
Valid-NEO: A Multi-Omics Platform for Neoantigen Detection and Quantification from Limited Clinical Samples




Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Buffers Used for Neoantigen Isolation
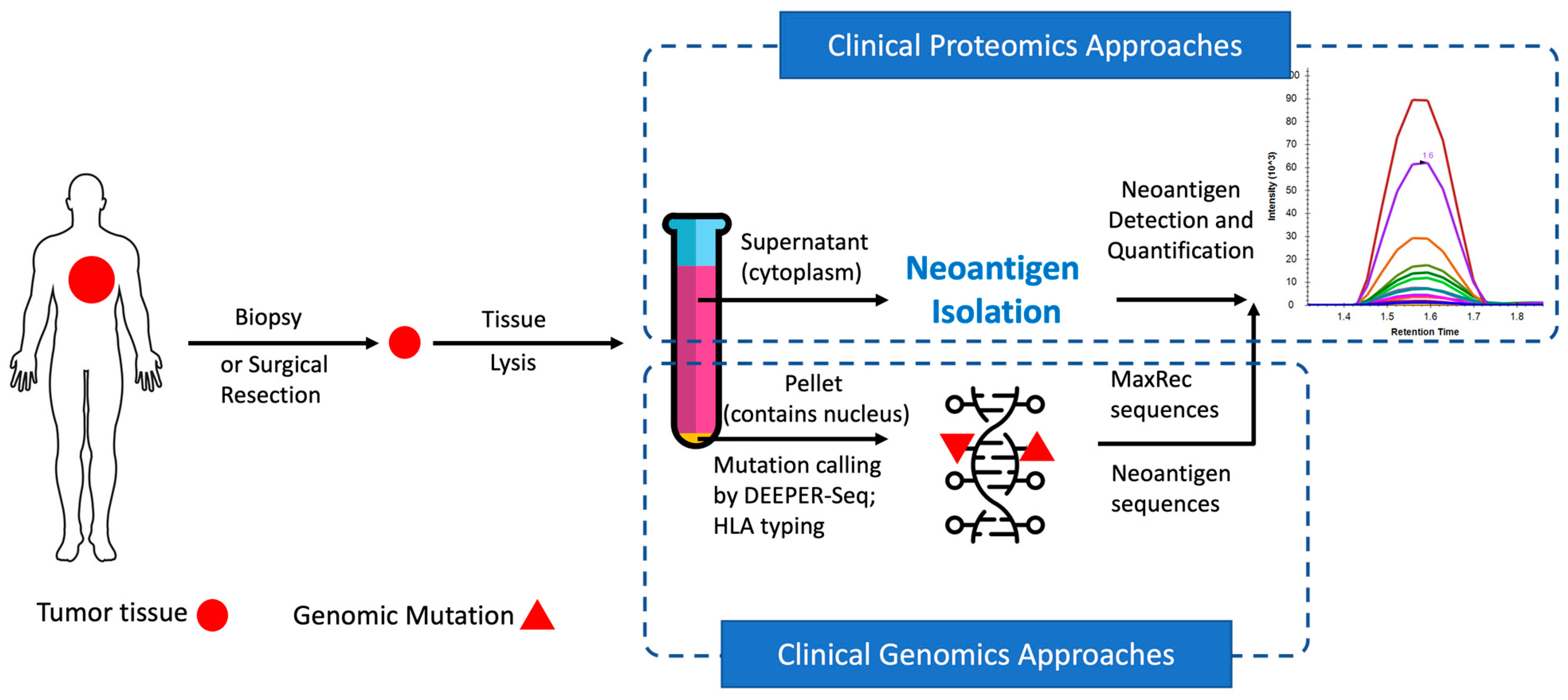
2.3. Tumor Sample Process and HLA Molecule Extraction
2.4. Construction of Valid-NEO
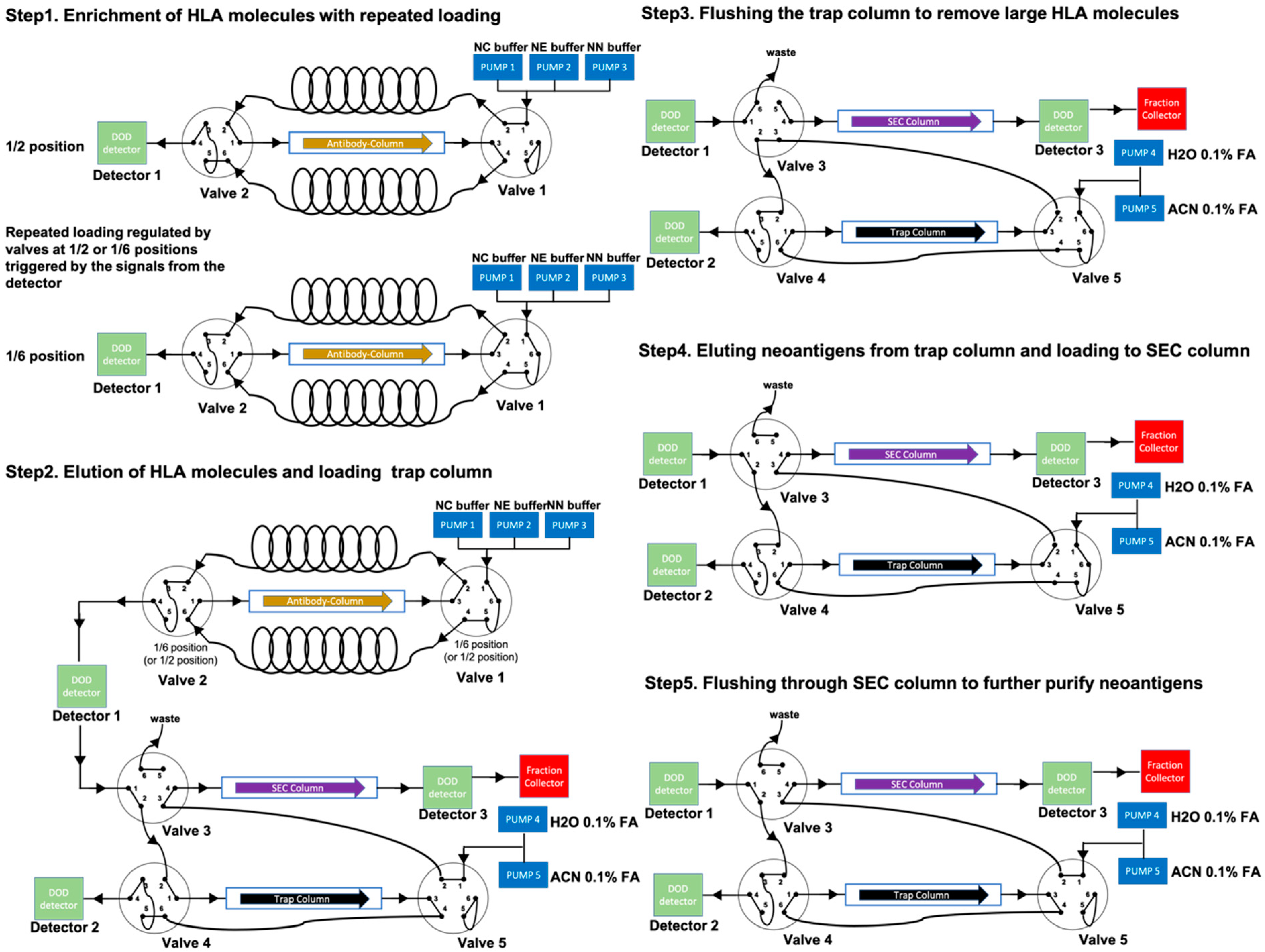
2.5. Online Enrichment of HLA Molecules through an Antibody-Column
2.6. Online Elution of Neoantigen Peptides and Antibody Column Regeneration
2.7. Online Isolation and Purification of Neoantigen Peptides
2.8. Mass Spectrometry Method Development
2.9. Pre-Conditioning the System to Ensure the Highest Sensitivity
2.10. Data Deposition
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlman, A.H.; Hwang, M.S.; Konig, M.F.; Hsiue, E.H.; Douglass, J.; DiNapoli, S.R.; Mog, B.J.; Bettegowda, C.; Pardoll, D.M.; Gabelli, S.B.; et al. Targeting public neoantigens for cancer immunotherapy. Nat. Cancer. 2021, 2, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Törngren, T.; Kvist, A.; et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1738. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 21, 13404. [Google Scholar] [CrossRef] [Green Version]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Douglass, J.; Hwang, M.S.; Hsiue, E.H.; Mog, B.J.; Zhang, M.; Papadopoulos, N.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. Direct Detection and Quantification of Neoantigens. Cancer Immunol. Res. 2019, 7, 1748–1754. [Google Scholar] [CrossRef]
- Hsiue, E.H.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef]
