
Multiomics Topic Modeling for Breast Cancer Classification


Abstract
:Simple Summary
Abstract
1. Introduction
- First, we will show how different layers of biological information can be efficiently integrated in the hSBM framework. We release the python package nSBM, inherited from hSBM [10], which is ready to install, easily executable, and can be used to infer the topic structure starting from different layers and types of biological data.
- Second, focusing on breast cancer, we will show that the combination of microRNA and protein-coding expression levels greatly improves the algorithm’s ability to identify cancer subtypes. These findings further confirm the important role previously recognized in several studies that miRNAs play in cancer development [19,20].
- Third, we use the inferred topic structure to select a few genes, miRNAs, and chromosomal duplications that seem to have a prognostic role in breast cancer and, thus, could be introduced as additional signatures of specific breast cancer subtypes. The extension of subtype signatures can help clinicians to fine-tune diagnostic protocols in the framework of a precision medicine approach to cancer [1].
2. Results
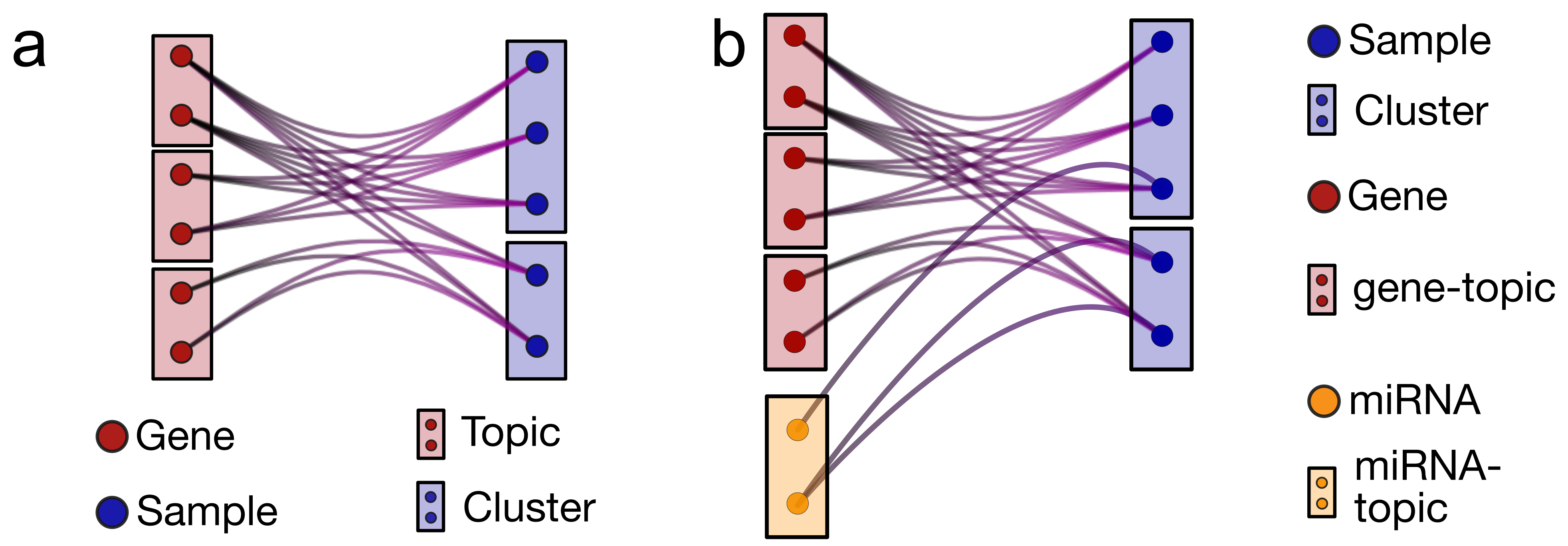
2.1. nSBM: A Multibranch Topic Modeling Algorithm
- Lack of a parametric prior.Thanks to the network-based approach and to the particular way links are used to update the block structure, this class of algorithms does not require a specific parametric assumption for the prior probability distribution of the latent variables. This is a major difference with respect to LDA and makes this class of algorithms particularly suited for biological systems in which long-tail distributions and hierarchical structures are ubiquitous (see the discussion on this point and the comparison with LDA in [3]).
- Probability distributions over latent variables of different types.The output of the algorithm is not deterministic but is instead a set of probabilities that associate a sample with latent variables of different types , and associate different features to topics, such as and . and represent the contribution of each miRNA- or gene-topic to each sample. On the other hand, and quantify how much each gene or miRNA contributes to a specific topic.As we will show in the following, these probability distributions capture relevant properties of the biological system.
- Hierarchical topic structure.Blocks and the probability distributions described above are available at different layers of resolution, from few large sets (clusters/gene-topics/miRNA-topics) at low resolution to many small sets at a higher resolution. The specific number of layers and their block composition are found by the algorithm optimization process and are not given as input. Therefore, the datasets can be organized in different ways depending on the resolution of interest. Note that not all possible resolutions are trivially present, as in standard hierarchical clustering.
- Concurrent and separate topic organization of the different network layers.Different ’omics have typically different normalization, and the numbers associated to different molecular features have often a very different meaning. A major advantage of nSBM with respect to other algorithms [26,27] is that each layer is independently contributing to the optimization process and a topic organization is given for each layer. Therefore, there is no need to reweight the different layers to balance their contributions since they are kept separate while concurrently contributing to the sample clustering. This makes the model suitable to be applied not only to genomics data, as we will discuss in this paper, but, ideally, to any combination and number of different concurrent ’omics.
