. Profiling of immune–cancer interactions at the single-cell level using a microfluidic well array. Analyst 2020, 145, 4138–4147. [Google Scholar] [CrossRef]
- Wang, X.; Scarfò, I.; Schmidts, A.; Toner, M.; Maus, M.V.; Irimia, D. Dynamic profiling of antitumor activity of CAR T cells using micropatterned tumor arrays. Adv. Sci. 2019, 6, 1901829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayuso, J.M.; Rehman, S.; Virumbrales-Munoz, M.; McMinn, P.H.; Geiger, P.; Fitzgerald, C.; Heaster, T.; Skala, M.C.; Beebe, D.J. Microfluidic tumor-on-a-chip model to evaluate the role of tumor environmental stress on NK cell exhaustion. Sci. Adv. 2021, 7, eabc2331. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Truttschel, R.; Gong, M.M.; Humayun, M.; Virumbrales-Munoz, M.; Vitek, R.; Felder, M.; Gillies, S.D.; Sondel, P.; Wisinski, K.B.; et al. Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model. Oncoimmunology 2019, 8, 1553477. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Son, K.; Hwang, Y.; Ko, J.; Lee, Y.; Doh, J.; Jeon, N.L. High-throughput microfluidic 3D cytotoxicity assay for cancer immunotherapy (CACI-IMPACT platform). Front. Immunol. 2019, 10, 1133. [Google Scholar] [CrossRef] [Green Version]
- Pavesi, A.; Tan, A.T.; Koh, S.; Chia, A.; Colombo, M.; Antonecchia, E.; Miccolis, C.; Ceccarello, E.; Adriani, G.; Raimondi, M.T.; et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight 2017, 2, e89762. [Google Scholar] [CrossRef]
- Ronteix, G.; Jain, S.; Angely, C.; Cazaux, M.; Khazen, R.; Bousso, P.; Baroud, C.N. A Multiscale Immuno-Oncology on-Chip System (MIOCS) establishes that collective T cell behaviors govern tumor regression. bioRxiv 2021. [Google Scholar] [CrossRef]
- Feder-Mengus, C.; Ghosh, S.; Weber, W.P.; Wyler, S.; Zajac, P.; Terracciano, L.; Oertli, D.; Heberer, M.; Martin, I.; Spagnoli, G.C.; et al. Multiple mechanisms underlie defective recognition of melanoma cells cultured in three-dimensional architectures by antigen-specific cytotoxic T lymphocytes. Br. J. Cancer 2007, 96, 1072–1082. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Kuang, Y.; Xing, X.; Simpson, R.J.; Huang, H.; Yang, T.; Chen, J.; Yang, L.; Liu, E.; He, W.; et al. Proteomic comparison of 3D and 2D glioma models reveals increased HLA-E expression in 3D models is associated with resistance to NK cell-mediated cytotoxicity. J. Proteome Res. 2014, 13, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Siegler, E.L.; Ta, H.P.; Cinay, G.E.; Zhou, H.; Gorrell, K.A.; Au, H.; Jarvis, B.M.; Wang, P.; Shen, K. Evaluating CAR-T Cell Therapy in a Hypoxic 3D Tumor Model. Adv. Healthc. Mater. 2019, 8, 1900001. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.M.; Gopalakrishnan, N.; Ibrahim, H.; Haug, M.; Halaas, Ø. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab Chip 2016, 16, 3728–3740. [Google Scholar] [CrossRef]