- Douglass, J.; Hsiue, E.H.; Mog, B.J.; Hwang, M.S.; DiNapoli, S.R.; Pearlman, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021, 6, eabd5515. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Martínez-Val, A.; Steigerwald, S.; Rüther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell Proteomics 2020, 19, 716–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell Proteomics 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubarev, R.A.; Makarov, A. Orbitrap mass spectrometry. Anal. Chem. 2013, 85, 5288–5296. [Google Scholar] [CrossRef]
- Bache, N.; Geyer, P.E.; Bekker-Jensen, D.B.; Hoerning, O.; Falkenby, L.; Treit, P.V.; Doll, S.; Paron, I.; Müller, J.B.; Meier, F.; et al. A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics. Mol. Cell Proteomics 2018, 17, 2284–2296. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, X.; Tang, P.S.; O’leary, G.M.; Zhang, M. Targeted sequencing of both DNA strands barcoded and captured individually by RNA probes to identify genome-wide ultra-rare mutations. Sci. Rep. 2017, 7, 3356. [Google Scholar] [CrossRef] [Green Version]
- Moser, A.C.; Hage, D.S. Immunoaffinity chromatography: An introduction to applications and recent developments. Bioanalysis 2010, 2, 769–790. [Google Scholar] [CrossRef] [Green Version]
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020, 38, 199–209. [Google Scholar] [CrossRef]
- Savage, P.; Stebbing, J.; Bower, M.; Crook, T. Why does cytotoxic chemotherapy cure only some cancers? Nat. Clin. Pract. Oncol. 2009, 6, 43–52. [Google Scholar] [CrossRef]
- Bonadonna, G.; Valagussa, P. Chemotherapy of breast cancer: Current views and results. Int. J. Radiat. Oncol. Biol. Phys. 1983, 9, 279–297. [Google Scholar] [CrossRef]
- Chan, D.L.; Morris, D.L.; Rao, A.; Chua, T.C. Intraperitoneal chemotherapy in ovarian cancer: A review of tolerance and efficacy. Cancer Manag. Res. 2012, 4, 413–422. [Google Scholar]