2.2. Subtype Classification of Breast Cancer Samples
2.3. Integrating microRNA Expression Profiles in a Topic Modeling Analysis
2.3.1. Including miRNAs in the Topic Modeling Analysis Modifies Both the Sample Clusters and the Gene-Topics
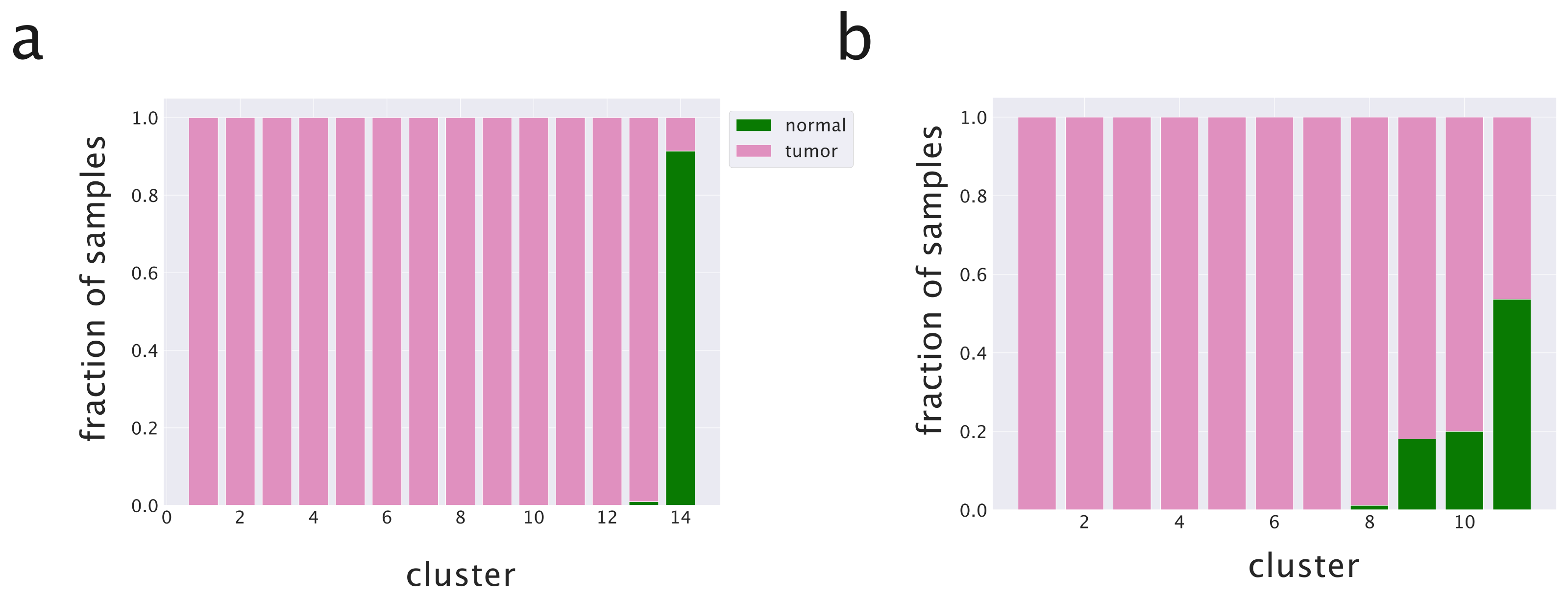
2.3.2. The Inclusion of miRNAs in the Topic Modeling Analysis Leads to a Better Separation of Healthy and Tumor Tissues
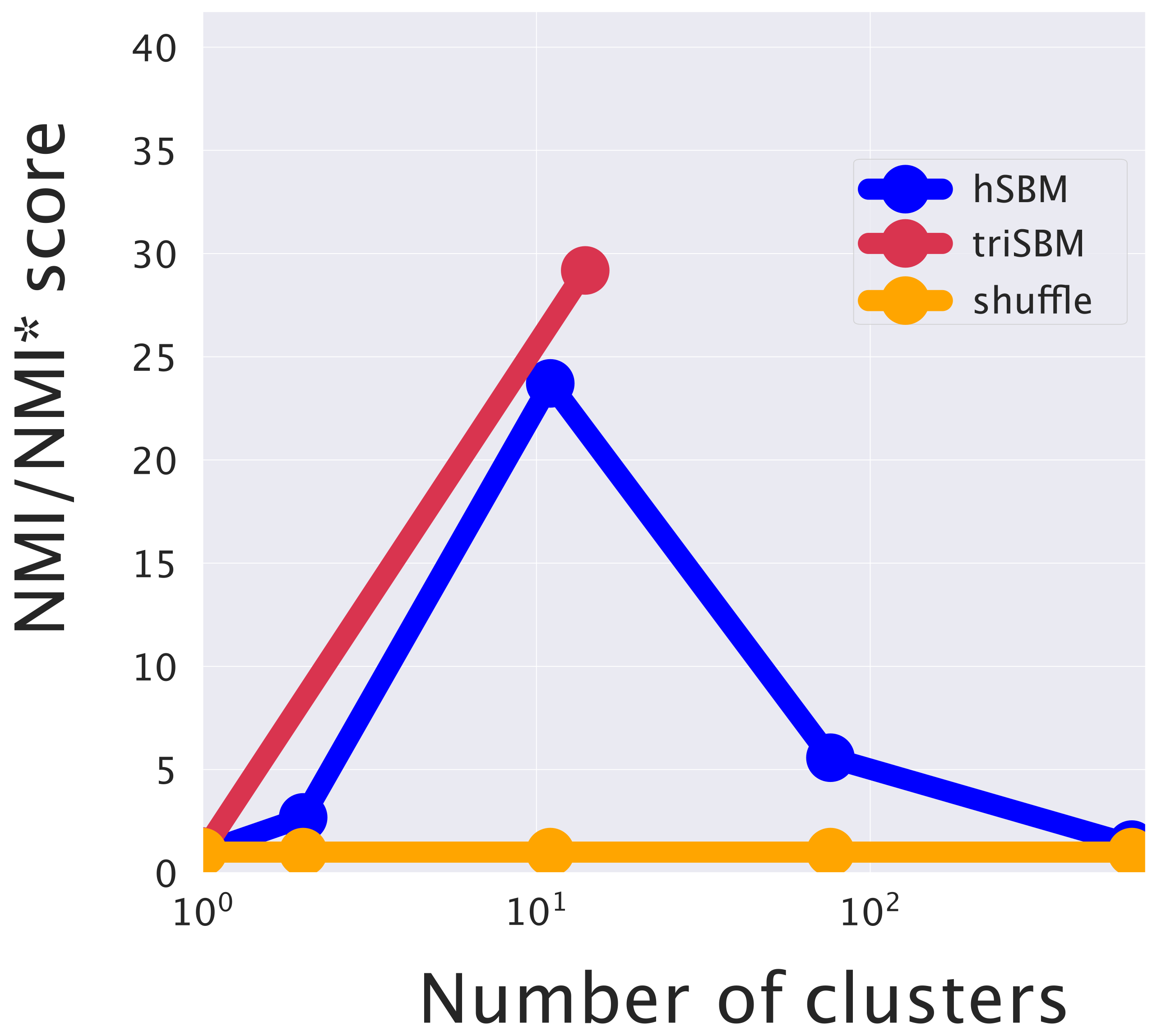
2.3.3. Including miRNAs in the Topic Modeling Analysis Improves the Identification of Cancer Subtypes
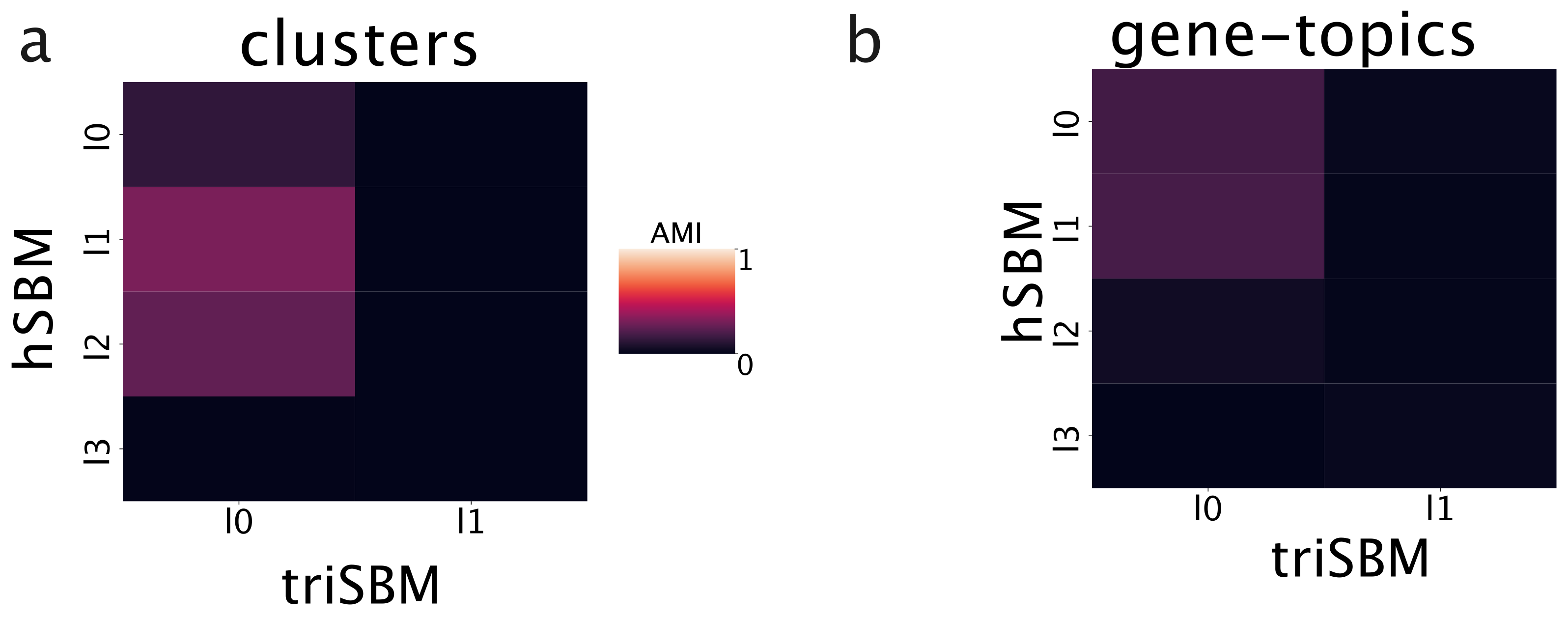
2.3.4. Check the Robustness of the Model with an Independent Labeling
2.3.5. Validation on an Independent Source of Data: METABRIC
2.4. triSBM Topics Can Be Used to Obtain Subtype-Specific Information
2.4.1. Analysis of Subtype-Specific Topics of Genes
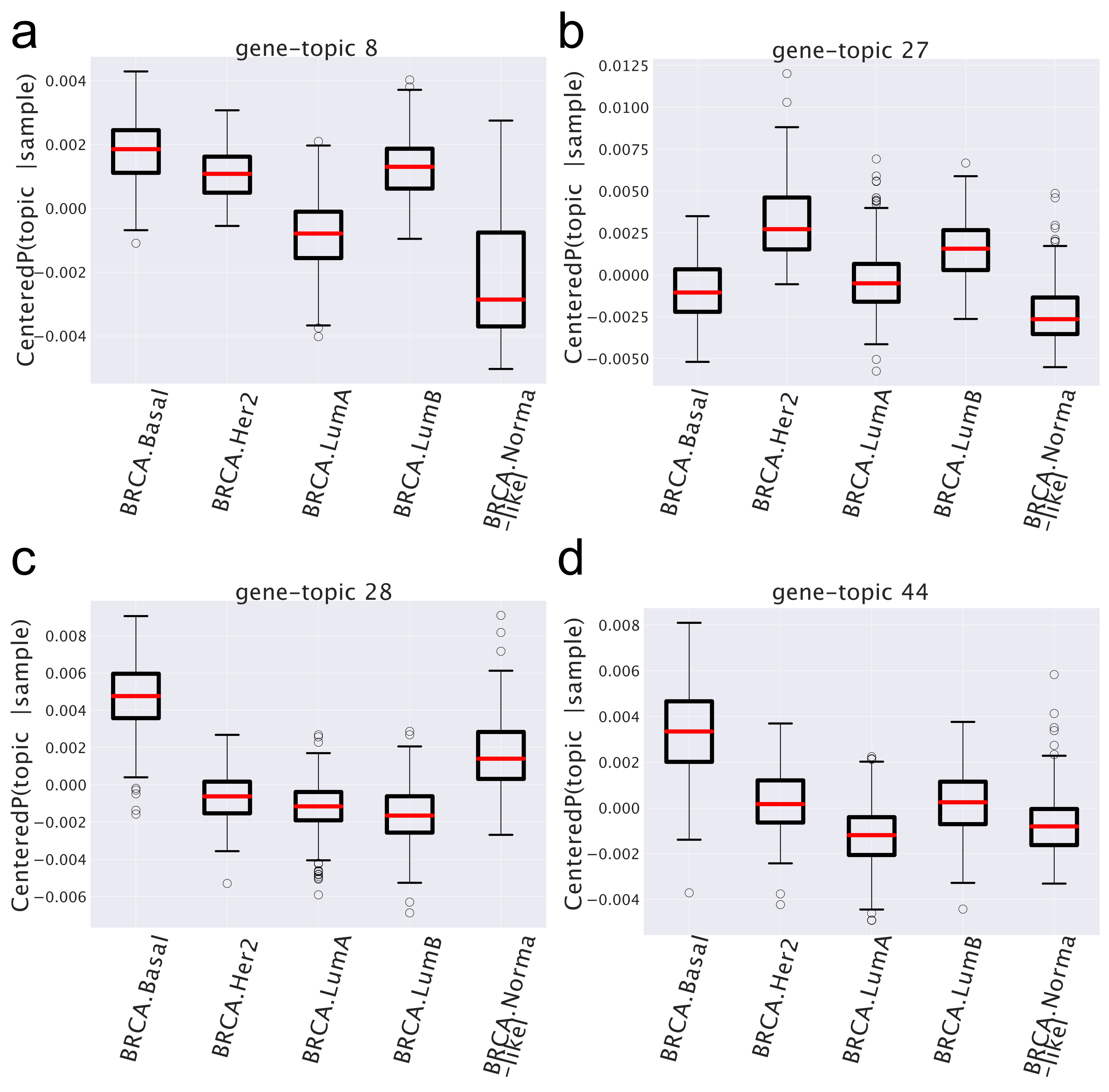
- There are topics, such as, for instance, topic 8 in Figure 6, which shows a similar behavior in all cancer subtypes and a different one (in the case of topic 8, it is depleted) in the normal tissues. These are the topics that allowed the algorithm to distinguish so accurately normal from cancer samples. In the case at hand, the functional analysis allows to easily understand the reason of this different behavior: the genes contained in topic 8 are strongly enriched in cell cycle keywords, which are likely to be associated to the proliferating nature of tumor tissues.
- Another interesting pattern is well-exemplified by topics 27, 28, and 44 in Figure 6. These are topics that are over-represented only in one particular subtype (in the example, topics 28 and 44 in the Basal subtype and topic 27 in the HER2 one) and can thus be used as signatures of these subtypes. This is in nice agreement with the finding of the gene enrichment analysis, which, for topics 28 and 44, provides a strong enrichment for the keyword SMID_BREAST_CANCER_BASAL_UP, which is known to be associated with the Basal subtype [44], while topic 27 is enriched in the keyword SMID_BREAST_CANCER_ERBB2_UP, which is in fact associated with the HER2 subtype [44]. These topics are the latent variables that allow the algorithm to distinguish among different subtypes.
