
Estrogen Receptor-Beta2 (ERβ2)–Mutant p53–FOXM1 Axis: A Novel Driver of Proliferation, Chemoresistance, and Disease Progression in High Grade Serous Ovarian Cancer (HGSOC)

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. siRNAs and Transfection
2.3. RNA Isolation and Quantitative Real Time PCR (RT-qPCR) Assays
2.4. Immunoblotting
2.5. Co-Immunoprecipitation (Co-IP) Assay
2.6. Chromatin Immunoprecipitation (ChIP) Assay
2.7. Flow Cytometry Analysis
2.8. Colony Formation Assay
2.9. Immunohistochemistry (IHC) on Patient Tissue Microarray (TMA)
2.9.1. Patient Characteristics and Tissue Procurement
2.9.2. TMA Construction
2.9.3. Antigen Retrieval
2.9.4. Quantitative Scoring of Nuclear ERβ2 and TP53 and FOXM1 Immunohistochemistry Signals
2.10. Proximity Ligation Assay (PLA)
2.10.1. Fluorescence-Based PLA
2.10.2. Bright-Field PLA
2.11. Statistical Analysis
3. Results
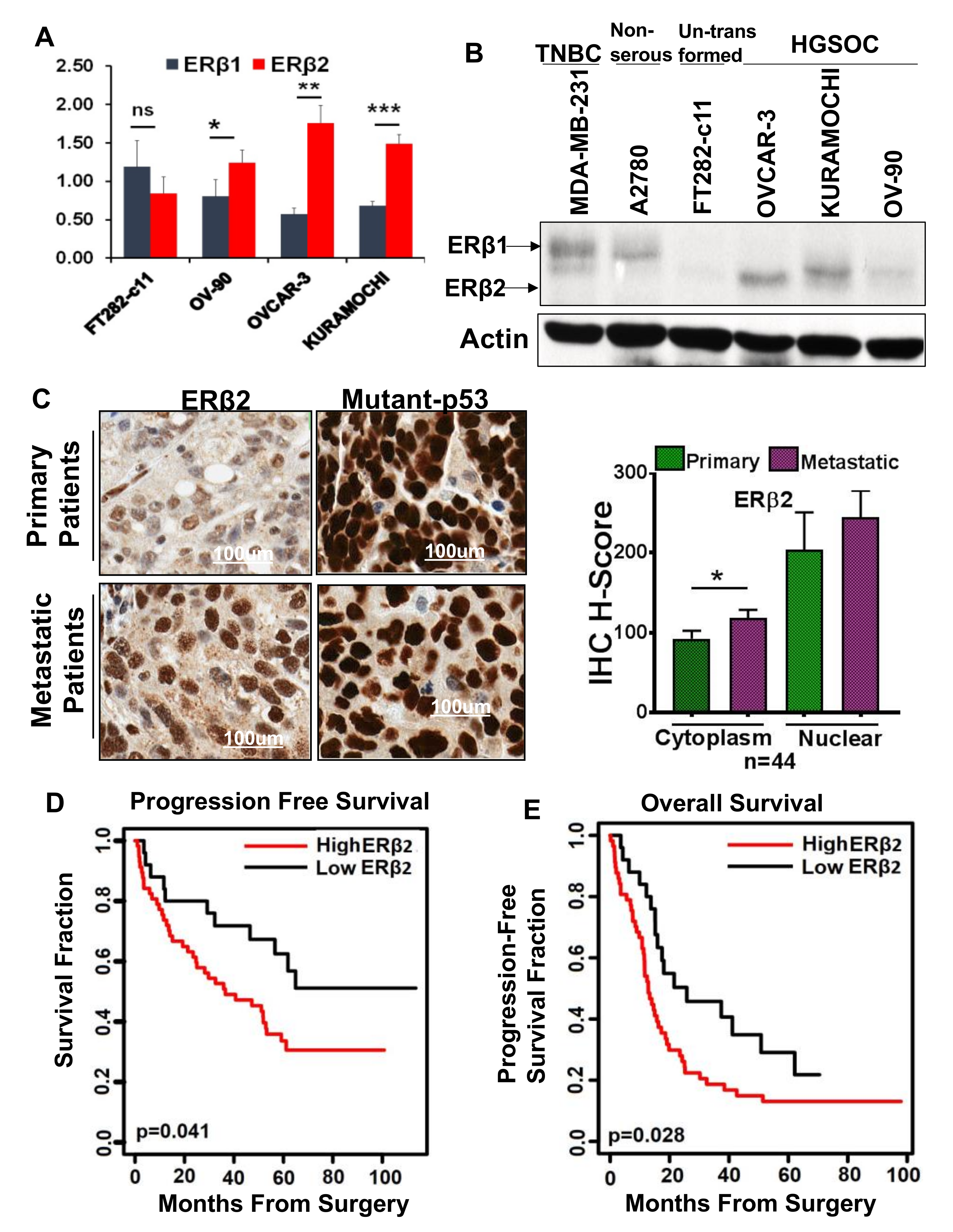
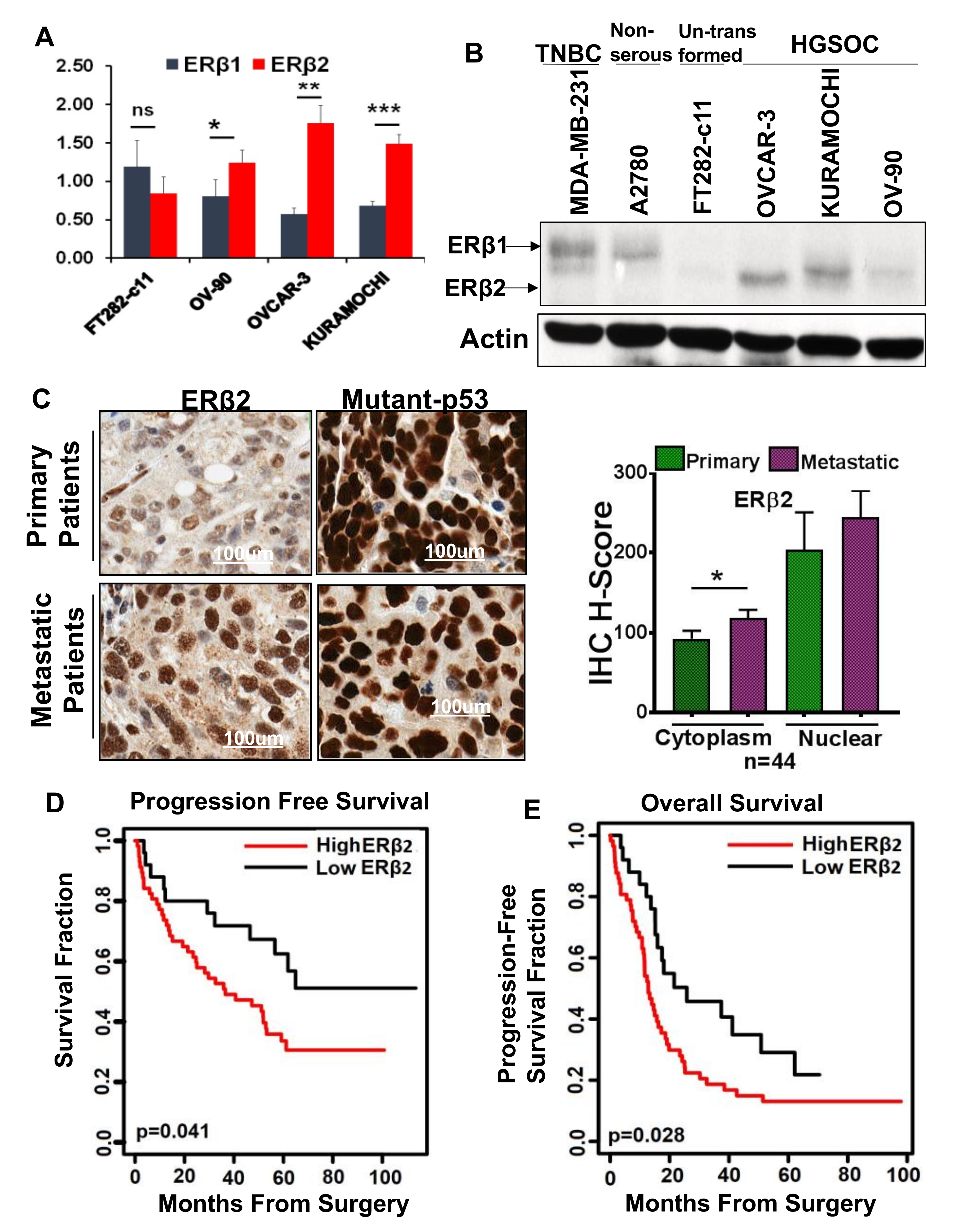
3.1. High ERβ2 Expression Is Associated with Tumor Progression in HGSOC
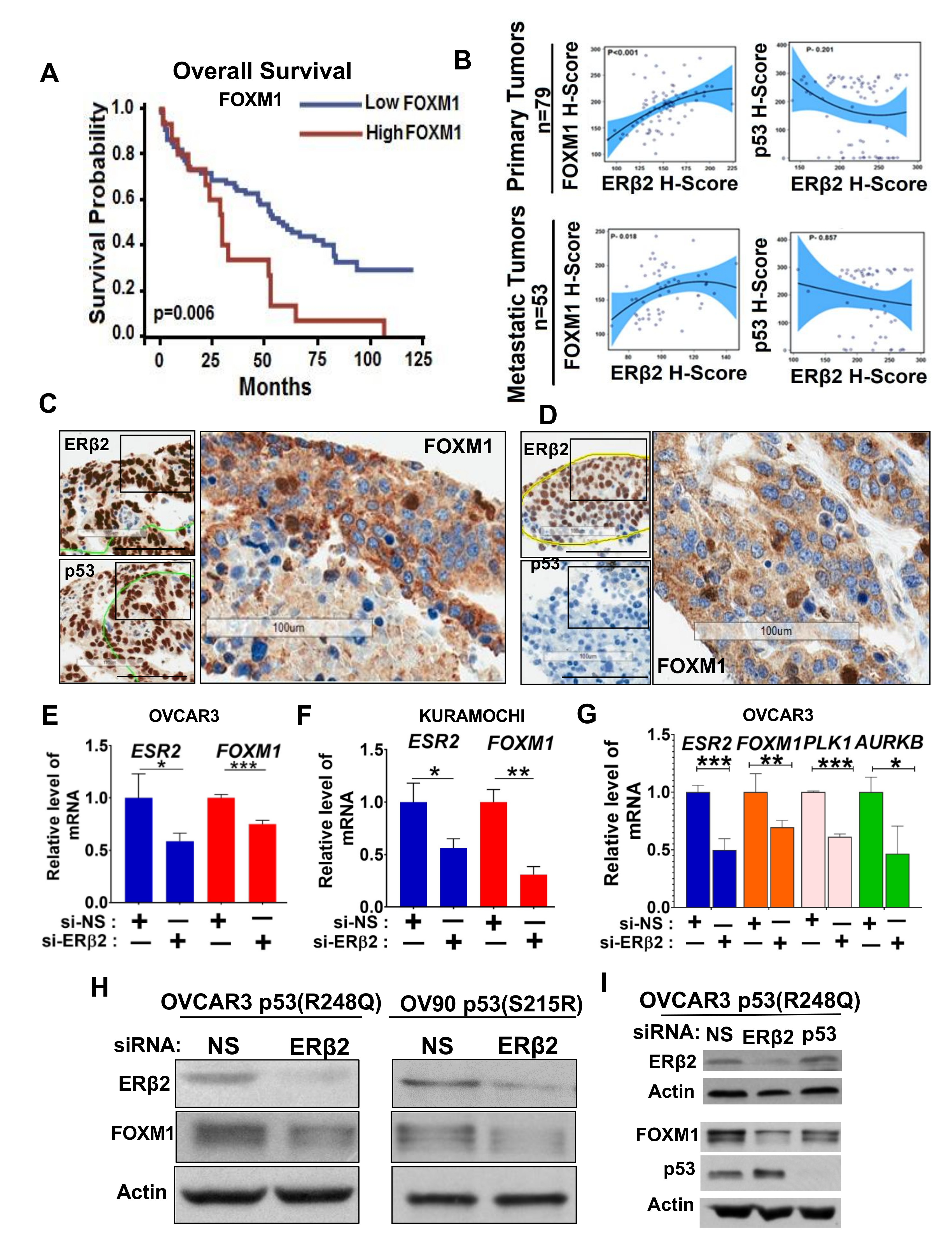
