
DNA Repair Enzyme Poly(ADP-Ribose) Polymerase 1/2 (PARP1/2)-Targeted Nuclear Imaging and Radiotherapy


Abstract
:Simple Summary
Abstract
1. Introduction to PARP Inhibitors
2. Introduction to Radiolabeled PARP Inhibitors
3. Preclinical Development and Recent Advances in PARP Imaging Agents
3.1. Olaparib-like Radiotracers
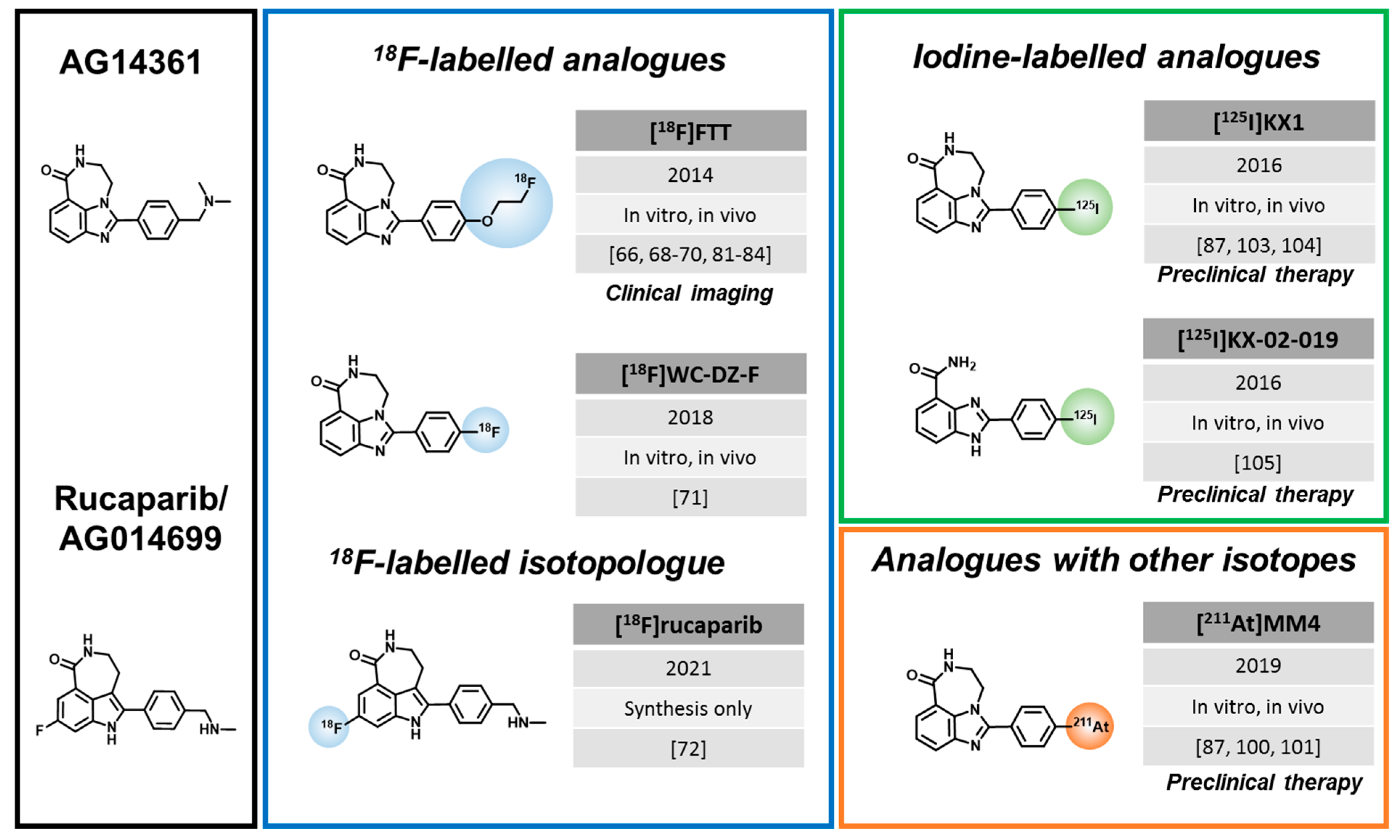
3.2. Rucaparib-like Radiotracers
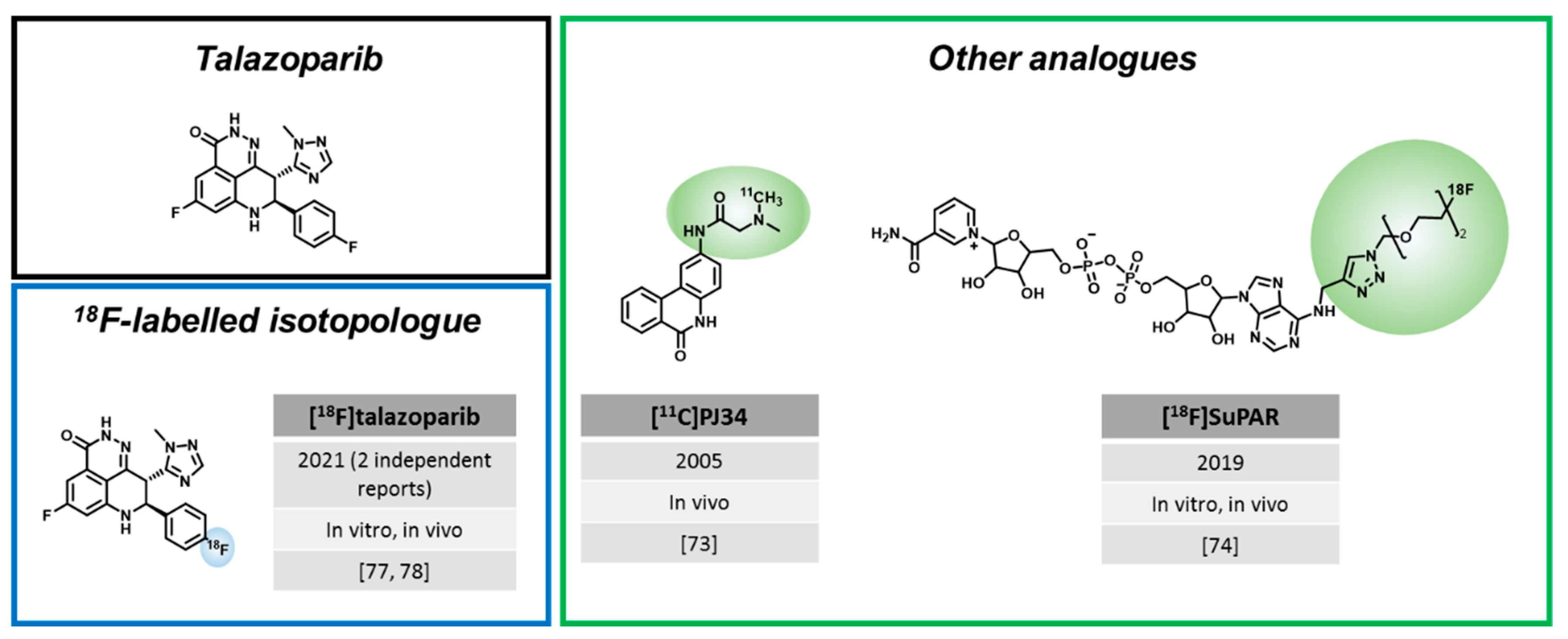
3.3. Radiotracers Based on Other PARPi
4. Clinical Evaluation of PARP Imaging Agents
4.1. [18F]PARPi


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tracer | Conditions | Planned/Final Cohort Size | Status (Clinicaltrials.Org) | NCT Number | Study Parameters | Results Published |
|---|---|---|---|---|---|---|
| [18F]PARPi | Head and neck cancer | 12 | Finished 1 | NCT03631017 | Static [18F]PARPi and [18F]FDG PET | [79] |
| New or recurrent brain tumors | 8 | Ongoing 1 | NCT04173104 | Static [18F]PARPi and [18F]FDG PET | [43] | |
| [18F]FTT | Head and neck, lung, ovarian, gastric, or pancreatic cancer | 50/16 | Finished 2 | NCT02469129 | Static [18F]FTT PET | [82,83] |
| Epithelial ovarian, fallopian tube, or primary peritoneal cancer | 30/20 | Ongoing 3 | NCT02637934 | Dynamic and static [18F]FTT and [18F]FDG PET, IF/IHC correlation | [70,84] | |
| Primary breast cancer | 30/30 | Finished 3 | NCT03083288 | Static [18F]FTT PET | [85] | |
| Primary or metastatic breast cancer | 30/4 | Ongoing 4 | NCT03846167 | [18F]FTT PET pre and post PARPi therapy | [81] | |
| Prostate cancer | 30 | Finished 3 | NCT03334500 | / | ||
| Pancreatic cancer | 30 | Ongoing 3 | NCT03492164 | / | ||
| Solid tumors | 120 | Ongoing 5 | NCT03604315 | / | ||
| Glioblastoma | 12 | Ongoing 4 | NCT04221061 | / | ||
| Breast Cancer | 36 | Not yet recruiting 6 | NCT05226663 | / |
4.2. [18F]FluorThanatrace
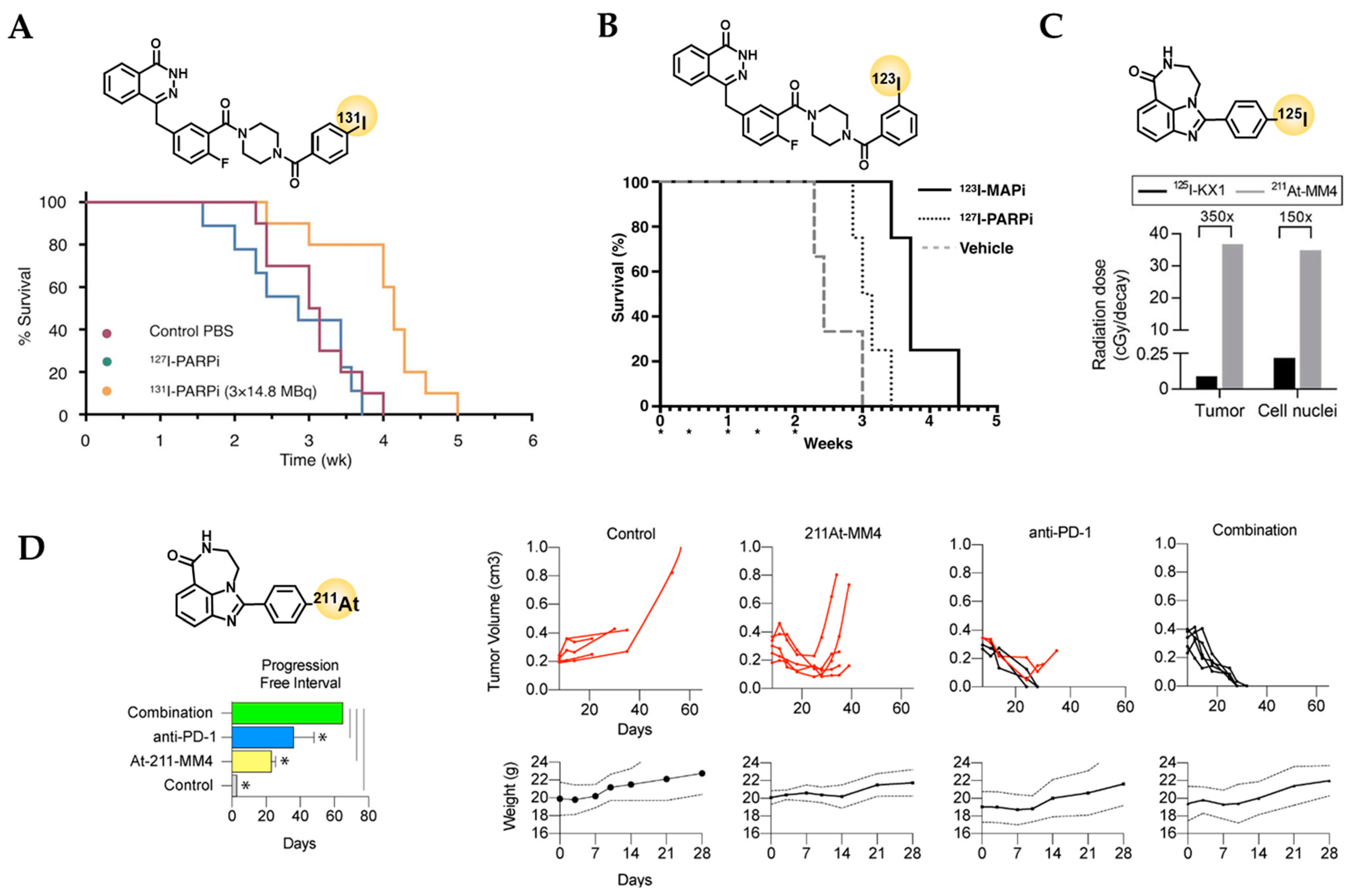
5. Current Status of PARP-Targeted Radiotherapy
5.1. Olaparib-like Radiotherapeutics
| Agent | Publication | Tumor Model | Mouse Strain | Treatment Groups | Median Survival |
|---|---|---|---|---|---|
| [211At]MM4 | [100] | GL26 (syngeneic) (Glioblastoma) | CB57BL/6J |
| PFI *:
|
| [101] | IMR-05 (Neuroblastoma) | SCID Hairless Congenic |
|
| |
| [123I]MAPi | [96] | U87-p53/tdTomato-CBRluc-Neo (Glioblastoma) | CrTac:NCr-Fo |
|
|
| [97] | TS543 (Glioblastoma) | CrTac:NCr-Fo |
|
| |
| [99] | HCT116 p53+/+ | CrTac:NCr-Fo |
|
| |
| [99] | HCT116 p53−/− (Colorectal cancer) | CrTac:NCr-Fo |
|
|
5.2. Rucaparib-like Radiotherapeutics
6. Considerations for Clinical Manufacturing
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, M.O.; Perry, N.J.S.; Poulogiannis, G. DNA damage, repair, and cancer metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, J.M. The comings and goings of parp-1 in response to DNA damage. DNA Repair. 2018, 71, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Haenni, S.S.; Elser, M.; Hottiger, M.O. Nuclear adp-ribosylation reactions in mammalian cells: Where are we today and where are we going? Microbiol. Mol. Biol. Rev. 2006, 70, 789–829. [Google Scholar] [CrossRef] [Green Version]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.E.; Malki, M.I. DNA damage/repair management in cancers. Cancers (Basel) 2020, 12, 1050. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A. Parp inhibitors as therapeutics: Beyond modulation of parylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef] [Green Version]
- Ferraris, D.V. Evolution of poly(adp-ribose) polymerase-1 (parp-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef]
- Lupo, B.; Trusolino, L. Inhibition of poly(adp-ribosyl)ation in cancer: Old and new paradigms revisited. Biochim. Biophys. Acta 2014, 1846, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Curtin, N.J.; Szabo, C. Therapeutic applications of parp inhibitors: Anticancer therapy and beyond. Mol. Asp. Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. Parp inhibition: Parp1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. Parp inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. Fda approval summary: Olaparib monotherapy or in combination with bevacizumab for the maintenance treatment of patients with advanced ovarian cancer. Oncologist 2021, 26, e164–e172. [Google Scholar] [CrossRef]