- Stockslager, M.A.; Bagnall, J.S.; Hecht, V.C.; Hu, K.; Aranda-Michel, E.; Payer, K.; Kimmerling, R.J.; Manalis, S.R. Microfluidic platform for characterizing TCR–pMHC interactions. Biomicrofluidics 2017, 11, 64103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, O.T.P.; Misun, P.M.; Lohasz, C.; Lee, J.; Wang, W.; Schroeder, T.; Hierlemann, A. An Immunocompetent Microphysiological System to Simultaneously Investigate Effects of Anti-Tumor Natural Killer Cells on Tumor and Cardiac Microtissues. Front. Immunol. 2021, 12, 781337. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.D.; Kratz, A.; Park, J.G.; Barrientos, E.S.; Saini, H.; Nguyen, T.; Pockaj, B.; Mouneimne, G.; LaBaer, J.; Nikkhah, M. A human organotypic microfluidic tumor model permits investigation of the interplay between patient-derived fibroblasts and breast cancer cells. Cancer Res. 2019, 79, 3139–3151. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.-Y.; Lee, J.-H.; Shin, Y.; Chung, S.; Kuh, H.-J. Co-culture of tumor spheroids and fibroblasts in a collagen matrix-incorporated microfluidic chip mimics reciprocal activation in solid tumor microenvironment. PLoS ONE 2016, 11, e0159013. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; De Ninno, A.; Mencattini, A.; Mermet-Meillon, F.; Fornabaio, G.; Evans, S.S.; Cossutta, M.; Khira, Y.; Han, W.; Sirven, P.; et al. Dissecting effects of anti-cancer drugs and cancer-associated fibroblasts by on-chip reconstitution of immunocompetent tumor microenvironments. Cell Rep. 2018, 25, 3884–3893.e3. [Google Scholar] [CrossRef] [Green Version]
- Fowell, D.J.; Kim, M. The spatio-temporal control of effector T cell migration. Nat. Rev. Immunol. 2021, 21, 582–596. [Google Scholar] [CrossRef]
- Rushdi, M.; Li, K.; Yuan, Z.; Travaglino, S.; Grakoui, A.; Zhu, C. Mechanotransduction in T Cell Development, Differentiation and Function. Cells 2020, 9, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’H, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T Cells Use Mechanical Force to Potentiate Target Cell Killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majedi, F.S.; Hasani-Sadrabadi, M.M.; Thauland, T.J.; Li, S.; Bouchard, L.-S.; Butte, M.J. T-cell activation is modulated by the 3D mechanical microenvironment. Biomaterials 2020, 252, 120058. [Google Scholar] [CrossRef]
- Rossy, J.; Laufer, J.M.; Legler, D.F. Role of Mechanotransduction and Tension in T Cell Function. Front. Immunol. 2018, 9, 2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.S.C.; Raychaudhuri, D.; Paul, B.; Chakrabarty, Y.; Ghosh, A.R.; Rahaman, O.; Talukdar, A.; Ganguly, D. Cutting Edge: Piezo1 Mechanosensors Optimize Human T Cell Activation. J. Immunol. 2018, 200, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef] [Green Version]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune–vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Haase, K.; Roger, D. Advances in on-chip vascularization. Regen. Med. 2017, 12, 285–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, W.; Lim, S.; Jeon, J.S. Vasculature-On-A-Chip for In Vitro Disease Models. Bioengineering 2017, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Paul, A.; Vrana, N.E.; Zhao, X.; Memic, A.; Hwang, Y.-S.; Dokmeci, M.R.; Khademhosseini, A. Microfluidic techniques for development of 3D vascularized tissue. Biomaterials 2014, 35, 7308–7325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.B.; Whisler, J.A.; Fröse, J.; Yu, C.; Shin, Y.; Kamm, R.D. On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc. 2017, 12, 865–880. [Google Scholar] [CrossRef]
- Sobrino, A.; Phan, D.T.T.; Datta, R.; Wang, X.; Hachey, S.J.; Romero-López, M.; Gratton, E.; Lee, A.P.; George, S.C.; Hughes, C.C.W. 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 2016, 6, 31589. [Google Scholar] [CrossRef] [Green Version]