- Yagoda, A.; Petrylak, D. Cytotoxic chemotherapy for advanced hormone-resistant prostate cancer. Cancer. 1993, 71 (Suppl. 3), 1098–1109. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Scaltriti, M.; Baselga, J. The epidermal growth factor receptor pathway: A model for targeted therapy. Clin Cancer Res. 2006, 12, 5268–5272. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, R.M.; Goldenberg, D.M. Targeted therapy of cancer: New prospects for antibodies and immunoconjugates. CA Cancer J. Clin. 2006, 56, 226–243. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Andersen, J.N.; Futreal, P.A. Cancer genomics: From discovery science to personalized medicine. Nat. Med. 2011, 17, 297–303. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Bagnyukova, T.V.; Serebriiskii, I.G.; Zhou, Y.; Hopper-Borge, E.A.; Golemis, E.A.; Astsaturov, I. Chemotherapy and signaling: How can targeted therapies supercharge cytotoxic agents? Cancer Biol. Ther. 2010, 10, 839–853. [Google Scholar] [CrossRef] [Green Version]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Neoantigen | MANA-SRM Detected Ratio to Standards | Valid-NEO (w/o MaxRec) Detected Ratio to Standards | Valid-NEO (w/MaxRec) | ||
|---|---|---|---|---|---|
| Detected Ratio to Standards | Detected Abundance (Unit: Atto mole) | Copy Number per Tumor Cell (Assuming 50 M Cells) | |||
| KRAS_Q61H | non-detectable ± N/A | 0.022 ± 0.006 | 1.045 ± 0.058 | 522.5 | 6.3 |
| KRAS_Q61L | 0.027 ± 0.002 | 0.053 ± 0.004 | 0.729 ± 0.037 | 364.5 | 4.4 |
| KRAS_Q61R | non-detectable ± N/A | 0.04 ± 0.004 | 1.495 ± 0.156 | 747.5 | 9.0 |
| IDH2_R140Q | non-detectable ± N/A | 0.015 ± 0.003 | 1.019 ± 0.057 | 509.5 | 6.1 |
| TP53_Y220C | non-detectable ± N/A | 0.054 ± 0.002 | 1.354 ± 0.014 | 677 | 8.2 |
| TP53_R248W | 0.011 ± 0.00006 | 0.086 ± 0.034 | 0.173 ± 0.031 | 86.5 | 1.0 |