- Deeks, E.D. Olaparib: First global approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Rucaparib: First global approval. Drugs 2017, 77, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Niraparib: First global approval. Drugs 2017, 77, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Talazoparib: First global approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.P.; Doroshow, J.H.; Ji, J.P.; Takeda, S.; Pommier, Y. Trapping of parp1 and parp2 by clinical parp inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific parp trapping by bmn 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Krastev, D.B.; Wicks, A.J.; Lord, C.J. Parp inhibitors—Trapped in a toxic love affair. Cancer Res. 2021, 81, 5605–5607. [Google Scholar] [CrossRef]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to parp inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density crispr-cas9 screens identify point mutations in parp1 causing parp inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.; Yang, J.C.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. Hpf1 completes the parp active site for DNA damage-induced adp-ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; Roberts, G.; Luger, K. Histone parylation factor 1 contributes to the inhibition of parp1 by cancer drugs. Nat. Commun. 2021, 12, 736. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Murai, J.; Pommier, Y. The evolving landscape of predictive biomarkers of response to parp inhibitors. J. Clin. Investig. 2018, 128, 1727–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossatz, S.; Carney, B.; Schweitzer, M.; Carlucci, G.; Miloushev, V.Z.; Maachani, U.B.; Rajappa, P.; Keshari, K.R.; Pisapia, D.; Weber, W.A.; et al. Biomarker-based pet imaging of diffuse intrinsic pontine glioma in mouse models. Cancer Res. 2017, 77, 2112–2123. [Google Scholar] [CrossRef] [Green Version]
- Demétrio de Souza França, P.; Roberts, S.; Kossatz, S.; Guru, N.; Mason, C.; Zanoni, D.K.; Abrahão, M.; Schöder, H.; Ganly, I.; Patel, S.G.; et al. Fluorine-18 labeled poly (adp-ribose) polymerase1 inhibitor as a potential alternative to 2-deoxy-2-[18f]fluoro-d-glucose positron emission tomography in oral cancer imaging. Nucl. Med. Biol. 2020, 84–85, 80–87. [Google Scholar] [CrossRef]
- Ambur Sankaranarayanan, R.; Kossatz, S.; Weber, W.; Beheshti, M.; Morgenroth, A.; Mottaghy, F.M. Advancements in parp1 targeted nuclear imaging and theranostic probes. J. Clin. Med. 2020, 9, 2130. [Google Scholar] [CrossRef]
- Knight, J.C.; Koustoulidou, S.; Cornelissen, B. Imaging the DNA damage response with pet and spect. Eur. J. Nucl. Med. Mol. Imaging 2017, 1065–1078. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.Y.; Tan, K.V.; Cornelissen, B. Parp inhibitors in cancer diagnosis and therapy. Clin. Cancer Res. 2021, 27, 1585–1594. [Google Scholar] [CrossRef]
- Puentes, L.N.; Makvandi, M.; Mach, R.H. Molecular imaging: Parp-1 and beyond. J. Nucl. Med. 2021, 62, 765–770. [Google Scholar] [CrossRef]
- Keliher, E.J.; Reiner, T.; Turetsky, A.; Hilderbrand, S.A.; Weissleder, R. High-yielding, two-step 18f labeling strategy for 18f-parp1 inhibitors. ChemMedChem 2011, 6, 424–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, T.; Keliher, E.J.; Earley, S.; Marinelli, B.; Weissleder, R. Synthesis and in vivo imaging of a 18f-labeled parp1 inhibitor using a chemically orthogonal scavenger-assisted high-performance method. Angew. Chem. 2011, 50, 1922–1925. [Google Scholar] [CrossRef] [PubMed]
