
Radiolabeled Somatostatin Analogs—A Continuously Evolving Class of Radiopharmaceuticals




Abstract
Simple Summary
Abstract
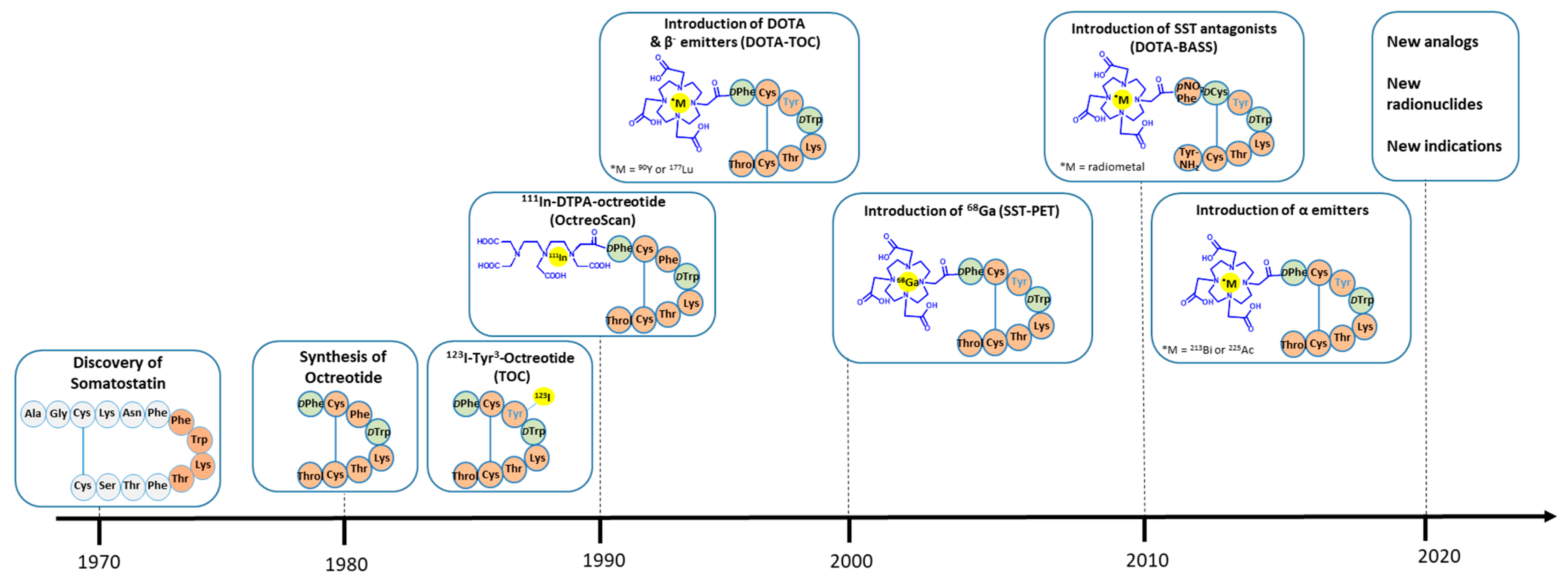
1. Introduction
2. Somatostatin Receptor Agonists: The Archetype and the Latest Developments
2.1. Peptide Sequences and Critical Amino Acid Positions
2.2. Clinical Studies and Approvals
2.3. Combination with Alpha-Emitters
2.4. Conjugates with Prolonged Circulation
3. Somatostatin Receptor Antagonists: Will They Make the Difference?
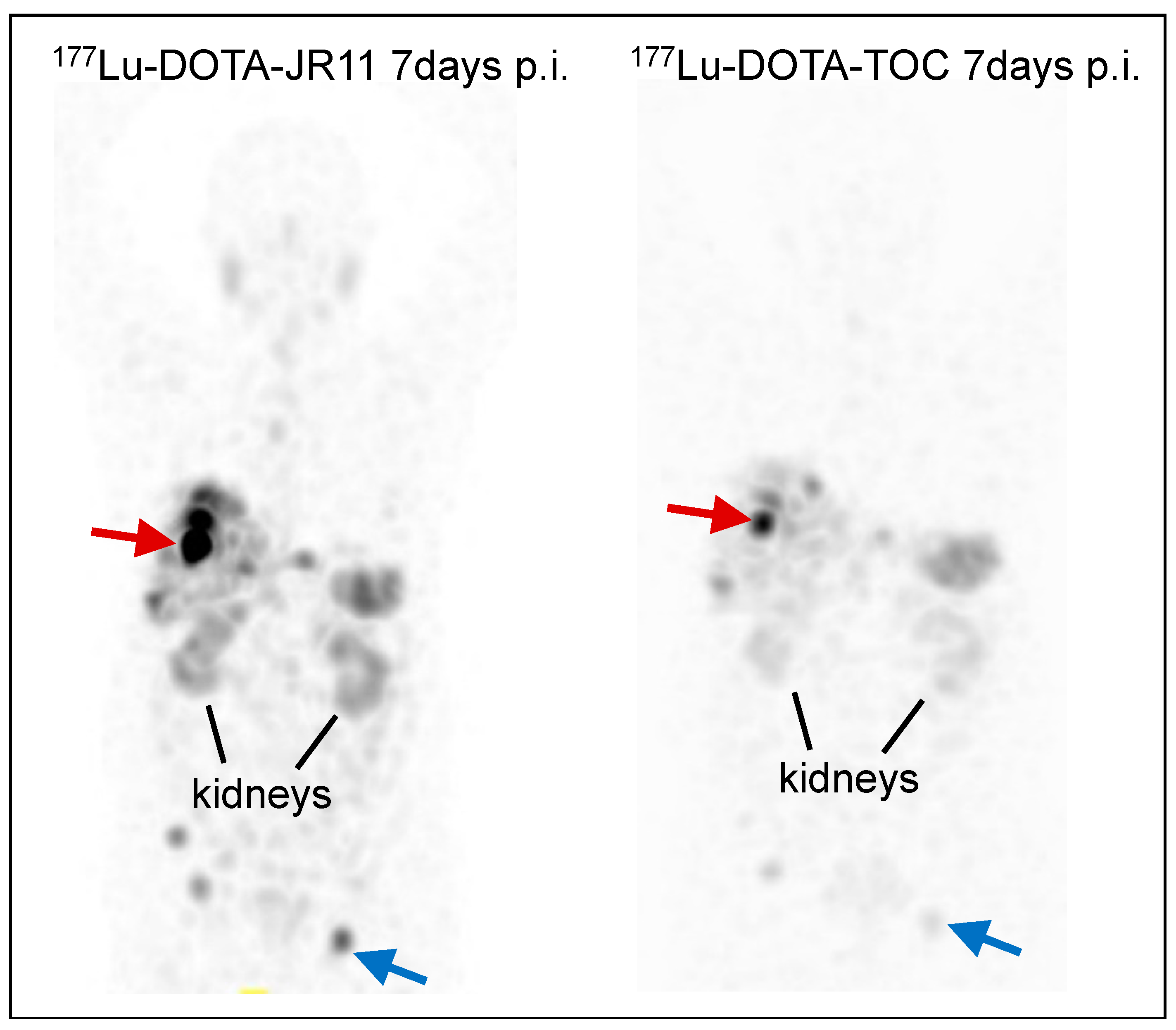
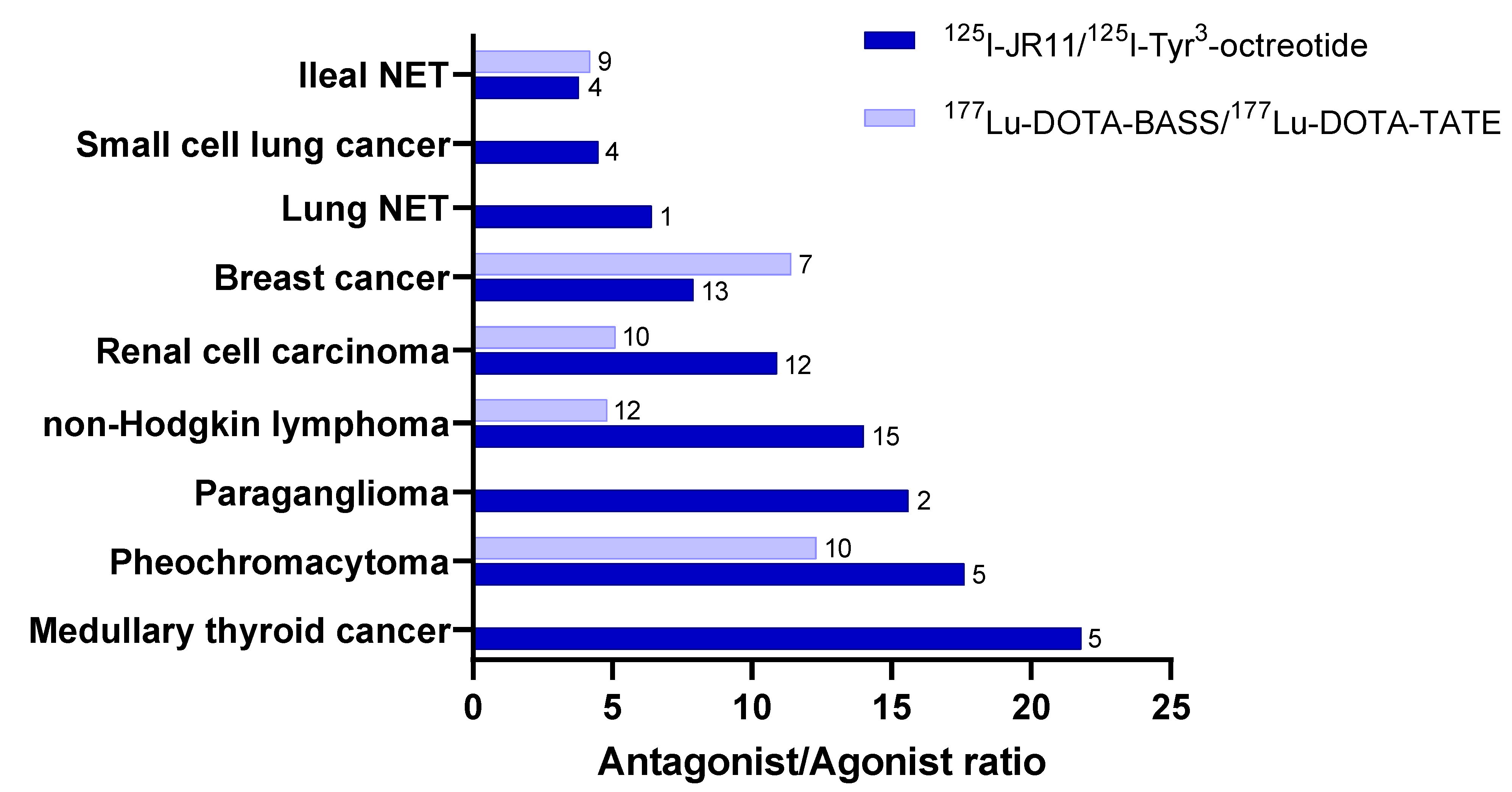
3.1. Preclinical Development
3.2. Clinical Translation
4. Novel Indications for Radiolabeled Somatostatin Analogs
5. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Günther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.-J.; Lupp, A.; Korbonits, M.; Castaño, J.P.; Wester, H.-J.; et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharmacol. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, F.; Bajetto, A.; Pattarozzi, A.; Gatti, M.; Würth, R.; Thellung, S.; Corsaro, A.; Villa, V.; Nizzari, M.; Florio, T. Peptide Receptor Targeting in Cancer: The Somatostatin Paradigm. Int. J. Pept. 2013, 2013, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: Molecular basis for in vivo multireceptor tumour targeting. Eur. J. Pediatr. 2003, 30, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.; Waser, B.; Schaer, J.-C.; Laissue, J.A. Somatostatin receptor sst1–sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur. J. Pediatr. 2001, 28, 836–846. [Google Scholar] [CrossRef]
- Volante, M.; Rosas, R.; Allìa, E.; Granata, R.; Baragli, A.; Muccioli, G.; Papotti, M. Somatostatin, cortistatin and their receptors in tumours. Mol. Cell. Endocrinol. 2008, 286, 219–229. [Google Scholar] [CrossRef]
- Ambrosini, V.; Kunikowska, J.; Baudin, E.; Bodei, L.; Bouvier, C.; Capdevila, J.; Cremonesi, M.; de Herder, W.W.; Dromain, C.; Falconi, M.; et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur. J. Cancer 2021, 146, 56–73. [Google Scholar] [CrossRef]
- Hicks, R.J.; Kwekkeboom, D.J.; Krenning, E.; Bodei, L.; Grozinsky-Glasberg, S.; Arnold, R.; Borbath, I.; Cwikla, J.B.; Toumpanakis, C.; Kaltsas, G.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasms: Peptide Receptor Radionuclide Therapy with Radiolabelled Somatostatin Analogues. Neuroendocrinology 2017, 105, 295–309. [Google Scholar] [CrossRef]
- Shah, M.H.; Goldner, W.S.; Benson, A.B.; Bergsland, E.; Blaszkowsky, L.S.; Brock, P.; Chan, J.; Das, S.; Dickson, P.V.; Fanta, P.; et al. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 839–868. [Google Scholar] [CrossRef]
- Veber, D.F.; Freidlinger, R.M.; Perlow, D.S.; Paleveda, W.J.; Holly, F.W.; Strachan, R.G.; Nutt, R.F.; Arison, B.H.; Homnick, C.; Randall, W.C.; et al. A potent cyclic hexapeptide analogue of somatostatin. Nature 1981, 292, 55–58. [Google Scholar] [CrossRef]