2.4.2. Analysis of Subtype-Specific Topics of miRNAs
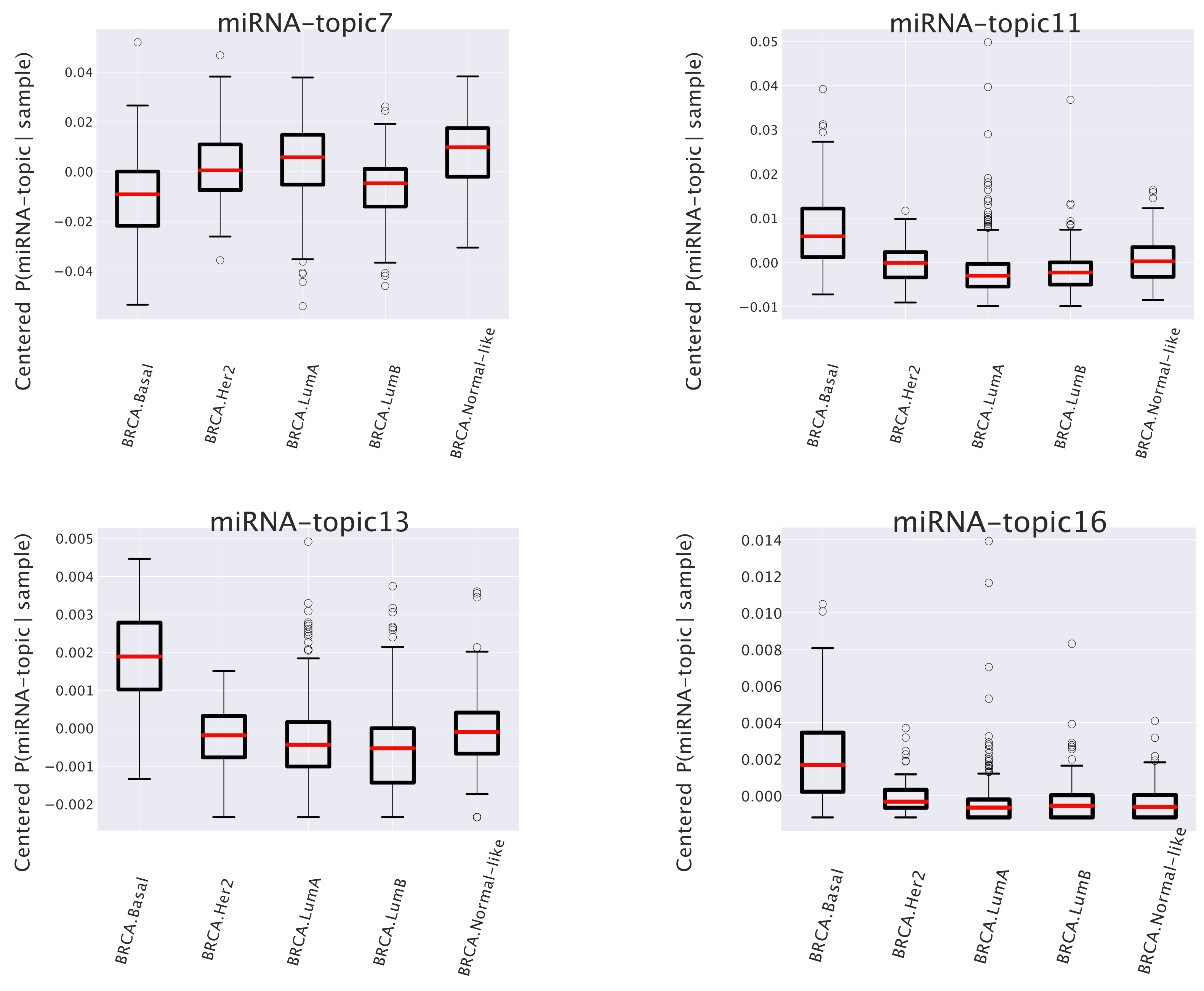
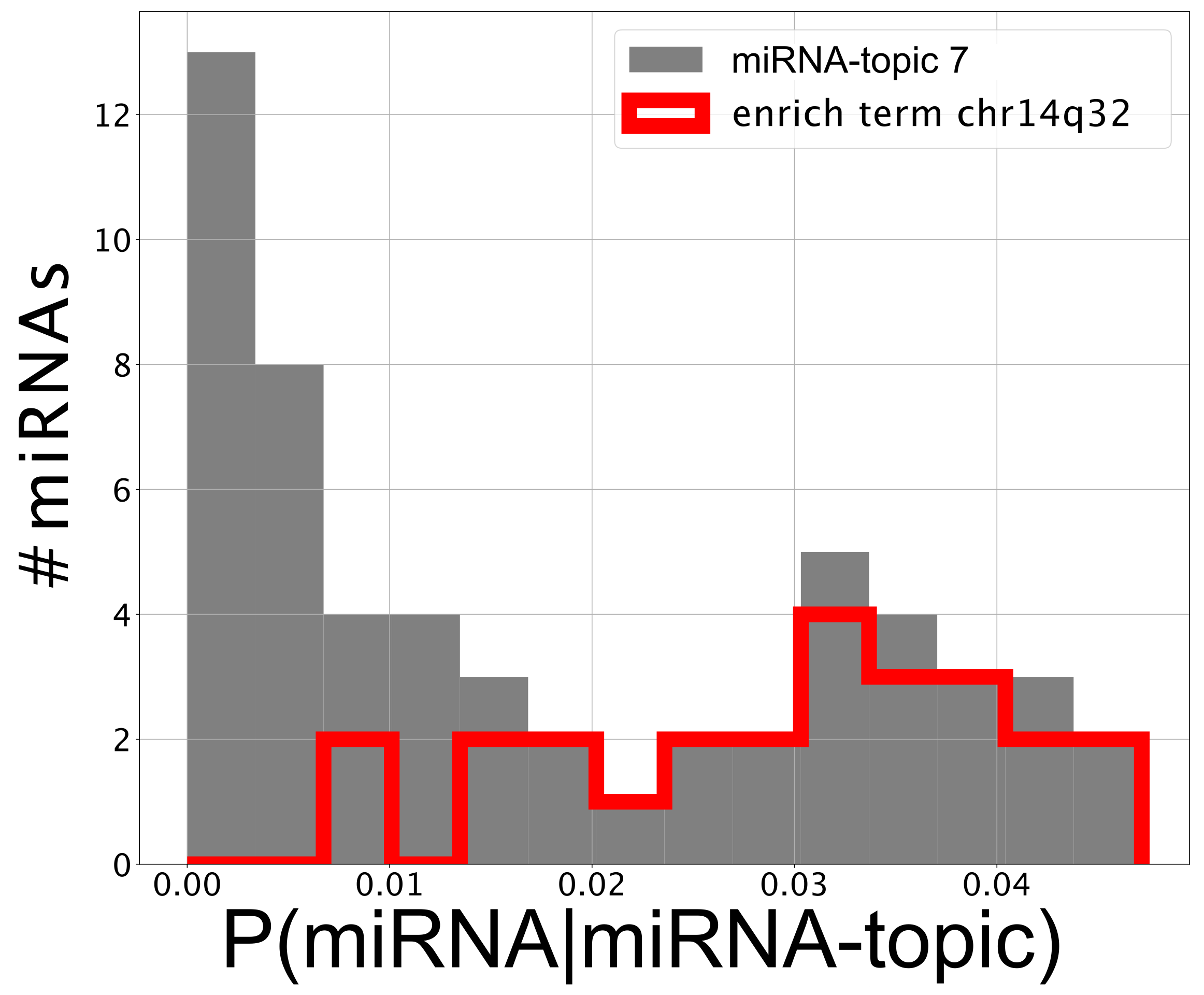
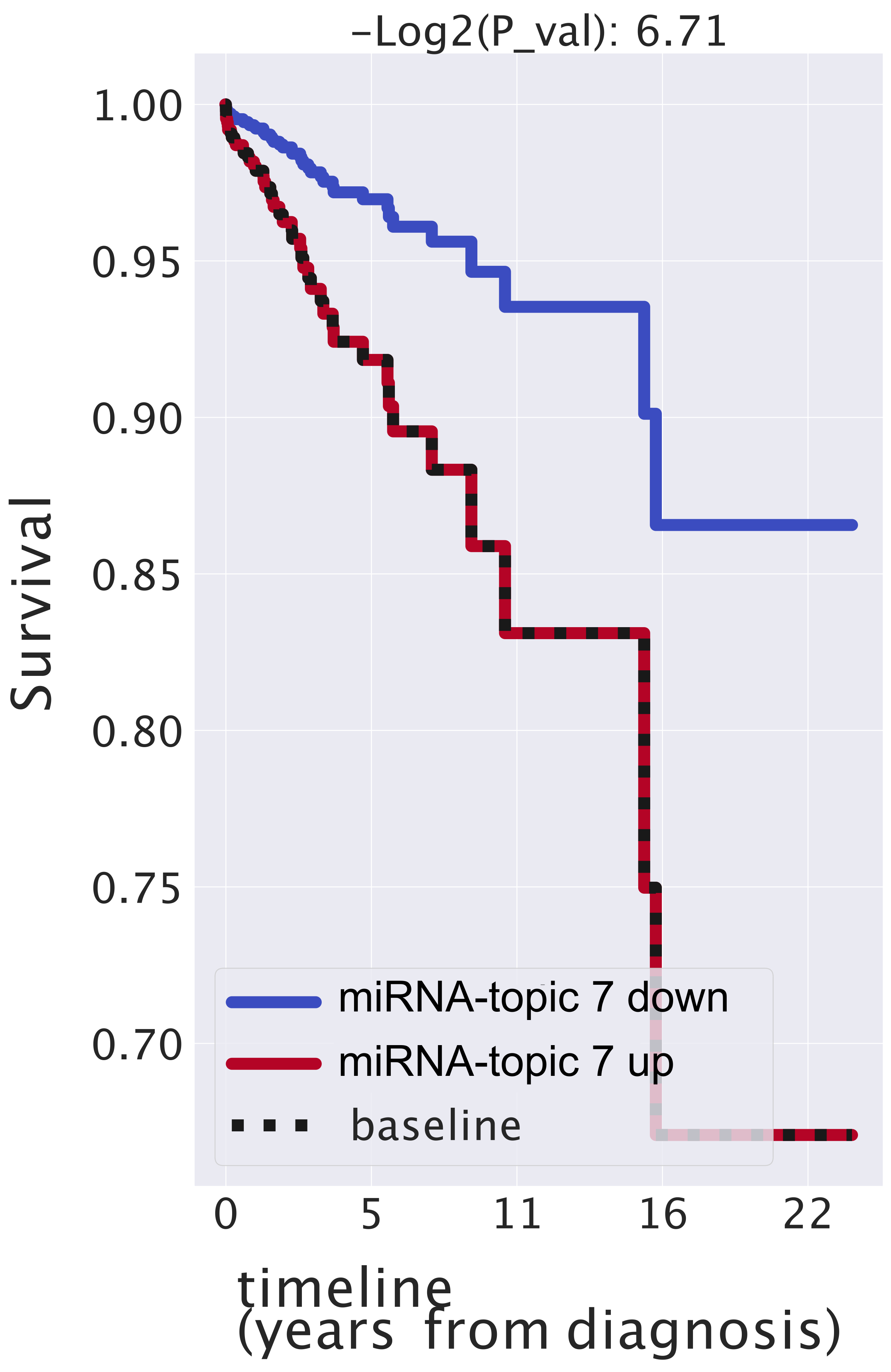
- The first one (named miRNA-topic 7 in our output, see https://github.com/BioPhys-Turin/keywordTCGA/blob/main/brca/trisbm/trisbm_level_0_topics.csv, accessed on 10 February 2022) is the typical example of a topic that shows no particular preference for a cancer subtype (see Figure 6) but shows a strong enrichment for a particular chromosomal locus: chr14q32 (see Table 2). This enrichment is due to the fact that most of the miRNAs of the topic are indeed contained in this locus. Moreover, looking at Figure 8, we see that these miRNAs are exactly those with the highest probability to belong to the topic. This strongly suggests that a somatic alteration (duplication or deletion) at this locus could be associated to the onset of cancer and could thus be used as a marker. Indeed, this locus is known to be associated with breast cancer [47]. Accordingly, if we perform a survival analysis between patients with this topic upregulated and patients with the topic downregulated (see next subsection), we find a remarkable increase in the survival probability of patients with the topic downregulated.However, this is not the end of the story. Looking at Table 2, we see that the same topic is also enriched in keywords associated to Alzheimer disease. Indeed, it is known that there is a sort of inverse comorbidity [48] between a few types of cancer (in particular, lung [49] and breast [50]) and Alzheimer’s disease. This association is confirmed and supported by our analysis, which also suggests that it could be mediated exactly by the microRNAs contained in miRNA-topic 7. Indeed, some of the miRNAs contained in the topic, such as mir-34c, are known oncosuppressors of