- Reiner, T.; Lacy, J.; Keliher, E.J.; Yang, K.S.; Ullal, A.; Kohler, R.H.; Vinegoni, C.; Weissleder, R. Imaging therapeutic parp inhibition in vivo through bioorthogonally developed companion imaging agents. Neoplasia 2012, 14, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Keliher, E.J.; Klubnick, J.A.; Reiner, T.; Mazitschek, R.; Weissleder, R. Efficient acid-catalyzed (18) f/(19) f fluoride exchange of bodipy dyes. ChemMedChem 2014, 9, 1368–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlucci, G.; Carney, B.; Brand, C.; Kossatz, S.; Irwin, C.P.; Carlin, S.D.; Keliher, E.J.; Weber, W.; Reiner, T. Dual-modality optical/pet imaging of parp1 in glioblastoma. Mol. Imaging Biol. 2015, 17, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Guru, N.; França, P.; Pirovano, G.; Huang, C.; Patel, S.; Reiner, T. [18f]parpi imaging is not affected by hpv status in vitro. Mol. Imaging 2021, 2021, 1–10. [Google Scholar] [CrossRef]
- Carney, B.; Carlucci, G.; Salinas, B.; Di Gialleonardo, V.; Kossatz, S.; Vansteene, A.; Longo, V.A.; Bolaender, A.; Chiosis, G.; Keshari, K.R.; et al. Non-invasive pet imaging of parp1 expression in glioblastoma models. Mol. Imaging Biol. 2016, 18, 386–392. [Google Scholar] [CrossRef] [Green Version]
- Carney, B.; Kossatz, S.; Lok, B.H.; Schneeberger, V.; Gangangari, K.K.; Pillarsetty, N.V.K.; Weber, W.A.; Rudin, C.M.; Poirier, J.T.; Reiner, T. Target engagement imaging of parp inhibitors in small-cell lung cancer. Nat. Commun. 2018, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Salloum, D.; Carney, B.; Brand, C.; Kossatz, S.; Sadique, A.; Lewis, J.S.; Weber, W.A.; Wendel, H.G.; Reiner, T. Targeted pet imaging strategy to differentiate malignant from inflamed lymph nodes in diffuse large b-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, e7441–e7449. [Google Scholar] [CrossRef] [Green Version]
- Donabedian, P.L.; Kossatz, S.; Engelbach, J.A.; Jannetti, S.A.; Carney, B.; Young, R.J.; Weber, W.A.; Garbow, J.R.; Reiner, T. Discriminating radiation injury from recurrent tumor with [(18)f]parpi and amino acid pet in mouse models. EJNMMI Res. 2018, 8, 59. [Google Scholar] [CrossRef]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2h-phthalazin- 1-one: A novel bioavailable inhibitor of poly(adp-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.C.; Pillarsetty, N.; Reiner, T. A one-pot radiosynthesis of [18f]parpi. J. Label. Compd. Radiopharm. 2020, 63, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Demétrio De Souza França, P.; Pirovano, G.; Piotrowski, A.F.; Nicklin, P.J.; Riedl, C.C.; Schwartz, J.; Bale, T.A.; Donabedian, P.L.; Kossatz, S.; et al. Preclinical and first-in-human-brain-cancer applications of [18f]poly (adp-ribose) polymerase inhibitor pet/mr. Neuro-Oncol. Adv. 2020, vdaa119. [Google Scholar] [CrossRef] [PubMed]
- Kossatz, S.; Pirovano, G.; Franca, P.D.D.; Strome, A.L.; Sunny, S.P.; Zanoni, D.K.; Mauguen, A.; Carney, B.; Brand, C.; Shah, V.; et al. Validation of the use of a fluorescent parp1 inhibitor for the detection of oral, oropharyngeal and oesophageal epithelial cancers. Nat. Biomed. Eng. 2020, 4, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Kossatz, S.; Brand, C.; Gutiontov, S.; Liu, J.T.; Lee, N.Y.; Gonen, M.; Weber, W.A.; Reiner, T. Detection and delineation of oral cancer with a parp1 targeted optical imaging agent. Sci. Rep. 2016, 6, 21371. [Google Scholar] [CrossRef] [Green Version]
- Franca, P.D.D.; Kossatz, S.; Brand, C.; Zanoni, D.K.; Roberts, S.; Guru, N.; Adilbay, D.; Mauguen, A.; Mayor, C.V.; Weber, W.A.; et al. A phase i study of a parp1-targeted topical fluorophore for the detection of oral cancer. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3618–3630. [Google Scholar] [CrossRef]
- Franca, P.D.D.; Guru, N.; Roberts, S.; Kossatz, S.; Mason, C.; Abrahao, M.; Ghossein, R.A.; Patel, S.G.; Reiner, T. Fluorescence-guided resection of tumors in mouse models of oral cancer. Sci. Rep. 2020, 10, 11175. [Google Scholar] [CrossRef]