- Veber, D.F.; Holly, F.W.; Nutt, R.F.; Bergstrand, S.J.; Brady, S.F.; Hisrschmann, R.; Glitzer, M.S.; Saperstein, R. Highly active cyclic and bicyclic somatostatin analogues of reduced ring size. Nat. 1979, 280, 512–514. [Google Scholar] [CrossRef]
- Bauer, W.; Briner, U.; Doepfner, W.; Haller, R.; Huguenin, R.; Marbach, P.; Petcher, T.J.; Pless, J. SMS 201–995: A very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982, 31, 1133–1140. [Google Scholar] [CrossRef]
- Narayanan, S.; Kunz, P.L. Role of Somatostatin Analogues in the Treatment of Neuroendocrine Tumors. J. Natl. Compr. Cancer Netw. 2015, 13, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Antunes, P.; Ginj, M.; Zhang, H.; Waser, B.; Baum, R.P.; Reubi, J.C.; Maecke, H. Are radiogallium-labelled DOTA-conjugated somatostatin analogues superior to those labelled with other radiometals? Eur. J. Nucl. Med. 2007, 34, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Ginj, M.; Schmitt, J.S.; Chen, J.; Waser, B.; Reubi, J.-C.; de Jong, M.; Schulz, S.; Maecke, H.R. Design, Synthesis, and Biological Evaluation of Somatostatin-Based Radiopeptides. Chem. Biol. 2006, 13, 1081–1090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Levine, R.; Krenning, E.P. Clinical History of the Theranostic Radionuclide Approach to Neuroendocrine Tumors and Other Types of Cancer: Historical Review Based on an Interview of Eric P. Krenning by Rachel Levine. J. Nucl. Med. 2017, 58, 3S–9S. [Google Scholar] [CrossRef]
- Ambrosini, V.; Fani, M.; Fanti, S.; Forrer, F.; Maecke, H.R. Radiopeptide Imaging and Therapy in Europe. J. Nucl. Med. 2011, 52 (Suppl. 2), S42–S55. [Google Scholar] [CrossRef]
- Eychenne, R.; Bouvry, C.; Bourgeois, M.; Loyer, P.; Benoist, E.; Lepareur, N. Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Mol. 2020, 25, 4012. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schär, J.-C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef]
- Schottelius, M.; Šimeček, J.; Hoffmann, F.; Willibald, M.; Schwaiger, M.; Wester, H.-J. Twins in spirit—Episode I: Comparative preclinical evaluation of [68Ga]DOTATATE and [68Ga]HA-DOTATATE. EJNMMI Res. 2015, 5, 22. [Google Scholar] [CrossRef]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Macke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected Sensitivity of sst2 Antagonists to N-Terminal Radiometal Modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.; Michael, M.; Hicks, R.J. 177Lu-Dotatate for Midgut Neuroendocrine Tumors. New Engl. J. Med. 2017, 376, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.; Wolin, E.; Chasen, B.; Kulke, M.; Bushnell, D.; Caplin, M.; Baum, R.P.; Kunz, P.; Hobday, T.; Hendifar, A.; et al. Health-Related Quality of Life in Patients with Progressive Midgut Neuroendocrine Tumors Treated With 177Lu-Dotatate in the Phase III NETTER-1 Trial. J. Clin. Oncol. 2018, 36, 2578–2584. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; E Pavel, M.; et al. 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef]