- Caballero, D.; Blackburn, S.M.; de Pablo, M.; Samitier, J.; Albertazzi, L. Tumour-vessel-on-a-chip models for drug delivery. Lab Chip 2017, 17, 3760–3771. [Google Scholar] [CrossRef]
- Dikici, S.; Claeyssens, F.; MacNeil, S. Bioengineering Vascular Networks to Study Angiogenesis and Vascularization of Physiologically Relevant Tissue Models in Vitro. ACS Biomater. Sci. Eng. 2020, 6, 3513–3528. [Google Scholar] [CrossRef]
- Chrobak, K.M.; Potter, D.R.; Tien, J. Formation of perfused, functional microvascular tubes in vitro. Microvasc. Res. 2006, 71, 185–196. [Google Scholar] [CrossRef]
- Bischel, L.L.; Young, E.W.; Mader, B.R.; Beebe, D.J. Tubeless microfluidic angiogenesis assay with three-dimensional endothelial-lined microvessels. Biomaterials 2012, 34, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.T.; Adriani, G.; Beyer, S.; Nhan, P.-T.; Kamm, R.D.; Kah, J. A Facile Method to Probe the Vascular Permeability of Nanoparticles in Nanomedicine Applications. Sci. Rep. 2017, 7, 707. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Kasuya, J.; Jeon, J.; Chung, S.; Kamm, R.D. A quantitative microfluidic angiogenesis screen for studying anti-angiogenic therapeutic drugs. Lab Chip 2015, 15, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.; Jeon, J.S.; Han, S.; Jung, G.-S.; Shin, S.; Lee, S.-H.; Sudo, R.; Kamm, R.D.; Chung, S. In vitro 3D collective sprouting angiogenesis under orchestrated ANG-1 and VEGF gradients. Lab Chip 2011, 11, 2175–2181. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.S.; Han, S.; Shin, Y.; Kwon, G.H.; Kamm, R.D.; Lee, S.-H.; Chung, S. Sprouting angiogenesis under a chemical gradient regulated by interactions with an endothelial monolayer in a microfluidic platform. Anal. Chem. 2011, 83, 8454–8459. [Google Scholar] [CrossRef] [PubMed]
- Kosyakova, N.; Kao, D.D.; Figetakis, M.; López-Giráldez, F.; Spindler, S.; Graham, M.; James, K.J.; Shin, J.W.; Liu, X.; Tietjen, G.T.; et al. Differential functional roles of fibroblasts and pericytes in the formation of tissue-engineered microvascular networks in vitro. NPJ Regen. Med. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Phan, D.T.T.; Sobrino, A.; George, S.C.; Hughes, C.C.W.; Lee, A.P. Engineering anastomosis between living capillary networks and endothelial cell-lined microfluidic channels. Lab Chip 2016, 16, 282–290. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Chung, M.; Jeon, N.L. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip 2013, 13, 1489–1500. [Google Scholar] [CrossRef]
- Whisler, J.A.; Chen, M.B.; Kamm, R.D. Control of perfusable microvascular network morphology using a multiculture microfluidic system. Tissue Eng. Part C Methods 2014, 20, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials 2018, 180, 117–129. [Google Scholar] [CrossRef]
- Campisi, M.; Lim, S.H.; Chiono, V.; Kamm, R.D. 3D Self-Organized Human Blood–Brain Barrier in a Microfluidic Chip. Program. Morphog. Methods Protoc. Methods Mol. Biol. 2021, 2258, 205–219. [Google Scholar]
- Chen, M.B.; Whisler, J.A.; Jeon, J.S.; Kamm, R.D. Mechanisms of tumor cell extravasation in an in vitro microvascular network platform. Integr. Biol. 2013, 5, 1262–1271. [Google Scholar] [CrossRef] [Green Version]