- Laird, J.; Lok, B.H.; Carney, B.; Kossatz, S.; de Stanchina, E.; Reiner, T.; Poirier, J.T.; Rudin, C.M. Positron-emission tomographic imaging of a fluorine 18-radiolabeled poly(adp-ribose) polymerase 1 inhibitor monitors the therapeutic efficacy of talazoparib in sclc patient-derived xenografts. J. Thorac. Oncol. 2019, 14, 1743–1752. [Google Scholar] [CrossRef]
- Zmuda, F.; Blair, A.; Liuzzi, M.C.; Malviya, G.; Chalmers, A.J.; Lewis, D.; Sutherland, A.; Pimlott, S.L. An (18)f-labeled poly(adp-ribose) polymerase positron emission tomography imaging agent. J. Med. Chem. 2018, 61, 4103–4114. [Google Scholar] [CrossRef]
- Stotz, S.; Kinzler, J.; Nies, A.T.; Schwab, M.; Maurer, A. Two experts and a newbie: [(18)f]parpi vs [(18)f]ftt vs [(18)f]fpyparp-a comparison of parp imaging agents. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 834–846. [Google Scholar] [CrossRef]
- Wilson, T.C.; Xavier, M.A.; Knight, J.; Verhoog, S.; Torres, J.B.; Mosley, M.; Hopkins, S.L.; Wallington, S.; Allen, P.D.; Kersemans, V.; et al. Pet imaging of parp expression using (18)f-olaparib. J. Nucl. Med. 2019, 60, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guibbal, F.; Isenegger, P.G.; Wilson, T.C.; Pacelli, A.; Mahaut, D.; Sap, J.B.I.; Taylor, N.J.; Verhoog, S.; Preshlock, S.; Hueting, R.; et al. Manual and automated cu-mediated radiosynthesis of the parp inhibitor [(18)f]olaparib. Nat. Protoc. 2020, 15, 1525–1541. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.D.; Chailanggar, N.; Pichler, B.J.; Maurer, A. Scalable 18f processing conditions for copper-mediated radiofluorination chemistry facilitate doe optimization studies and afford an improved synthesis of [18f]olaparib. Org. Biomol. Chem. 2021, 19, 6995–7000. [Google Scholar] [CrossRef] [PubMed]
- Oplustil, O.; Connor, L.; Rulten, S.L.; Cranston, A.N.; Odedra, R.; Brown, H.; Jaspers, J.E.; Jones, L.; Knights, C.; Evers, B.; et al. The parp inhibitor azd2461 provides insights into the role of parp3 inhibition for both synthetic lethality and tolerability with chemotherapy in preclinical models. Cancer Res. 2016, 76, 6084. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.W.; Puentes, L.N.; Schmitz, A.; Hsieh, C.J.; Weng, C.C.; Hou, C.; Li, S.; Kuo, Y.M.; Padakanti, P.; Lee, H.; et al. Synthesis and evaluation of an azd2461 [(18)f]pet probe in non-human primates reveals the parp-1 inhibitor to be non-blood-brain barrier penetrant. Bioorg. Chem. 2019, 83, 242–249. [Google Scholar] [CrossRef]
- Guibbal, F.; Hopkins, S.L.; Pacelli, A.; Isenegger, P.G.; Mosley, M.; Torres, J.B.; Dias, G.M.; Mahaut, D.; Hueting, R.; Gouverneur, V.; et al. [18f]azd2461, an insight on difference in parp binding profiles for DNA damage response pet imaging. Mol. Imaging Biol. 2020, 22, 1226–1234. [Google Scholar] [CrossRef]
- Salinas, B.; Irwin, C.P.; Kossatz, S.; Bolaender, A.; Chiosis, G.; Pillarsetty, N.; Weber, W.A.; Reiner, T. Radioiodinated parp1 tracers for glioblastoma imaging. EJNMMI Res. 2015, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- Zmuda, F.; Malviya, G.; Blair, A.; Boyd, M.; Chalmers, A.J.; Sutherland, A.; Pimlott, S.L. Synthesis and evaluation of a radioiodinated tracer with specificity for poly(adp-ribose) polymerase-1 (parp-1) in vivo. J. Med. Chem. 2015, 58, 8683–8693. [Google Scholar] [CrossRef] [Green Version]
- Andersen, T.L.; Friis, S.D.; Audrain, H.; Nordeman, P.; Antoni, G.; Skrydstrup, T. Efficient 11c-carbonylation of isolated aryl palladium complexes for pet: Application to challenging radiopharmaceutical synthesis. J. Am. Chem. Soc. 2015, 137, 1548–1555. [Google Scholar] [CrossRef]
- Ferrat, M.; Dahl, K.; Halldin, C.; Schou, M. “In-loop” carbonylation—A simplified method for carbon-11 labelling of drugs and radioligands. J. Label. Compd. Radiopharm. 2020, 63, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Hu, P.; Banizs, A.B.; He, J. Initial evaluation of cu-64 labeled parpi-dota pet imaging in mice with mesothelioma. Bioorg Med. Chem. Lett. 2017, 27, 3472–3476. [Google Scholar] [CrossRef]