- Partelli, S.; Bertani, E.; Bartolomei, M.; Perali, C.; Muffatti, F.; Grana, C.M.; Lena, M.S.; Doglioni, C.; Crippa, S.; Fazio, N.; et al. Peptide receptor radionuclide therapy as neoadjuvant therapy for resectable or potentially resectable pancreatic neuroendocrine neoplasms. Surgery 2018, 163, 761–767. [Google Scholar] [CrossRef]
- Parghane, R.V.; Bhandare, M.; Chaudhari, V.; Ostwal, V.; Ramaswamy, A.; Talole, S.; Shrikhande, S.V.; Basu, S. Surgical Feasibility, Determinants, and Overall Efficacy of Neoadjuvant 177Lu-DOTATATE PRRT for Locally Advanced Unresectable Gastroenteropancreatic Neuroendocrine Tumors. J. Nucl. Med. 2021, 62, 1558–1563. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Pediatr. 2014, 41, 2106–2119. [Google Scholar] [CrossRef]
- Delpassand, E.S.; Tworowska, I.; Esfandiari, R.; Torgue, J.; Hurt, J.; Shafie, A.; Núñez, R. Targeted Alpha-Emitter Therapy With 212Pb-DOTAMTATE for the Treatment of Metastatic SSTR-Expressing Neuroendocrine Tumors: First-in-Human, Dose-Escalation Clinical Trial. J. Nucl. Med. 2022. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. Ac-225-DOTATOC—An empiric dose finding for alpha particle emitter based radionuclide therapy of neuroendocrine tumors. J. Nucl. Med. 2015, 56, 1232. [Google Scholar]
- Ballal, S.; Yadav, M.P.; Bal, C.; Sahoo, R.K.; Tripathi, M. Broadening horizons with 225Ac-DOTATATE targeted alpha therapy for gastroenteropancreatic neuroendocrine tumour patients stable or refractory to 177Lu-DOTATATE PRRT: First clinical experience on the efficacy and safety. Eur. J. Pediatr. 2020, 47, 934–946. [Google Scholar] [CrossRef]
- Kratochwil, C.; Apostolidis, L.; Rathke, H.; Apostolidis, C.; Bicu, F.; Bruchertseifer, F.; Choyke, P.L.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Dosing 225Ac-DOTATOC in patients with somatostatin-receptor-positive solid tumors: 5-year follow-up of hematological and renal toxicity. Eur. J. Pediatr. 2021, 49, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Bandara, N.; Jacobson, O.; Mpoy, C.; Chen, X.; Rogers, B.E. Novel Structural Modification Based on Evans Blue Dye to Improve Pharmacokinetics of a Somastostatin-Receptor-Based Theranostic Agent. Bioconjugate Chem. 2018, 29, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Jacobson, O.; Niu, G.; Kiesewetter, D.O.; Wang, Z.; Zhu, G.; Ma, Y.; Liu, G.; Chen, X. Evans Blue Attachment Enhances Somatostatin Receptor Subtype-2 Imaging and Radiotherapy. Theranostics 2018, 8, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, H.; Jacobson, O.; Cheng, Y.; Niu, G.; Li, F.; Bai, C.; Zhu, Z.; Chen, X. Safety, Pharmacokinetics, and Dosimetry of a Long-Acting Radiolabeled Somatostatin Analog 177Lu-DOTA-EB-TATE in Patients with Advanced Metastatic Neuroendocrine Tumors. J. Nucl. Med. 2018, 59, 1699–1705. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cheng, Y.; Zang, J.; Sui, H.; Wang, H.; Jacobson, O.; Zhu, Z.; Chen, X. Dose escalation of an Evans blue–modified radiolabeled somatostatin analog 177Lu-DOTA-EB-TATE in the treatment of metastatic neuroendocrine tumors. Eur. J. Pediatr. 2019, 47, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Hänscheid, H.; Hartrampf, P.E.; Schirbel, A.; Buck, A.K.; Lapa, C. Intraindividual comparison of [177Lu]Lu-DOTA-EB-TATE and [177Lu]Lu-DOTA-TOC. Eur. J. Pediatr. 2021, 48, 2566–2572. [Google Scholar] [CrossRef]
- Perrin, M.H.; Sutton, S.W.; Cervini, L.A.; Rivier, J.E.; Vale, W.W. Comparison of an agonist, urocortin, and an antagonist, astressin, as radioligands for characterization of corticotropin-releasing factor receptors. J. Pharmacol. Exp. Ther. 1999, 288, 729–734. [Google Scholar]
- Bass, R.T.; Buckwalter, B.L.; Patel, B.P.; Pausch, M.H.; Price, L.A.; Strnad, J.; Hadcock, J.R. Identification and characterization of novel somatostatin antagonists. Mol. Pharmacol. 1996, 50, 709–715. [Google Scholar]
- Cescato, R.; Waser, B.; Fani, M.; Reubi, J.C. Evaluation of 177Lu-DOTA-sst2 Antagonist Versus 177Lu-DOTA-sst2 Agonist Binding in Human Cancers In Vitro. J. Nucl. Med. 2011, 52, 1886–1890. [Google Scholar] [CrossRef]
- Wang, X.; Fani, M.; Schulz, S.; Rivier, J.; Reubi, J.C.; Maecke, H.R. Comprehensive evaluation of a somatostatin-based radiolabelled antagonist for diagnostic imaging and radionuclide therapy. Eur. J. Pediatr. 2012, 39, 1876–1885. [Google Scholar] [CrossRef]
- Cescato, R.; Erchegyi, J.; Waser, B.; Piccand, V.; Maecke, H.R.; Rivier, J.E.; Reubi, J.C.; Mäcke, H.R. Design and in Vitro Characterization of Highly sst2-Selective Somatostatin Antagonists Suitable for Radiotargeting. J. Med. Chem. 2008, 51, 4030–4037. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of Somatostatin Receptor–Positive Tumors Using 64Cu- and 68Ga-Somatostatin Antagonists: The Chelate Makes the Difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Mansi, R.; McDougall, L.; Kaufmann, J.; Bouterfa, H.; Wild, D.; Fani, M. Biodistribution, Pharmacokinetics, and Dosimetry of 177Lu-, 90Y-, and 111In-Labeled Somatostatin Receptor Antagonist OPS201 in Comparison to the Agonist 177Lu-DOTATATE: The Mass Effect. J. Nucl. Med. 2017, 58, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Dalm, S.U.; Nonnekens, J.; Doeswijk, G.N.; de Blois, E.; van Gent, D.C.; Konijnenberg, M.W.; de Jong, M. Comparison