- Boussommier-Calleja, A.; Atiyas, Y.; Haase, K.; Headley, M.; Lewis, C.; Kamm, R.D. The effects of monocytes on tumor cell extravasation in a 3D vascularized microfluidic model. Biomaterials 2019, 198, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Moses, J.C.; Bandyopadhyay, A.; Mandal, B.B. 3D Printing/Bioprinting Based Tailoring of in Vitro Tissue Models: Recent Advances and Challenges. ACS Appl. Bio. Mater. 2019, 2, 1385–1405. [Google Scholar] [CrossRef] [PubMed]
- Miri, A.K.; Khalilpour, A.; Cecen, B.; Maharjan, S.; Shin, S.R.; Khademhosseini, A. Multiscale bioprinting of vascularized models. Biomaterials 2019, 198, 204–216. [Google Scholar] [CrossRef]
- Ewart, L.; Roth, A. Opportunities and challenges with microphysiological systems: A pharma end-user perspective. Nat. Rev. Drug Discov. 2020, 20, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Kim, H.; Sung, J.H.; Choi, N.; Lee, K.; Kim, H.N. Microphysiological systems for recapitulating physiology and function of blood-brain barrier. Biomaterials 2020, 232, 119732. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Votanopoulos, K.I.; Forsythe, S.; Msc, H.S.; Mazzocchi, A.; Aleman, J.; Miller, L.; Levine, E.; Triozzi, P.; Skardal, A. Model of Patient-Specific Immune-Enhanced Organoids for Immunotherapy Screening: Feasibility Study. Ann. Surg. Oncol. 2019, 27, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Pernik, M.; Bird, C.; Traylor, J.; Shi, D.; Richardson, T.; McBrayer, S.; Abdullah, K. Patient-Derived Cancer Organoids for Precision Oncology Treatment. J. Pers. Med. 2021, 11, 423. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, S.D.; Erali, R.A.; Sasikumar, S.; Laney, P.; Shelkey, E.; D’Agostino, R.; Miller, L.D.; Shen, P.; Levine, E.A.; Soker, S.; et al. Organoid Platform in Preclinical Investigation of Personalized Immunotherapy Efficacy in Appendiceal Cancer: Feasibility Study. Clin. Cancer Res. 2021, 27, 5141–5150. [Google Scholar] [CrossRef]
- Campisi, M.; Sundararaman, S.K.; Shelton, S.E.; Knelson, E.H.; Mahadevan, N.R.; Yoshida, R.; Tani, T.; Ivanova, E.; Cañadas, I.; Osaki, T.; et al. Tumor-Derived cGAMP Regulates Activation of the Vasculature. Front. Immunol. 2020, 11, 2090. [Google Scholar] [CrossRef] [PubMed]
- Nayyar, G.; Chu, Y.; Cairo, M.S. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 2019, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Herland, A.; Maoz, B.M.; Das, D.; Somayaji, M.R.; Prantil-Baun, R.; Novak, R.; Cronce, M.; Huffstater, T.; Jeanty, S.S.F.; Ingram, M.; et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat. Biomed. Eng. 2020, 4, 421–436. [Google Scholar] [CrossRef]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef]
- Brassard, J.A.; Nikolaev, M.; Hübscher, T.; Hofer, M.; Lutolf, M.P. Recapitulating macro-scale tissue self-organization through organoid bioprinting. Nat. Mater. 2020, 20, 22–29. [Google Scholar] [CrossRef]
- Novak, R.; Ingram, M.; Marquez, S.; Das, D.; Delahanty, A.; Herland, A.; Maoz, B.M.; Jeanty, S.S.F.; Somayaji, M.R.; Burt, M.; et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng. 2020, 4, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Offeddu, G.S.; Serrano, J.C.; Chen, S.W.; Shelton, S.E.; Shin, Y.; Floryan, M.; Kamm, R.D. Microheart: A microfluidic pump for functional vascular culture in microphysiological systems. J. Biomech. 2021, 119, 110330. [Google Scholar] [CrossRef] [PubMed]