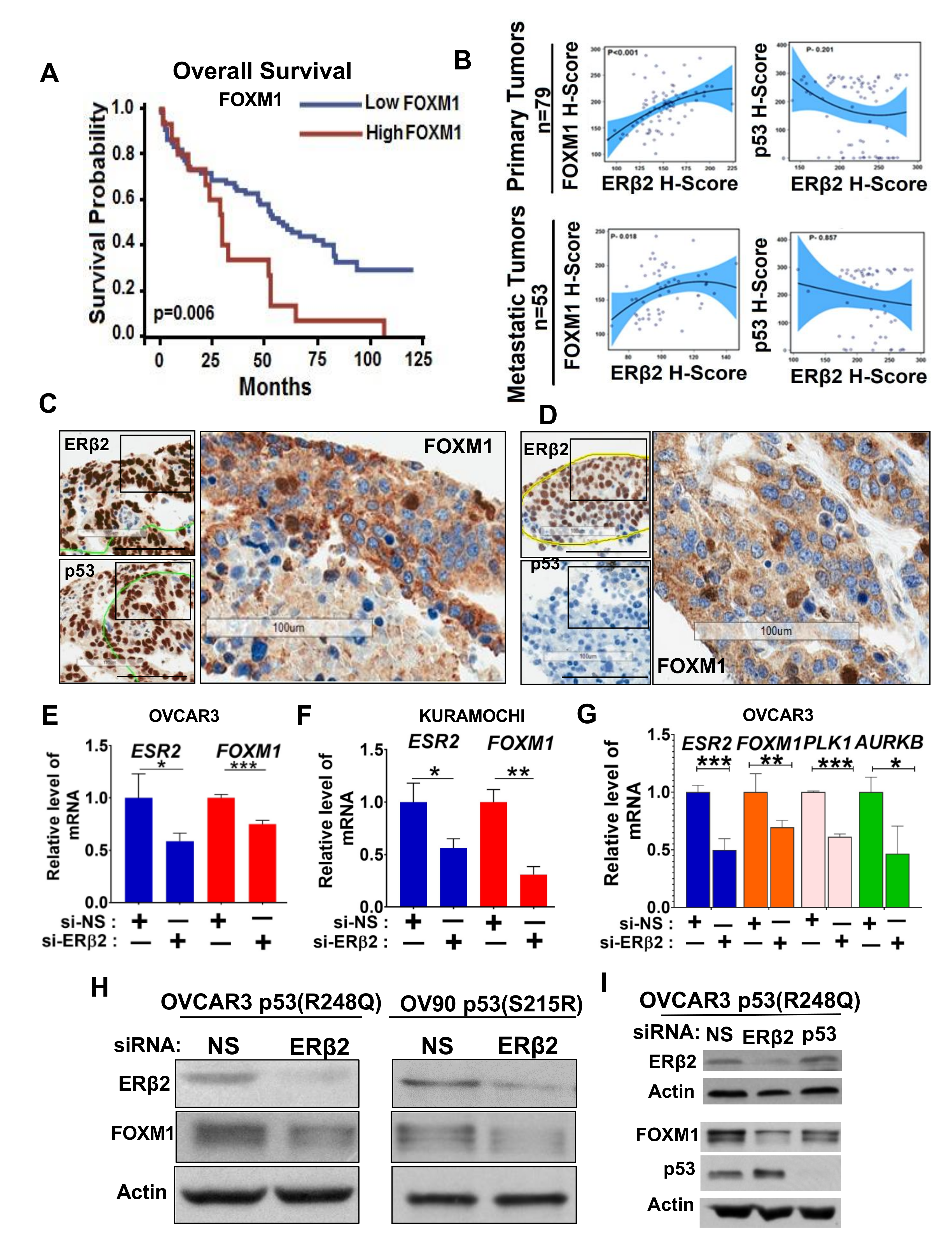
3.2. Upregulation of FOXM1 by ERβ2 Is Mutant p53-Dependent
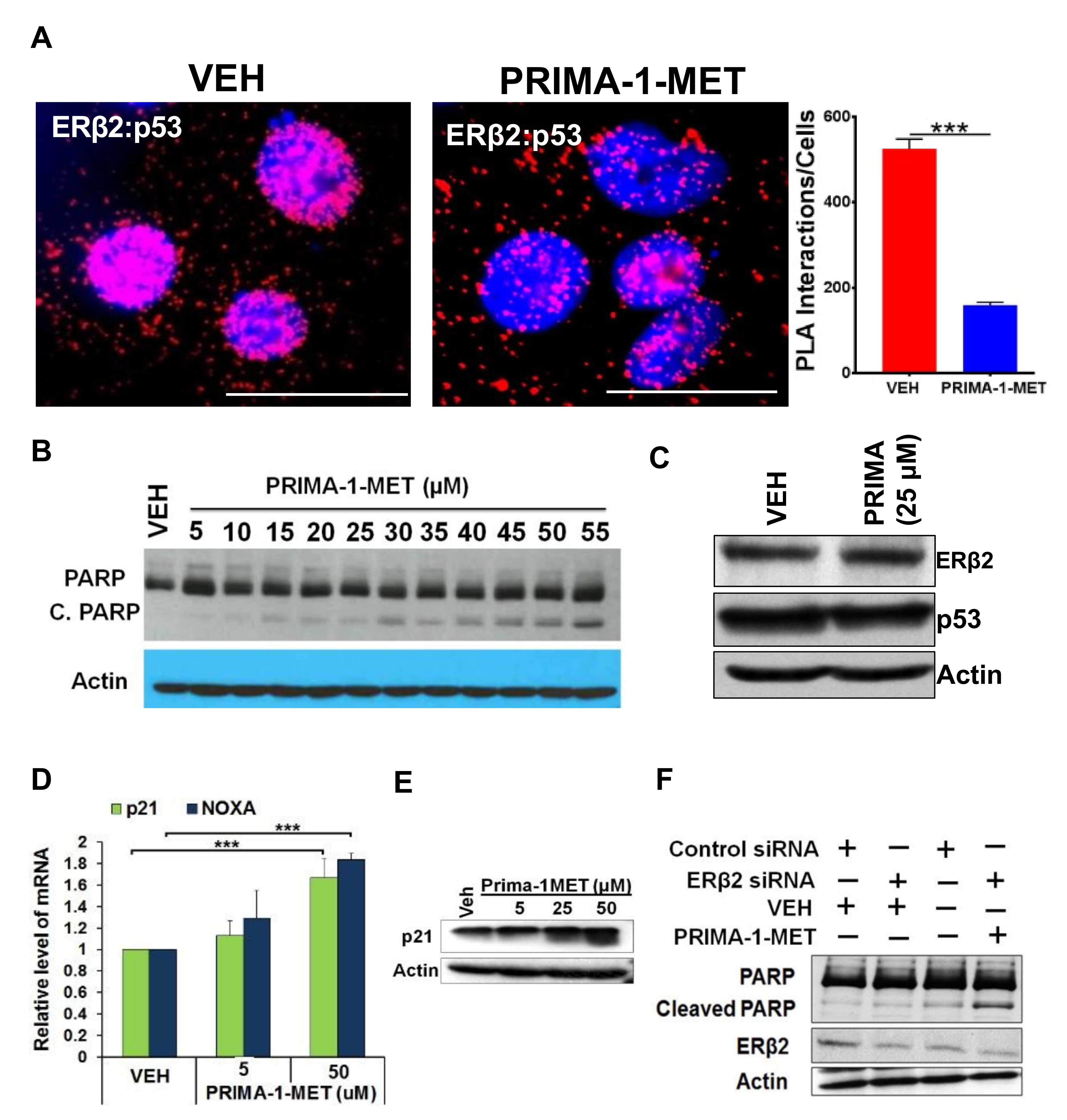
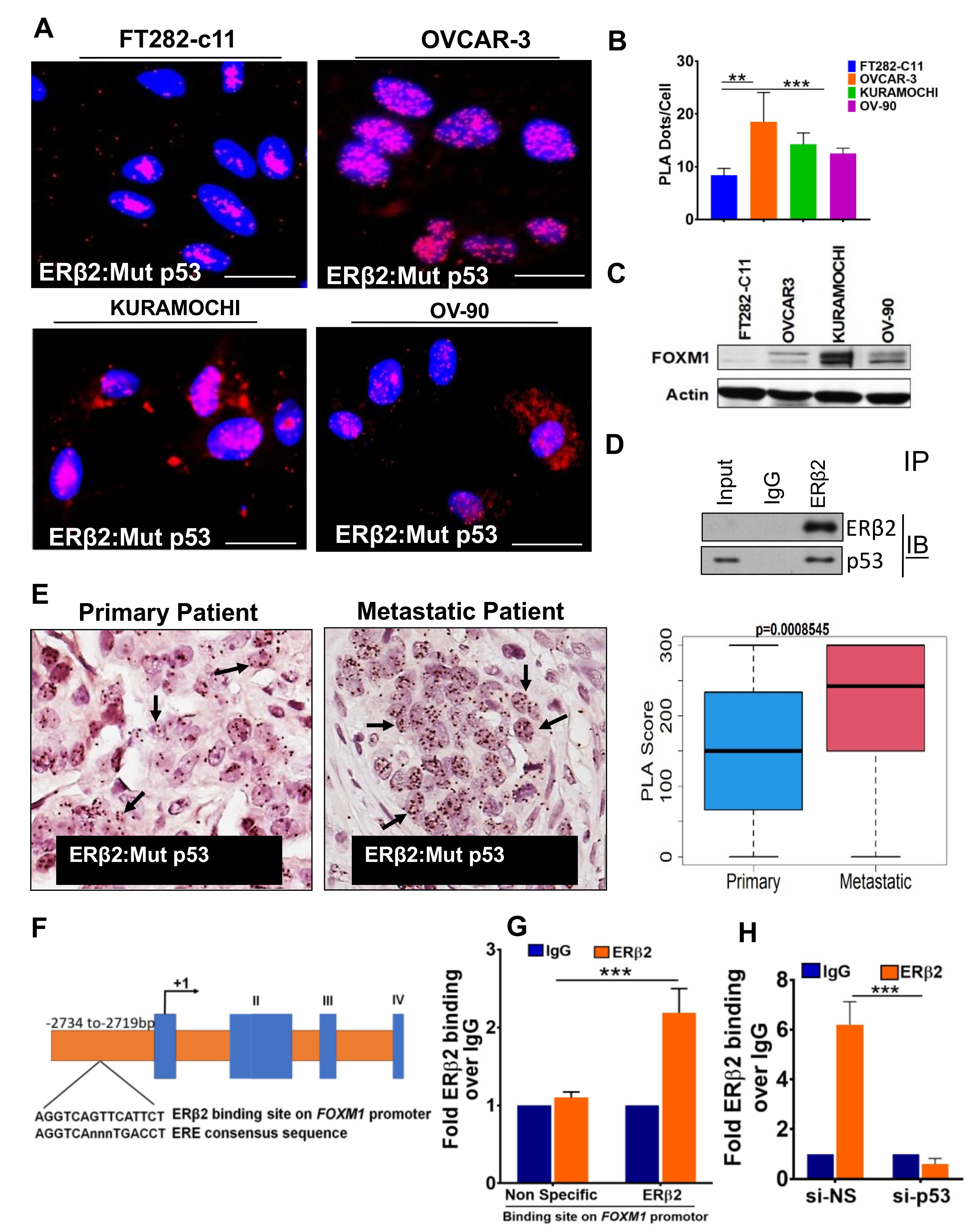