- Thomas, H.D.; Calabrese, C.R.; Batey, M.A.; Canan, S.; Hostomsky, Z.; Kyle, S.; Maegley, K.A.; Newell, D.R.; Skalitzky, D.; Wang, L.-Z.; et al. Preclinical selection of a novel poly(adp-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther. 2007, 6, 945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, R.; Jones, C.; Middleton, M.; Wilson, R.; Evans, J.; Olsen, A.; Curtin, N.; Boddy, A.; McHugh, P.; Newell, D.; et al. Phase i study of the poly(adp-ribose) polymerase inhibitor, ag014699, in combination with temozolomide in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7917–7923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berna, M.J.; Tapia, J.A.; Sancho, V.; Jensen, R.T. Progress in developing cholecystokinin (cck)/gastrin receptor ligands that have therapeutic potential. Curr. Opin. Pharmacol. 2007, 7, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Blaker, M.; Koch, J.; Lohse, A.W.; Pace, A. Cck1 and cck2 receptors are expressed on pancreatic stellate cells and induce collagen production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Chu, W.; Xu, J.; Jones, L.A.; Peng, X.; Li, S.; Chen, D.L.; Mach, R.H. Synthesis, [¹⁸f] radiolabeling, and evaluation of poly (adp-ribose) polymerase-1 (parp-1) inhibitors for in vivo imaging of parp-1 using positron emission tomography. Bioorg. Med. Chem. 2014, 22, 1700–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalitzky, D.J.; Marakovits, J.T.; Maegley, K.A.; Ekker, A.; Yu, X.H.; Hostomsky, Z.; Webber, S.E.; Eastman, B.W.; Almassy, R.; Li, J.K.; et al. Tricyclic benzimidazoles as potent poly(adp-ribose) polymerase-1 inhibitors. J. Med. Chem. 2003, 46, 210–213. [Google Scholar] [CrossRef]
- Edmonds, C.E.; Makvandi, M.; Lieberman, B.P.; Xu, K.; Zeng, C.; Li, S.; Hou, C.; Lee, H.; Greenberg, R.A.; Mankoff, D.A.; et al. [(18)f]fluorthanatrace uptake as a marker of parp1 expression and activity in breast cancer. Am. J. Nucl. Med. Mol. Imaging 2016, 6, 94–101. [Google Scholar]
- Sander Effron, S.; Makvandi, M.; Lin, L.; Xu, K.; Li, S.; Lee, H.; Hou, C.; Pryma, D.A.; Koch, C.; Mach, R.H. Parp-1 expression quantified by [(18)f]fluorthanatrace: A biomarker of response to parp inhibition adjuvant to radiation therapy. Cancer Biother. Radiopharm. 2017, 32, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Makvandi, M.; Pantel, A.; Schwartz, L.; Schubert, E.; Xu, K.; Hsieh, C.J.; Hou, C.; Kim, H.; Weng, C.C.; Winters, H.; et al. A pet imaging agent for evaluating parp-1 expression in ovarian cancer. J. Clin. Investig. 2018, 128, 2116–2126. [Google Scholar] [CrossRef]
- Zhou, D.; Xu, J.; Mpoy, C.; Chu, W.; Kim, S.H.; Li, H.; Rogers, B.E.; Katzenellenbogen, J.A. Preliminary evaluation of a novel 18f-labeled parp-1 ligand for pet imaging of parp-1 expression in prostate cancer. Nucl. Med. Biol. 2018, 66, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Destro, G.; Guibbal, F.; Chan, C.Y.; Cornelissen, B.; Gouverneur, V. Copper-mediated radiosynthesis of [(18)f]rucaparib. Org. Lett. 2021, 23, 7290–7294. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Chu, W.; Zhang, J.; Dence, C.S.; Welch, M.J.; Mach, R.H. Synthesis and in vivo evaluation of [11c]pj34, a potential radiotracer for imaging the role of parp-1 in necrosis. Nucl. Med. Biol. 2005, 32, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Shuhendler, A.J.; Cui, L.; Chen, Z.; Shen, B.; Chen, M.; James, M.L.; Witney, T.H.; Bazalova-Carter, M.; Gambhir, S.S.; Chin, F.T.; et al. [(18)f]-supar: A radiofluorinated probe for noninvasive imaging of DNA damage-dependent poly(adp-ribose) polymerase activity. Bioconjug. Chem. 2019, 30, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. Bmn 673, a novel and highly potent parp1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.Q.; Aoyagi-Scharber, M.; Post, L.E. Discovery and characterization of (8s,9r)-5-fluoro-8-(4-fluorophenyl)-9-(1-methyl-1h-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3h-pyrido[4,3,2-de]phthalazin-3-one (bmn 673, talazoparib), a novel, highly potent, and orally efficacious poly(adp-ribose) polymerase-1/2 inhibitor, as an anticancer agent. J. Med. Chem. 2016, 59, 335–357. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, H.; Mpoy, C.; Afrin, S.; Rogers, B.E.; Garbow, J.R.; Katzenellenbogen, J.A.; Xu, J. Radiosynthesis and evaluation of talazoparib and its derivatives as parp-1-targeting agents. Biomedicines 2021, 9, 565. [Google Scholar] [CrossRef]