of the Therapeutic Response to Treatment with a 177Lu-Labeled Somatostatin Receptor Agonist and Antagonist in Preclinical Models. J. Nucl. Med. 2016, 57, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Plas, P.; Vauquelin, G.; Fani, M. Distinct In Vitro Binding Profile of the Somatostatin Receptor Subtype 2 Antagonist [177Lu]Lu-OPS201 Compared to the Agonist [177Lu]Lu-DOTA-TATE. Pharmaceuticals 2021, 14, 1265. [Google Scholar] [CrossRef]
- Abiraj, K.; Ursillo, S.; Tamma, M.L.; Rylova, S.N.; Waser, B.; Constable, E.C.; Fani, M.; Nicolas, G.P.; Reubi, J.C.; Maecke, H.R. The tetraamine chelator outperforms HYNIC in a new technetium-99m-labelled somatostatin receptor 2 antagonist. EJNMMI Res. 2018, 8, 75. [Google Scholar] [CrossRef]
- Gaonkar, R.; Wiesmann, F.; Del Pozzo, L.; McDougall, L.; Zanger, S.; Mikołajczak, R.; Mansi, R.; Fani, M. SPECT Imaging of SST2-Expressing Tumors with 99mTc-Based Somatostatin Receptor Antagonists: The Role of Tetraamine, HYNIC, and Spacers. Pharm. 2021, 14, 300. [Google Scholar] [CrossRef]
- Fani, M.; Weingaertner, V.; Kolenc-Peitl, P.K.; Mansi, R.; Gaonkar, R.H.; Garnuszek, P.; Mikolajczak, R.; Novak, D.; Simoncic, U.; Hubalewska-Dydejczyk, A.; et al. Selection of the First 99mTc-Labelled Somatostatin Receptor Subtype 2 Antagonist for Clinical Translation—Preclinical Assessment of Two Optimized Candidates. Pharmaceuticals 2020, 14, 19. [Google Scholar] [CrossRef]
- Wild, D.; Fani, M.; Behe, M.; Brink, I.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; Weber, W.A. First Clinical Evidence That Imaging with Somatostatin Receptor Antagonists Is Feasible. J. Nucl. Med. 2011, 52, 1412–1417. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Beykan, S.; Bouterfa, H.; Kaufmann, J.; Bauman, A.; Lassmann, M.; Reubi, J.C.; Rivier, J.E.; Maecke, H.R.; Fani, M.; et al. Safety, Biodistribution, and Radiation Dosimetry of 68Ga-OPS202 in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase I Imaging Study. J. Nucl. Med. 2018, 59, 909–914. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Schreiter, N.; Kaul, F.; Uiters, J.; Bouterfa, H.; Kaufmann, J.; Erlanger, T.E.; Cathomas, R.; Christ, E.; Fani, M.; et al. Sensitivity Comparison of68Ga-OPS202 and68Ga-DOTATOC PET/CT in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase II Imaging Study. J. Nucl. Med. 2018, 59, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Krebs, S.; O’Donoghue, J.A.; Biegel, E.; Beattie, B.J.; Reidy, D.; Lyashchenko, S.K.; Lewis, J.S.; Bodei, L.; Weber, W.A.; Pandit-Taskar, N. Comparison of 68Ga-DOTA-JR11 PET/CT with dosimetric 177Lu-satoreotide tetraxetan (177Lu-DOTA-JR11) SPECT/CT in patients with metastatic neuroendocrine tumors undergoing peptide receptor radionuclide therapy. Eur. J. Pediatr. 2020, 47, 3047–3057. [Google Scholar] [CrossRef] [PubMed]