| TP53_R213L | non-detectable ± N/A | 0.373 ± 0.057 | 0.662 ± 0.03 | 331 | 4.0 |
| KRAS_G12V_9mer | non-detectable ± N/A | 0.146 ± 0.022 | 5.458 ± 1.206 | 2729 | 32.9 |
| KRAS_G12V_10mer | non-detectable ± N/A | 0.141 ± 0.024 | 6.381 ± 1.693 | 3190.5 | 38.4 |
| KRAS_G12D_9mer | 0.0719 ± 0.012 | 0.145 ± 0.067 | 2.933 ± 1.227 | 1466.5 | 17.7 |
| KRAS_G12D_10mer | 0.113 ± 0.012 | 0.244 ± 0.104 | 6.518 ± 3.748 | 3259 | 39.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terai, Y.L.; Huang, C.; Wang, B.; Kang, X.; Han, J.; Douglass, J.; Hsiue, E.H.-C.; Zhang, M.; Purohit, R.; deSilva, T.; et al. Valid-NEO: A Multi-Omics Platform for Neoantigen Detection and Quantification from Limited Clinical Samples. Cancers 2022, 14, 1243. https://doi.org/10.3390/cancers14051243
Terai YL, Huang C, Wang B, Kang X, Han J, Douglass J, Hsiue EH-C, Zhang M, Purohit R, deSilva T, et al. Valid-NEO: A Multi-Omics Platform for Neoantigen Detection and Quantification from Limited Clinical Samples. Cancers. 2022; 14(5):1243. https://doi.org/10.3390/cancers14051243
Chicago/Turabian StyleTerai, Yuri Laguna, Chun Huang, Baoli Wang, Xiaonan Kang, Jing Han, Jacqueline Douglass, Emily Han-Chung Hsiue, Ming Zhang, Raj Purohit, Taylor deSilva, and et al. 2022. "Valid-NEO: A Multi-Omics Platform for Neoantigen Detection and Quantification from Limited Clinical Samples" Cancers 14, no. 5: 1243. https://doi.org/10.3390/cancers14051243
APA StyleTerai, Y. L., Huang, C., Wang, B., Kang, X., Han, J., Douglass, J., Hsiue, E. H.-C., Zhang, M., Purohit, R., deSilva, T., & Wang, Q. (2022). Valid-NEO: A Multi-Omics Platform for Neoantigen Detection and Quantification from Limited Clinical Samples. Cancers, 14(5), 1243. https://doi.org/10.3390/cancers14051243