- Bowden, G.D.; Stotz, S.; Kinzler, J.; Geibel, C.; Lämmerhofer, M.; Pichler, B.J.; Maurer, A. Doe optimization empowers the automated preparation of enantiomerically pure [18f]talazoparib and its in vivo evaluation as a parp radiotracer. J. Med. Chem. 2021, 64, 15690–15701. [Google Scholar] [CrossRef]
- Schöder, H.; França, P.D.D.S.; Nakajima, R.; Burnazi, E.; Roberts, S.; Brand, C.; Grkovski, M.; Mauguen, A.; Dunphy, M.P.; Ghossein, R.A.; et al. Safety and feasibility of parp1/2 imaging with [18f]-parpi in patients with head and neck cancer. Clin. Cancer Res. 2020, 13, 3110–3116. [Google Scholar] [CrossRef] [Green Version]
- Quinn, B.; Dauer, Z.; Pandit-Taskar, N.; Schoder, H.; Dauer, L.T. Radiation dosimetry of 18f-fdg pet/ct: Incorporating exam-specific parameters in dose estimates. Bmc Med. Imaging 2016, 16, 41. [Google Scholar] [CrossRef]
- McDonald, E.S.; Pantel, A.R.; Shah, P.D.; Farwell, M.D.; Clark, A.S.; Doot, R.K.; Pryma, D.A.; Carlin, S.D. In vivo visualization of parp inhibitor pharmacodynamics. JCI Insight 2021, 6, e146592. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.S.; Dyroff, S.; Brooks, F.J.; Spayd, K.J.; Lim, S.; Engle, J.T.; Phillips, S.; Tan, B.; Wang-Gillam, A.; Bognar, C.; et al. Pet of poly (adp-ribose) polymerase activity in cancer: Preclinical assessment and first in-human studies. Radiology 2017, 282, 453–463, Erratum in Radiology 2019, 291, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.J.; Pantel, A.R.; Viswanath, V.; Dominguez, T.L.; Makvandi, M.; Lee, H.; Li, S.; Schubert, E.K.; Pryma, D.A.; Farwell, M.D.; et al. Kinetic and static analysis of poly-(adenosine diphosphate-ribose) polymerase-1 (parp-1) targeted (18)f-fluorthanatrace ((18)f-ftt) pet images of ovarian cancer. J. Nucl. Med. 2021, 44–50. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.S.; Doot, R.K.; Pantel, A.R.; Farwell, M.D.; Mach, R.H.; Maxwell, K.N.; Mankoff, D.A. Positron emission tomography imaging of poly–(adenosine diphosphate–ribose) polymerase 1 expression in breast cancer: A nonrandomized clinical trial. JAMA Oncol. 2020, 6, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Blower, P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015, 44, 4819–4844. [Google Scholar] [CrossRef]
- Morphis, M.; van Staden, J.A.; du Raan, H.; Ljungberg, M. Evaluation of iodine-123 and iodine-131 spect activity quantification: A monte carlo study. EJNMMI Phys. 2021, 8, 61. [Google Scholar] [CrossRef]
- Lee, H.; Riad, A.; Martorano, P.; Mansfield, A.; Samanta, M.; Batra, V.; Mach, R.H.; Maris, J.M.; Pryma, D.A.; Makvandi, M. Parp-1-targeted auger emitters display high-let cytotoxic properties in vitro but show limited therapeutic utility in solid tumor models of human neuroblastoma. J. Nucl. Med. 2019, 61, 850–856. [Google Scholar] [CrossRef]
- Guérard, F.; Gestin, J.-F.; Brechbiel, M.W. Production of [(211)at]-astatinated radiopharmaceuticals and applications in targeted α-particle therapy. Cancer Biother. Radiopharm. 2013, 28, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Poty, S.; Francesconi, L.C.; McDevitt, M.R.; Morris, M.J.; Lewis, J.S. A-emitters for radiotherapy: From basic radiochemistry to clinical studies-part 1. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 878–884. [Google Scholar] [CrossRef] [Green Version]