- Krebs, S.; Pandit-Taskar, N.; Reidy, D.; Beattie, B.J.; Lyashchenko, S.K.; Lewis, J.S.; Bodei, L.; Weber, W.A.; O’Donoghue, J.A. Biodistribution and radiation dose estimates for 68Ga-DOTA-JR11 in patients with metastatic neuroendocrine tumors. Eur. J. Pediatr. 2018, 46, 677–685. [Google Scholar] [CrossRef]
- Zhu, W.; Cheng, Y.; Wang, X.; Yao, S.; Bai, C.; Zhao, H.; Jia, R.; Xu, J.; Huo, L. Head-to-Head Comparison of 68Ga-DOTA-JR11 and 68Ga-DOTATATE PET/CT in Patients with Metastatic, Well-Differentiated Neuroendocrine Tumors: A Prospective Study. J. Nucl. Med. 2020, 61, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B.; Cescato, R.; Gloor, B.; Stettler, C.; Christ, E. Internalized Somatostatin Receptor Subtype 2 in Neuroendocrine Tumors of Octreotide-Treated Patients. J. Clin. Endocrinol. Metab. 2010, 95, 2343–2350. [Google Scholar] [CrossRef]
- Wild, D.; Fani, M.; Fischer, R.; Del Pozzo, L.; Kaul, F.; Krebs, S.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; Weber, W.A. Comparison of Somatostatin Receptor Agonist and Antagonist for Peptide Receptor Radionuclide Therapy: A Pilot Study. J. Nucl. Med. 2014, 55, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Reidy-Lagunes, D.; Pandit-Taskar, N.; O’Donoghue, J.A.; Krebs, S.; Staton, K.D.; Lyashchenko, S.K.; Lewis, J.S.; Raj, N.; Gönen, M.; Lohrmann, C.; et al. Phase I Trial of Well-Differentiated Neuroendocrine Tumors (NETs) with Radiolabeled Somatostatin Antagonist 177Lu-Satoreotide Tetraxetan. Clin. Cancer Res. 2019, 25, 6939–6947. [Google Scholar] [CrossRef]
- Nicolas, G.; Ansquer, C.; Lenzo, N.; Grønbæk, H.; Haug, A.; Navalkissoor, S.; Beauregard, J.-M.; Germann, N.; McEwan, S.; Wild, D.; et al. 1160O An international open-label study on safety and efficacy of 177Lu-satoreotide tetraxetan in somatostatin receptor positive neuroendocrine tumours (NETs): An interim analysis. Ann. Oncol. 2020, 31, S771. [Google Scholar] [CrossRef]
- Singh, A.; Kulkarni, H.; Langbein, T.; Mueller, D.; Senftleben, S.; Fani, M.; Maecke, H.; Baum, R. PET/CT imaging of somatostatin receptor expressing solid tumors with the novel somatotostatin receptor antagonist 68Ga-NODAGA-LM3. J. Nucl. Med. 2018, 59, 42. [Google Scholar]
- Singh, A.; Zhang, J.; Kulkarni, H.; Langbein, T.; Baum, R. First-in-human study of a novel somatostatin receptor antagonist 68Ga-NODAGA-LM3 for molecular imaging of paraganglioma patients. J. Nucl. Med. 2019, 60, 339. [Google Scholar]