breast cancer [51,52] and, at the same time, are recognized markers of Alzheimer’s disease [53,54]. The most important of these is the abovementioned mir-34c, which is in fact, strongly associated with miRNA-topic 7, being the only miRNA in the topic with not belonging to the locus chr14q32 (see Figure 8).
- A second class of topics is represented by the other three entries of Figure 7 (miRNA-topics 11, 13, and 16 in our output), which show a different behavior in one of the subtypes with respect to the others (in the present case, these topics are upregulated in samples belonging to the basal subtype). Out of these, only miRNA-topic 11 shows a significant entry in the table of enriched keywords: Table 2. The enrichment is for another chromosomal locus: chr19q13. What is interesting is that this locus has been associated in the past to other types of cancer [55]. Our analysis suggests that it could also play a role in breast cancer and, in particular, in the Basal subtype.
2.5. miRNAs Contained in miRNA-Topic 7 Are Strongly Associated with Breast Cancer and May Affect the Survival of Patients
3. Discussion
3.1. Including Regulatory Interactions in the TriSBM Framework
3.2. Adding Further Layers of Information: The Case of Copy Number Variation
4. Materials and Methods
4.1. The Cancer Genome Atlas Data
4.2. Metadata and Cancer Subtypes
4.3. METABRIC miRNA Landscape Data
4.4. nSBM: A Multibranch Stochastic Block Modeling Algorithm
- The search for optimal allocation of the latent variables is performed by inheriting and expanding [25] hierarchical Stochastic Block Modeling (hSBM) introduced in [10]. Note that the training process is performed simultaneously in all branches of the network: this means that all the types of data contribute to the learning process at the same time, without, in principle, any preference at the beginning.