- Fourie, H.; Nair, S.; Muller, X.; Rossouw, D.; Beukes, P.; Newman, R.; Zeevaart, J.; Vandevoorde, C.; Slabbert, J. Estimating the relative biological effectiveness of auger electron emitter 123i in human lymphocytes. Front. Phys. 2020, 8, 567732. [Google Scholar] [CrossRef]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy—A review. EJNMMI Radiopharm. Chem. 2019, 4, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalutsky, M.R.; Reardon, D.A.; Pozzi, O.R.; Vaidyanathan, G.; Bigner, D.D. Targeted alpha-particle radiotherapy with 211at-labeled monoclonal antibodies. Nucl. Med. Biol. 2007, 34, 779–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becquerel, L.N.H. Atomic and Nuclear Data Base. Available online: http://www.lnhb.fr/nuclear-data/nuclear-data-table/http://nucleardata.nuclear.lu.se/toi/nuclide.asp?iZA=90018&sortG=E&sortA=E (accessed on 31 January 2022).
- Zanglis, A. [111in-dtpa0-d-phe1]-octreotide: The ligand—the receptor—the label. In Liver Intra-Arterial Prrt with 111in-Octreotide: The Tumoricidal Efficacy of 111in Auger Electron Emission; Limouris, G.S., Ed.; Springer: Cham, Switzerland, 2021; pp. 29–63. [Google Scholar]
- Mody, V.V.; Singh, A.N.; Deshmukh, R.; Shah, S. Chapter 40—Thyroid hormones, iodine and iodides, and antithyroid drugs. In Side Effects of Drugs Annual; Ray, S.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 37, pp. 513–519. [Google Scholar]
- Jannetti, S.A.; Carlucci, G.; Carney, B.; Kossatz, S.; Shenker, L.; Carter, L.M.; Salinas, B.; Brand, C.; Sadique, A.; Donabedian, P.L.; et al. Parp-1-targeted radiotherapy in mouse models of glioblastoma. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1225–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirovano, G.; Jannetti, S.A.; Carter, L.M.; Sadique, A.; Kossatz, S.; Guru, N.; Demétrio De Souza França, P.; Maeda, M.; Zeglis, B.M.; Lewis, J.S.; et al. Targeted brain tumor radiotherapy using an auger emitter. Clin. Cancer Res. 2020, 26, 2871–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.C.; Jannetti, S.A.; Guru, N.; Pillarsetty, N.; Reiner, T.; Pirovano, G. Improved radiosynthesis of (123)i-mapi, an auger theranostic agent. Int. J. Radiat. Biol. 2020, 1–7. [Google Scholar] [CrossRef]
- Wilson, T.; Pirovano, G.; Xiao, G.; Samuels, Z.; Roberts, S.; Viray, T.; Guru, N.; Zanzonico, P.; Gollub, M.; Pillarsetty, N.V.K.; et al. Parp-targeted auger therapy in p53 mutant colon cancer xenograft mouse models. Mol. Pharm. 2021, 18, 3418–3428. [Google Scholar] [CrossRef]
- Dabagian, H.; Taghvaee, T.; Martorano, P.; Martinez, D.; Samanta, M.; Watkins, C.M.; Chai, R.; Mansfield, A.; Graham, T.J.; Maris, J.M.; et al. Parp targeted alpha-particle therapy enhances response to pd-1 immune-checkpoint blockade in a syngeneic mouse model of glioblastoma. ACS Pharmacol. Transl. Sci. 2021, 4, 344–351. [Google Scholar] [CrossRef]
- Makvandi, M.; Lee, H.; Puentes, L.N.; Reilly, S.W.; Rathi, K.S.; Weng, C.C.; Chan, H.S.; Hou, C.; Raman, P.; Martinez, D.; et al. Targeting parp-1 with alpha-particles is potently cytotoxic to human neuroblastoma in preclinical models. Mol. Cancer Ther. 2019, 18, 1195–1204. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, R.A.; Peil, J.; Vogg, A.T.J.; Bolm, C.; Terhorst, S.; Classen, A.; Bauwens, M.; Maurer, J.; Mottaghy, F.; Morgenroth, A. Auger emitter conjugated parp inhibitor for therapy in triple negative breast cancers: A comparative in-vitro study. Cancers 2022, 14, 230. [Google Scholar] [CrossRef]
- Makvandi, M.; Xu, K.; Lieberman, B.P.; Anderson, R.C.; Effron, S.S.; Winters, H.D.; Zeng, C.; McDonald, E.S.; Pryma, D.A.; Greenberg, R.A.; et al. A radiotracer strategy to quantify parp-1 expression in vivo provides a biomarker that can enable patient selection for parp inhibitor therapy. Cancer Res. 2016, 76, 4516–4524. [Google Scholar] [CrossRef] [Green Version]