- Haase, K.; Piatti, F.; Marcano, M.; Shin, Y.; Visone, R.; Redaelli, A.; Rasponi, M.; Kamm, R.D. Physiologic flow-conditioning limits vascular dysfunction in engineered human capillaries. Biomaterials 2021, 280, 121248. [Google Scholar] [CrossRef] [PubMed]
- Appelt-Menzel, A.; Cubukova, A.; Günther, K.; Edenhofer, F.; Piontek, J.; Krause, G.; Stüber, T.; Walles, H.; Neuhaus, W.; Metzger, M. Establishment of a Human Blood-Brain Barrier Co-culture Model Mimicking the Neurovascular Unit Using Induced Pluri- and Multipotent Stem Cells. Stem Cell Rep. 2017, 8, 894–906. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.S.; Zhang, Y.N.; Zhang, W. Cancer-on-a-chip systems at the frontier of nanomedicine. Drug Discov. Today 2017, 22, 1392–1399. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Garreta, E.; Kamm, R.D.; Lopes, S.M.C.D.S.; Lancaster, M.A.; Weiss, R.; Trepat, X.; Hyun, I.; Montserrat, N. Rethinking organoid technology through bioengineering. Nat. Mater. 2020, 20, 145–155. [Google Scholar] [CrossRef]
- Jang, K.J.; Otieno, M.A.; Ronxhi, J.; Lim, H.K.; Ewart, L.; Kodella, K.; Petropolis, D.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Liver-Chip: Reproducing Human and Cross-Species Toxicities. BioRxiv 2019, 631002. [Google Scholar] [CrossRef] [Green Version]
- Ewart, L.; Fabre, K.; Chakilam, A.; Dragan, Y.; Duignan, D.B.; Eswaraka, J.; Gan, J.; Guzzie-Peck, P.; Otieno, M.; Jeong, C.G.; et al. Navigating tissue chips from development to dissemination: A pharmaceutical industry perspective. Exp. Biol. Med. 2017, 242, 1579–1585. [Google Scholar] [CrossRef] [Green Version]
- Franzen, N.; van Harten, W.H.; Retèl, V.P.; Loskill, P.; Raaij, J.va.; Jzerman, M.I. Impact of organ-on-a-chip technology on pharmaceutical R&D costs. Drug Discov. Today 2019, 24, 1720–1724. [Google Scholar]
- International Consortium for Innovation an Quality. Available online: https://iqconsortium.org (accessed on 28 May 2022).
- European Society for Alternatives for Animal Testing (EUSAAT). Available online: http://www.eusaat-congress.eu (accessed on 28 May 2022).
- European Organ on Chip Society. Available online: https://euroocs.eu/annual-meeting/ (accessed on 29 May 2022).
- Available online: https://mpsworldsummit.com/ (accessed on 29 May 2022).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campisi, M.; Shelton, S.E.; Chen, M.; Kamm, R.D.; Barbie, D.A.; Knelson, E.H. Engineered Microphysiological Systems for Testing Effectiveness of Cell-Based Cancer Immunotherapies. Cancers 2022, 14, 3561. https://doi.org/10.3390/cancers14153561
Campisi M, Shelton SE, Chen M, Kamm RD, Barbie DA, Knelson EH. Engineered Microphysiological Systems for Testing Effectiveness of Cell-Based Cancer Immunotherapies. Cancers. 2022; 14(15):3561. https://doi.org/10.3390/cancers14153561
Chicago/Turabian StyleCampisi, Marco, Sarah E. Shelton, Minyue Chen, Roger D. Kamm, David A. Barbie, and Erik H. Knelson. 2022. "Engineered Microphysiological Systems for Testing Effectiveness of Cell-Based Cancer Immunotherapies" Cancers 14, no. 15: 3561. https://doi.org/10.3390/cancers14153561
APA StyleCampisi, M., Shelton, S. E., Chen, M., Kamm, R. D., Barbie, D. A., & Knelson, E. H. (2022). Engineered Microphysiological Systems for Testing Effectiveness of Cell-Based Cancer Immunotherapies. Cancers, 14(15), 3561. https://doi.org/10.3390/cancers14153561