- As mentioned in the main text, nSBM attempts to maximize the posterior probability that the model describes the datain a completely nonparametric [68] way. Instead of maximizing the probability of the model, as usual, it minimizes the Description Length . We used the minimise_nested_blockmodel_dl function from graph-tool [69]. In our setting, is a block matrix in which each block is a “Bag of Features” (i.e., genes, miRNAs, …). It can be seen as a two-dimensional matrix whose entries are the weights mentioned above. The probability of accepting the move of a node with a neighbor t from group r to group s is [65]where is the number of edges between groups t and s; is the total number of edges connected to group t. From this, another advantage of a multibranch approach should be clear: different ’omics may have their own normalization. In fact, when moving a sample from r to s, the probability is estimated considering only the branch to which t belongs. If the node t is a gene, is normalized, taking only into account the mRNA expression values.
- We set the algorithm so as to do a sort of model selection minimizing the Description Length times and then choosing the model with the shortest Description Length.
- The intrinsic complexity of typical Stochastic Block Modeling algorithms is (, , and are hyperparameters of the model), which equals if the graph is sparse () [71], where V is the number of vertices (samples, genes, and microRNAs) and E is the number of edges. If , the complexity is not logarithmic and the CPU time needed to minimize the description length increases as well. In this case, to reduce the CPU bottleneck, one can apply a log-transformation to the data, which strongly reduces the number of edges E. We ran the model on a 48-core machine with 768 GB of memory [72].
4.5. Gene and miRNA Selection
4.6. Evaluation Metrics
Description Length How Well the Model Describes the Data
4.7. Construction of the Distributions
4.8. Survival Analysis
4.9. Code and nSBM Software Package
5. Conclusions
- Using the python package: nSBM, inherited from hSBM [10], ready to install and easily executable on n-partite networks, will be straightforward to address different types of biological data.
- Second, the integration of multiple sources of data, such as microRNA expression levels and the protein-coding mRNA ones, greatly improves the ability of the algorithm to identify breast cancer subtypes.
- Third, we use our results to identify a few genes and miRNAs and characterize a few chromosomal duplications that seem to have a particular prognostic role in breast cancer and could be used as signatures to predict the particular breast cancer subtypes.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SBM | Stochastic Block Modeling |
| TCGA | The Cancer Genome Atlas |
| GSEA | Gene Set Enrichment Analysis |
| FDR | False Discovery Rate |
| FPKM | Fragments Per Kilobase of transcript per Million mapped reads |
References
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; Hsiao, C.J.; Stephens, M. Visualizing the structure of RNA-seq expression data using grade of membership models. PLoS Genet. 2017, 13, e1006599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, F.; Osella, M.; Caselle, M. A Topic Modeling Analysis of TCGA Breast and Lung Cancer Transcriptomic Data. Cancers 2020, 12, 3799. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T. Probabilistic latent semantic indexing. In Proceedings of the 22nd annual international ACM SIGIR Conference on Research and Development in Information Retrieval, Association for Computing Machinery, Berkeley, CA, USA, 1 August 1999; pp. 50–57. [Google Scholar] [CrossRef]
- Blei, D.M.; Ng, A.Y.; Jordan, M.I. Latent dirichlet allocation. J. Mach. Learn. Res. 2003, 3, 993–1022. [Google Scholar] [CrossRef]
- Lancichinetti, A.; Sirer, M.I.; Wang, J.X.; Acuna, D.; Körding, K.; Amaral, L.A.N. High-reproducibility and high-accuracy method for automated topic classification. Phys. Rev. X 2015, 5, 011007. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yao, S.; Liu, L.; Tang, L.; Dong, W. An overview of topic modeling and its current applications in bioinformatics. SpringerPlus 2016, 5, 1608. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, C.; Kaneko, K. Zipf’s Law in Gene Expression. Phys. Rev. Lett. 2003, 90, 088102. [Google Scholar] [CrossRef] [Green Version]
- Mazzolini, A.; Gherardi, M.; Caselle, M.; Cosentino Lagomarsino, M.; Osella, M. Statistics of Shared Components in Complex Component Systems. Phys. Rev. X 2018, 8, 021023. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, M.; Peixoto, T.P.; Altmann, E.G. A network approach to topic models. Sci. Adv. 2018, 4, 1360. [Google Scholar] [CrossRef] [Green Version]
- Lazzardi, S.; Valle, F.; Mazzolini, A.; Scialdone, A.; Caselle, M.; Osella, M. Emergent Statistical Laws in Single-Cell Transcriptomic Data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fortunato, S. Community detection in graphs. Phys. Rep. 2010, 486, 75–174. [Google Scholar] [CrossRef] [Green Version]
- Fortunato, S.; Hric, D. Community detection in networks: A user guide. Phys. Rep. 2016, 659, 1–44. [Google Scholar] [CrossRef] [Green Version]
- Morelli, L.; Giansanti, V.; Cittaro, D. Nested Stochastic Block Models applied to the analysis of single cell data. BMC Bioinform. 2021, 22, 576. [Google Scholar] [CrossRef]
- Holland, P.; Laskey, K.B.; Leinhardt, S. Stochastic blockmodels: First steps. Soc. Netw. 1983, 5, 109–137. [Google Scholar] [CrossRef]
- Chang, K.; Creighton, C.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrot, K.; Shmulevich, I.; The Cancer Genome Atlas Research Network. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar]
- Berger, A.C.; Korkut, A.; Kanchi, E. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Cantini, L.; Medico, E.; Fortunato, S.; Caselle, M. Detection of gene communities in multi-networks reveals cancer drivers. Sci. Rep. 2015, 5, 17386. [Google Scholar] [CrossRef] [Green Version]
- Cantini, L.; Caselle, M.; Forget, A.; Zinovyev, A.; Barillot, E.; Martignetti, L. A review of computational approaches detecting microRNAs involved in cancer. Front. Biosci. Landmark 2017, 22, 1774–1791. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E.J.; Clauset, A. Structure and inference in annotated networks. Nat. Commun. 2016, 7, 11863. [Google Scholar] [CrossRef] [Green Version]
- Mcauliffe, J.; Blei, D. Supervised Topic Models. Adv. Neural Inf. Process. Syst. 2007, 20, 121–128. [Google Scholar]
- Hyland, C.C.; Tao, Y.; Azizi, L.; Gerlach, M.; Peixoto, T.P.; Altmann, E.G. Multilayer networks for text analysis with multiple data types. EPJ Data Sci. 2021, 10, 1–16. [Google Scholar] [CrossRef]
- Fajardo-Fontiveros, O.; Guimerà, R.; Sales-Pardo, M. Node Metadata Can Produce Predictability Crossovers in Network Inference Problems. Phys. Rev. X 2022, 12, 011010. [Google Scholar] [CrossRef]
- Valle, F. nSBM: Multi Branch Topic Modeling. Zenodo 2021. Available online: https://zenodo.org/record/6120683 (accessed on 30 June 2021).