- Riad, A.; Gitto, S.B.; Lee, H.; Winters, H.D.; Martorano, P.M.; Hsieh, C.-J.; Xu, K.; Omran, D.K.; Powell, D.J., Jr.; Mach, R.H.; et al. Parp theranostic auger emitters are cytotoxic in brca mutant ovarian cancer and viable tumors from ovarian cancer patients enable ex-vivo screening of tumor response. Molecules 2020, 25, 6029. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.C.; Makvandi, M.; Xu, K.; Lieberman, B.P.; Zeng, C.; Pryma, D.A.; Mach, R.H. Iodinated benzimidazole parp radiotracer for evaluating parp1/2 expression in vitro and in vivo. Nucl. Med. Biol. 2016, 43, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Boersma, H.H.; Sturkenboom, M.G.; Lub-de Hooge, M.N.; Elsinga, P.H.; Luurtsema, G.; Dierckx, R.A.; Kosterink, J.G. Basic aspects of good manufacturing practice for pet-radiopharmaceuticals. In Trends on the Role of Pet in Drug Development; Elsinga, P.H., van Waarde, A., Paans, A.M.J., Dierckx, R.A.J.O., Eds.; World Scientific: Singapore, 2012; pp. 727–749. [Google Scholar]
- Vermeulen, K.; Verbruggen, A.; Bormans, G.; Cleeren, F. Moving a radiotracer from bench to bedside in europe. In Handbook of Radiopharmaceuticals: Methodology and Applications; Scott, P., Kilbourn, M., Eds.; Wiley: Hoboken, NJ, USA, 2021; pp. 515–532. [Google Scholar]
- Sandle, T. A review of cleanroom microflora: Types, trends, and patterns. PDA J. Pharm. Sci. Tech. 2011, 65, 392–403. [Google Scholar] [CrossRef]
- Baudhuin, H.; Cousaert, J.; Vanwolleghem, P.; Raes, G.; Caveliers, V.; Keyaerts, M.; Lahoutte, T.; Xavier, C. 68ga-labeling: Laying the foundation for an anti-radiolytic formulation for nota-sdab pet tracers. Pharmaceuticals 2021, 14, 448. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.J.H.; Hockley, B.G.; Kung, H.F.; Manchanda, R.; Zhang, W.; Kilbourn, M.R. Studies into radiolytic decomposition of fluorine-18 labeled radiopharmaceuticals for positron emission tomography. Appl. Radiat. Isot. 2009, 67, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia Wang, R.M.v.D. High-efficiency production of radiopharmaceuticals via droplet radiochemistry: A review of recent progress. Mol. Imaging 2020, 19, 1–21. [Google Scholar] [CrossRef]
- Roy, J. Pharmaceutical impurities—A mini-review. AAPS PharmSciTech 2002, 3, 1–8. [Google Scholar] [CrossRef]
- Elsinga, P.; Todde, S.; Penuelas, I.; Meyer, G.; Farstad, B.; Faivre-Chauvet, A.; Mikolajczak, R.; Westera, G.; Gmeiner-Stopar, T.; Decristoforo, C.; et al. Guidance on current good radiopharmacy practice (cgrpp) for the small-scale preparation of radiopharmaceuticals. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1049–1062. [Google Scholar] [CrossRef] [Green Version]

| Isotopes | ||||
|---|---|---|---|---|
| 123I | 125I | 131I | 211At | |
| Half-life | 13.2 h | 59.3 d | 8.0 d | 7.2 h |
| Major decay mode | EC | EC | ß− decay | α decay |
| SPECT imaging (abundance) | ƴ: 159 keV (abundance: 83%) | ƴ: 35.5 keV (abundance: 7%) | ƴ: 364.5 keV (abundance: 82%) (high energy coll.) | K x-rays (77–92 keV) |
| ß− energy | none | none | 606 keV (abundance: 90%) | none |
| α energy | none | none | none | 1 α/decay (5.9 MeV–7.5 MeV) |
| Auger energy | 11 AE/decay | 21 AE/decay | none | 6.3 AE/decay |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.T.; Pacelli, A.; Nader, M.; Kossatz, S. DNA Repair Enzyme Poly(ADP-Ribose) Polymerase 1/2 (PARP1/2)-Targeted Nuclear Imaging and Radiotherapy. Cancers 2022, 14, 1129. https://doi.org/10.3390/cancers14051129
Nguyen NT, Pacelli A, Nader M, Kossatz S. DNA Repair Enzyme Poly(ADP-Ribose) Polymerase 1/2 (PARP1/2)-Targeted Nuclear Imaging and Radiotherapy. Cancers. 2022; 14(5):1129. https://doi.org/10.3390/cancers14051129
Chicago/Turabian StyleNguyen, Nghia T., Anna Pacelli, Michael Nader, and Susanne Kossatz. 2022. "DNA Repair Enzyme Poly(ADP-Ribose) Polymerase 1/2 (PARP1/2)-Targeted Nuclear Imaging and Radiotherapy" Cancers 14, no. 5: 1129. https://doi.org/10.3390/cancers14051129
APA StyleNguyen, N. T., Pacelli, A., Nader, M., & Kossatz, S. (2022). DNA Repair Enzyme Poly(ADP-Ribose) Polymerase 1/2 (PARP1/2)-Targeted Nuclear Imaging and Radiotherapy. Cancers, 14(5), 1129. https://doi.org/10.3390/cancers14051129