- Zhu, W.; Cheng, Y.; Jia, R.; Zhao, H.; Bai, C.; Xu, J.; Yao, S.; Huo, L. A Prospective, Randomized, Double-Blind Study to Evaluate the Safety, Biodistribution, and Dosimetry of 68Ga-NODAGA-LM3 and 68Ga-DOTA-LM3 in Patients with Well-Differentiated Neuroendocrine Tumors. J. Nucl. Med. 2021, 62, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.P.; Zhang, J.; Schuchardt, C.; Mueller, D.; Maecke, H. First-in-Humans Study of the SSTR Antagonist 177Lu-DOTA-LM3 for Peptide Receptor Radionuclide Therapy in Patients with Metastatic Neuroendocrine Neoplasms: Dosimetry, Safety, and Efficacy. J. Nucl. Med. 2021, 62, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B.; Mäcke, H.; Rivier, J.E. Highly Increased 125I-JR11 Antagonist Binding In Vitro Reveals Novel Indications for sst2 Targeting in Human Cancers. J. Nucl. Med. 2017, 58, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Cuccurullo, V.; Di Stasio, G.D.; Prisco, M.R.; Mansi, L. Is there a clinical usefulness for radiolabeled somatostatin analogues beyond the consolidated role in NETs? Indian J. Radiol. Imaging 2017, 27, 509–516. [Google Scholar] [CrossRef]
- Anzola, L.K.; Rivera, J.N.; Ramirez, J.C.; Signore, A.; Mut, F. Molecular Imaging of Vulnerable Coronary Plaque with Radiolabeled Somatostatin Receptors (SSTR). J. Clin. Med. 2021, 10, 5515. [Google Scholar] [CrossRef]
- Meester, E.J.; Krenning, B.J.; de Blois, E.; de Jong, M.; van der Steen, A.F.W.; Bernsen, M.R.; van der Heiden, K. Imaging inflammation in atherosclerotic plaques, targeting SST2 with [111In]In-DOTA-JR. J. Nucl. Cardiol. 2020, 28, 2506–2513. [Google Scholar] [CrossRef]
- Schatka, I.; Wollenweber, T.; Haense, C.; Brunz, F.; Gratz, K.F.; Bengel, F.M. Peptide Receptor–Targeted Radionuclide Therapy Alters Inflammation in Atherosclerotic Plaques. J. Am. Coll. Cardiol. 2013, 62, 2344–2345. [Google Scholar] [CrossRef]
- Lapa, C.; Kircher, M.; Hänscheid, H.; Schirbel, A.; Grigoleit, G.U.; Klinker, E.; Böck, M.; Samnick, S.; Pelzer, T.; Buck, A.K. Peptide receptor radionuclide therapy as a new tool in treatment-refractory sarcoidosis—Initial experience in two patients. Theranostics 2018, 8, 644–649. [Google Scholar] [CrossRef]
- Borgna, F.; Haller, S.; Rodriguez, J.M.M.; Ginj, M.; Grundler, P.V.; Zeevaart, J.R.; Köster, U.; Schibli, R.; van der Meulen, N.P.; Müller, C. Combination of terbium-161 with somatostatin receptor antagonists—a potential paradigm shift for the treatment of neuroendocrine neoplasms. Eur. J. Pediatr. 2021, 1–14. [Google Scholar] [CrossRef]
- Kulaksiz, H.; Eissele, R.; Rössler, D.; Schulz, S.; Höllt, V.; Cetin, Y.; Arnold, R. Identification of somatostatin receptor subtypes 1, 2A, 3, and 5 in neuroendocrine tumours with subtype specific antibodies. Gut 2002, 50, 52–60. [Google Scholar] [CrossRef]