- Ward, J.H., Jr. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.; Sorlie, T.; Eisen, M.; van de Rijn, M.; Jeffrey, S.; Rees, C.; Pollack, J.; Ross, D.; Johnsen, H.; Akslen, L.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Harbeck Nadia, G.M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.; van de Rijn, M.; Jeffrey, S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; Malta, T.M.; Bontempi, G.; Ceccarelli, M.; Berman, B.P.; Noushmehr, H. TCGAbiolinksGUI: A graphical user interface to analyze cancer molecular and clinical data. F1000Research 2018, 7, 439. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Parker, J.; Fan, C.; Perou, C. PAM50 assay and the three-gene model for identifying the major and clinically relevant molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 135, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantini, L.; Caselle, M. Hope4Genes: A Hopfield-like class prediction algorithm for transcriptomic data. Sci. Rep. 2019, 9, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Li, W.X.; Guan, D.; Gong, M.; Ye, S.; Fang, Z.; Huang, J.F.; Lu, A. Regulatory network reconstruction of five essential microRNAs for survival analysis in breast cancer by integrating miRNA and mRNA expression datasets. Funct. Integr. Genom. 2019, 19, 645–658. [Google Scholar] [CrossRef]
- Bertoli, G.; Cava, C.; Castiglioni, I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics 2015, 5, 1122–1143. [Google Scholar] [CrossRef]
- Rosenberg, A.; Hirschberg, J. V-measure: A conditional entropy-based external cluster evaluation measure. In Proceedings of the 2007 Joint Conference on Empirical Methods in Natural Language Processing and Computational Natural Language Learning, Prague, Czech Republic, 28–30 June 2007; pp. 410–420. [Google Scholar]
- Shi, H.; Gerlach, M.; Diersen, I.; Downey, D.; Amaral, L. A new evaluation framework for topic modeling algorithms based on synthetic corpora. In Proceedings of the 22nd International Conference on Artificial Intelligence and Statistics, Okinawa, Japan, 16–18 April 2019; pp. 816–826. [Google Scholar]
- Horr, C.; Buechler, S.A. Breast Cancer Consensus Subtypes: A system for subtyping breast cancer tumors based on gene expression. NPJ Breast Cancer 2021, 7, 136. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.M.; Foekens, J.A.; Martens, J.W.M. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [Green Version]
- van ’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adélaïde, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006, 25, 2273–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drago-García, D.; Espinal-Enríquez, J.; Hernández-Lemus, E. Network analysis of EMT and MET micro-RNA regulation in breast cancer. Sci. Rep. 2017, 7, 13534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalá-López, F.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Valderas, J.M.; Gómez-Beneyto, M.; Martinez, S.; Balanzá-Martínez, V.; Climent, J.; Valencia, A.; McGrath, J.; et al. Inverse and Direct Cancer Comorbidity in People with Central Nervous System Disorders: A Meta-Analysis of Cancer Incidence in 577,013 Participants of 50 Observational Studies. Psychother. Psychosom. 2014, 83, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Sanchez Valle, J.; Pancaldi, V.; Baudot, A.; Barillot, E.; Caselle, M.; Valencia, A.; Zinovyev, A.; Cantini, L. Molecular Inverse Comorbidity between Alzheimer’s Disease and Lung Cancer: New Insights from Matrix Factorization. Int. J. Mol. Sci. 2019, 20, 3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forés-Martos, J.; Boullosa, C.; Rodrigo-Domínguez, D.; Sánchez-Valle, J.; Suay-García, B.; Climent, J.; Falcó, A.; Valencia, A.; Puig-Butillé, J.A.; Puig, S.; et al. Transcriptomic and Genetic Associations between Alzheimer’s Disease, Parkinson’s Disease, and Cancer. Cancers 2021, 13, 2990. [Google Scholar] [CrossRef] [PubMed]
- Achari, C.; Winslow, S.; Ceder, Y.; Larsson, C. Expression of miR-34c induces G2/M cell cycle arrest in breast cancer cells. BMC Cancer 2014, 14, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Li, Y.; Gao, J.; Zhang, T.; Li, S.; Luo, A.; Chen, H.; Ding, F.; Wang, X.; Liu, Z. MicroRNA-34 suppresses breast cancer invasion and metastasis by directly targeting Fra-1. Oncogene 2013, 32, 4294–4303. [Google Scholar] [CrossRef]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Lee, K.F.; Lu, Y.; Clarke, I.; Shih, D.; Eberhart, C.; Collins, V.P.; Van Meter, T.; Picard, D.; Zhou, L.; et al. Frequent Amplification of a chr19q13.41 MicroRNA Polycistron in Aggressive Primitive Neuroectodermal Brain Tumors. Cancer Cell 2009, 16, 533–546. [Google Scholar] [CrossRef] [Green Version]
- Cantini, L.; Bertoli, G.; Cava, C.; Dubois, T.; Zinovyev, A.; Caselle, M.; Castiglioni, I.; Barillot, E.; Martignetti, L. Identification of microRNA clusters cooperatively acting on epithelial to mesenchymal transition in triple negative breast cancer. Nucleic Acids Res. 2019, 47, 2205–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease–gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef]
- Cox, D.R. Regression models and life-tables. J. R. Stat. Soc. 1972, 34, 187–202. [Google Scholar] [CrossRef]
- Osella, M.; Riba, A.; Testori, A.; Corà, D.; Caselle, M. Interplay of microRNA and epigenetic regulation in the human regulatory network. Front. Genet. 2014, 5, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reale, E.; Taverna, D.; Cantini, L.; Martignetti, L.; Osella, M.; De Pittà, C.; Virga, F.; Orso, F.; Caselle, M. Investigating the epi-miRNome: Identification of epi-miRNAs using transfection experiments. Epigenomics 2019, 11, 1581–1599. [Google Scholar] [CrossRef] [PubMed]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1—integrative database of human microRNA target predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef]
- Papadopoulos, G.L.; Reczko, M.; Simossis, V.A.; Sethupathy, P.; Hatzigeorgiou, A.G. The database of experimentally supported targets: A functional update of TarBase. Nucleic Acids Res. 2009, 37, D155–D158. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, T.P. Merge-split Markov chain Monte Carlo for community detection. Phys. Rev. E 2020, 102, 012305. [Google Scholar] [CrossRef]