- Papotti, M.; Bongiovanni, M.; Volante, M.; Allìa, E.; Landolfi, S.; Helboe, L.; Schindler, M.; Cole, S.L.; Bussolati, G. Expression of somatostatin receptor types 1–5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. A correlative immunohistochemical and reverse-transcriptase polymerase chain reaction analysis. Virchows Arch. Int. J. Pathol. 2002, 440, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Maecke, H.R. Approaches to Multireceptor Targeting: Hybrid Radioligands, Radioligand Cocktails, and Sequential Radioligand Applications. J. Nucl. Med. 2017, 58, S10–S16. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Amino Acid Sequence | Radiotracer | IC50 (nM ± SEM) | Clinical Status | ||
|---|---|---|---|---|---|
| SST2 | SST3 | SST5 | |||
| Agonists | |||||
| D-Phe-c(Cys-Phe-D-Trp-Lys-Thr-Cys)Thr(ol) | 111In-DTPA-OC * | 22 ± 3.6 | 182 ± 13 | 237 ± 52 | FDA/EMA approved |
| D-Phe-c(Cys-Tyr-D-Trp-Lys-Thr-Cys)Thr(ol) | 68Ga-DOTA-TOC * | 2.5 ± 0.5 | 613 ± 140 | 73 ± 21 | Prospective phase II |
| 90Y-DOTA-TOC * | 11 ± 1.7 | 389 ± 135 | 114 ± 29 | Clinical data | |
| 177Lu-DOTA-TOC | n.r. | n.r. | n.r. | Prospective phase III | |
| D-Phe-c(Cys-Tyr-D-Trp-Lys-Thr-Cys)Thr | 68Ga-DOTA-TATE * | 0.2 ± 0.04 | >1′000 | 377 ± 18 | FDA/EMA approved |
| 177Lu-DOTA-TATE # | 2.0 ± 0.8 | 162 ± 16 | >1′000 | FDA/EMA approved | |
| D-Phe-c(Cys-1-Nal-D-Trp-Lys-Thr-Cys)Thr(ol) | 68Ga-DOTA-NOC & | 1.9 ± 0.4 | 40 ± 5.8 | 7.2 ± 1.6 | Prospective phase II |
| Antagonists | |||||
| p-NO2-Phe-c(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)Tyr-NH2 | 111In-DOTA-BASS ¥ | 9.4 ± 0.4 | >1000 | >1000 | Preliminary clinical data |
| p-Cl-Phe-c(D-Cys-Tyr-D-Aph(Cbm)-Lys-Thr-Cys)Tyr-NH2 | 68Ga-DOTA-LM3 ¶ | 12.5 ± 4.3 | >1000 | >1000 | Prospective phase I/II |
| 68Ga-NODAGA-LM3 ¶ | 1.3 ± 0.3 | >1000 | >1000 | Prospective phase I/II | |
| 177Lu-DOTA-LM3 | n.r. | n.r. | n.r. | Preliminary clinical data | |
| p-Cl-Phe-c(D-Cys-Aph(Hor)-D-Aph(Cbm)-Lys-Thr-Cys)Tyr-NH2 | 68Ga-NODAGA-JR11 ¶ | 1.2 ± 0.2 | >1000 | >1000 | Prospective phase I/II |
| 68Ga-DOTA-JR11 ¶ | 29 ± 2.7 | >1000 | >1000 | Prospective theranostic | |
| 177Lu-DOTA-JR11 ¶ | 0.7 ± 0.2 | >1000 | >1000 | Prospective phase I/II | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fani, M.; Mansi, R.; Nicolas, G.P.; Wild, D. Radiolabeled Somatostatin Analogs—A Continuously Evolving Class of Radiopharmaceuticals. Cancers 2022, 14, 1172. https://doi.org/10.3390/cancers14051172
Fani M, Mansi R, Nicolas GP, Wild D. Radiolabeled Somatostatin Analogs—A Continuously Evolving Class of Radiopharmaceuticals. Cancers. 2022; 14(5):1172. https://doi.org/10.3390/cancers14051172
Chicago/Turabian StyleFani, Melpomeni, Rosalba Mansi, Guillaume P. Nicolas, and Damian Wild. 2022. "Radiolabeled Somatostatin Analogs—A Continuously Evolving Class of Radiopharmaceuticals" Cancers 14, no. 5: 1172. https://doi.org/10.3390/cancers14051172
APA StyleFani, M., Mansi, R., Nicolas, G. P., & Wild, D. (2022). Radiolabeled Somatostatin Analogs—A Continuously Evolving Class of Radiopharmaceuticals. Cancers, 14(5), 1172. https://doi.org/10.3390/cancers14051172