- Nikolsky, Y.; Sviridov, E.; Yao, J.; Dosymbekov, D.; Ustyansky, V.; Kaznacheev, V.; Dezso, Z.; Mulvey, L.; Macconaill, L.E.; Winckler, W.; et al. Genome-wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res. 2008, 68, 9532–9540. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, T.P. Model Selection and Hypothesis Testing for Large-Scale Network Models with Overlapping Groups. Physical Review X 2015, 5, 011033. [Google Scholar] [CrossRef] [Green Version]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.; Fulton, R.; McLellan, M. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, T.P. Nonparametric Bayesian inference of the microcanonical stochastic block model. Phys. Rev. E 2017, 95, 12317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, T.P. The graph-tool python library. Figshare 2014. [Google Scholar] [CrossRef]
- Peixoto, T.P. Hierarchical Block Structures and High-Resolution Model Selection in Large Networks. Phys. Rev. X 2014, 4, 011047. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, T.P. Efficient Monte Carlo and greedy heuristic for the inference of stochastic block models. Phys. Rev. E 2014, 89, 012804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinucci, M.; Bagnasco, S.; Lusso, S.; Pasteris, P.; Rabellino, S.; Vallero, S. OCCAM: A flexible, multi-purpose and extendable HPC cluster. J. Physics Conf. Ser. 2017, 898, 082039. [Google Scholar] [CrossRef] [Green Version]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Yen, T.C.; Larremore, D.B. Community detection in bipartite networks with stochastic block models. Phys. Rev. E 2020, 102, 032309. [Google Scholar] [CrossRef]
- Kass, R.E.; Raftery, A.E. Bayes Factors; American Statistical Association: Boston, MA, USA, 1995; Volume 90, pp. 773–795. [Google Scholar] [CrossRef]
- Lucchetta, M.; da Piedade, I.; Mounir, M.; Vabistsevits, M.; Terkelsen, T.; Papaleo, E. Distinct signatures of lung cancer types: Aberrant mucin O-glycosylation and compromised immune response. BMC Cancer 2019, 19, 824. [Google Scholar] [CrossRef]
- Davidson-Pilon, C. lifelines: Survival analysis in Python. J. Open Source Softw. 2019, 4, 1317. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | False Discovery Rate |
|---|---|
| gene-topic 6 (55) | |
| SMID_BREAST_CANCER_BASAL_DN | × |
| FARMER_BREAST_CANCER_APOCRINE_VS_LUMIN MINAL | × |
| gene-topic 8 (19) | |
| MODULE_54 (cell cycle) | × |
| gene-topic 12 (13) | |
| MODULE_1 (ovary genes) | × |
| SMID_BREAST_CANCER_BASAL_DN | |
| gene-topic 15 (26) | |
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSINSITION | × |
| gene-topic 25 (40) | |
| CHARAFE_BREAST_CANCER_LUMINAL_VS_MESEN SENCHYMAL_UP | × |
| SMID_BREAST_CANCER_BASAL_DN | × |
| VANTVEER_BREAST_CANCER_ESR1_UP | × |
| gene-topic 27 (44) | |
| SMID_BREAST_CANCER_ERBB2_UP | × |
| gene-topic 28 (53) | |
| SMID_BREAST_CANCER_BASAL_UP | × |
| gene-topic 37 (54) | |
| FAN_OVARY_CL13_MONOCYTE_MACROPHAGE | × |
| VANTVEER_BREAST_CANCER_ESR1_DN | × |
| gene-topic 44 (37) | |
| SMID_BREAST_CANCER_BASAL_UP | × |
| gene-topic 53 (39) | |
| SMID_BREAST_CANCER_BASAL_DN | × |
| gene-topic 55 (58) | |
| SMID_BREAST_CANCER_BASAL_DN | × |
| FARMER_BREAST_CANCER_BASAL_VS_LULMINAL | × |
| VANTVEER_BREAST_CANCER_ESR1_UP | × |
| SMID_BREAST_CANCER_LUMINAL_B_UP | × |
| gene-topic 68 (54) | |
| SMID_BREAST_CANCER_BASAL_UP | × |
| CHARAFE_BREAST_CANCER_LUMINAL_VS_BASAL_DN | × |
| SMID_BREAST_CANCER_LUMINAL_B_DN | × |
| CHARAFE_BREAST_CANCER_LUMINAL_VS_MESENCHYMAL_DN | × |
| Term | False Discovery Rate |
|---|---|
| miRNA-topic 7 (57) | |
| chr14q32 | × |
| WP_ALZHEIMERS_DISEASE | × |
| miRNA-topic 11 (60) | |
| chr19q13 | × |
| microRNA | P(miRNA|miRNA-topic 7) |
|---|---|
| hsa-mir-654 | 0.047 |
| hsa-mir-758 | 0.046 |
| hsa-mir-493 | 0.042 |
| hsa-mir-889 | 0.041 |
| hsa-mir-34c | 0.041 |
| hsa-mir-431 | 0.039 |
| hsa-mir-369 | 0.039 |
| hsa-mir-370 | 0.039 |
| hsa-mir-410 | 0.037 |
| hsa-mir-154 | 0.035 |
| hsa-mir-495 | 0.035 |
| hsa-mir-511 | 0.035 |
| hsa-mir-411 | 0.033 |
| hsa-mir-432 | 0.032 |
| hsa-mir-31 | 0.031 |
| hsa-mir-487b | 0.030 |
| hsa-mir-376c | 0.030 |
| hsa-mir-412 | 0.030 |
| … | <0.030 |
| Term | False Discovery Rate |
|---|---|
| CNV-topic 1 (41) | |
| chr20q13 | × |
| NIKOLSKY_BREAST_CANCER_20Q12_Q13_AMPLICON | × |
| CNV-topic 3 (50) | |
| chr17q23 | × |
| NIKOLSKY_BREAST_CANCER_17Q21_Q25_AMPLICON | × |
| CNV-topic 4 (17) | |
| chr8q24 | × |
| NIKOLSKY_BREAST_CANCER_8Q23_Q24_AMPLICON | × |
| CNV-topic 6 (53) | |
| chr8q12 | × |
| chr8q11 | × |
| chr8q13 | × |
| NIKOLSKY_BREAST_CANCER_8Q12_Q22_AMPLICON | × |
| CNV-topic 7 (47) | |
| chr1q32 | × |
| chr1q41 | × |
| CNV-topic 13 (14) | |
| chr8q11 | × |
| NIKOLSKY_BREAST_CANCER_8P12_P11_AMPLICON | × |
| CNV-topic 18 (16) | |
| NIKOLSKY_BREAST_CANCER_17Q21_Q25_AMPLICON | × |
| chr17q23 | × |
| CNV-topic 22 (11) | |
| chr8p11 | × |
| NIKOLSKY_BREAST_CANCER_8P12_P11_AMPLICON | × |
| CNV-topic 25 (11) | |
| NIKOLSKY_BREAST_CANCER_8P12_P11_AMPLICON | × |
| chr8p11 | × |
| CNV-topic 26 (21) | |
| chr20q13 | × |
| NIKOLSKY_BREAST_CANCER_20Q12_Q13_AMPLICON | × |
| CNV-topic 28 (5) | |
| NIKOLSKY_BREAST_CANCER_17Q11_Q21_AMPLICON | × |
| chr17q21 | × |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valle, F.; Osella, M.; Caselle, M. Multiomics Topic Modeling for Breast Cancer Classification. Cancers 2022, 14, 1150. https://doi.org/10.3390/cancers14051150
Valle F, Osella M, Caselle M. Multiomics Topic Modeling for Breast Cancer Classification. Cancers. 2022; 14(5):1150. https://doi.org/10.3390/cancers14051150
Chicago/Turabian StyleValle, Filippo, Matteo Osella, and Michele Caselle. 2022. "Multiomics Topic Modeling for Breast Cancer Classification" Cancers 14, no. 5: 1150. https://doi.org/10.3390/cancers14051150
APA StyleValle, F., Osella, M., & Caselle, M. (2022). Multiomics Topic Modeling for Breast Cancer Classification. Cancers, 14(5), 1150. https://doi.org/10.3390/cancers14051150