3.3. ERβ2 and Mutant p53 Physically Interact and Are Recruited to the FOXM1 Gene Promoter Leading to Activation of Transcription
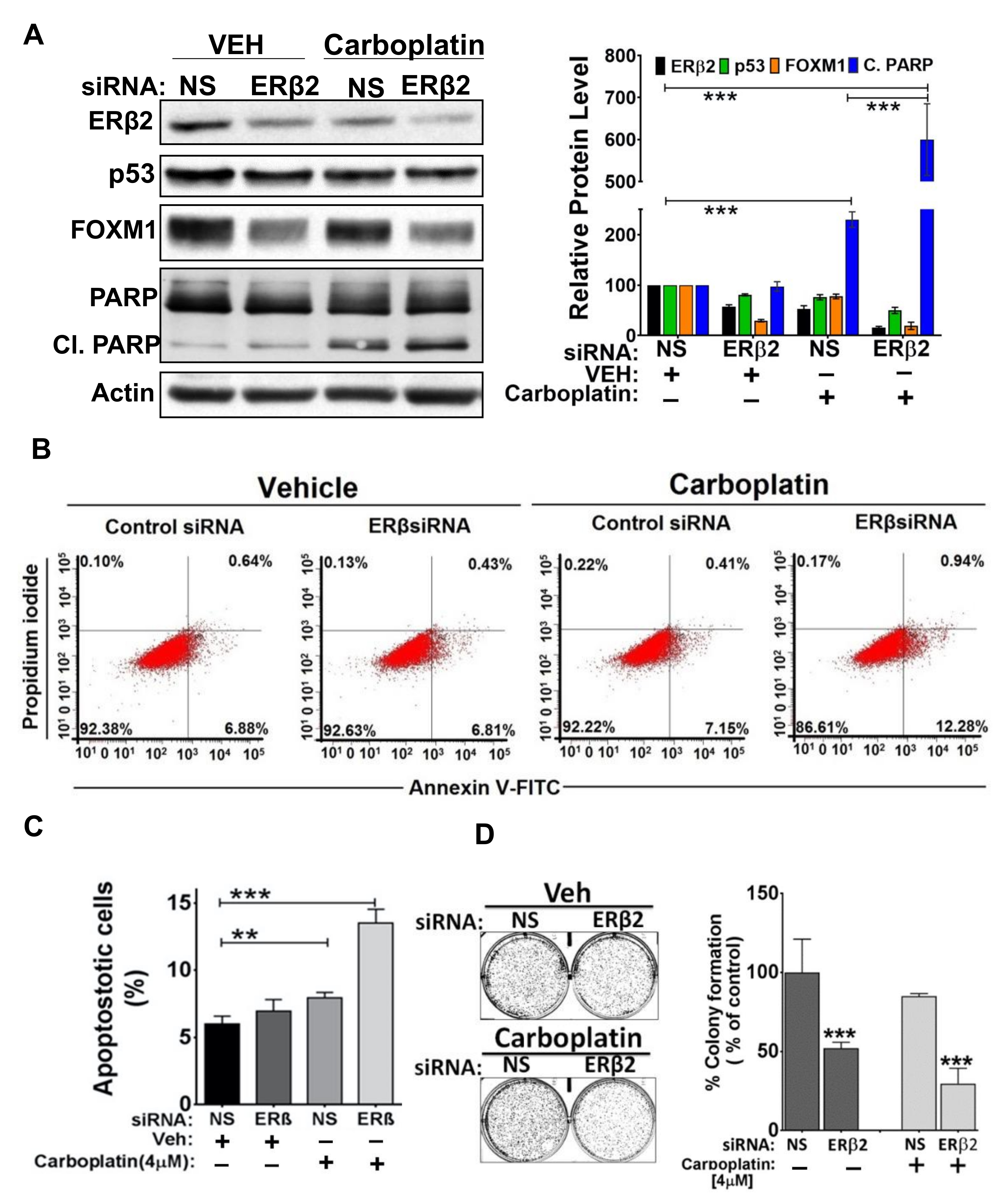
3.4. ERβ2 Activates FOXM1 Expression and Confers Resistance to Carboplatin
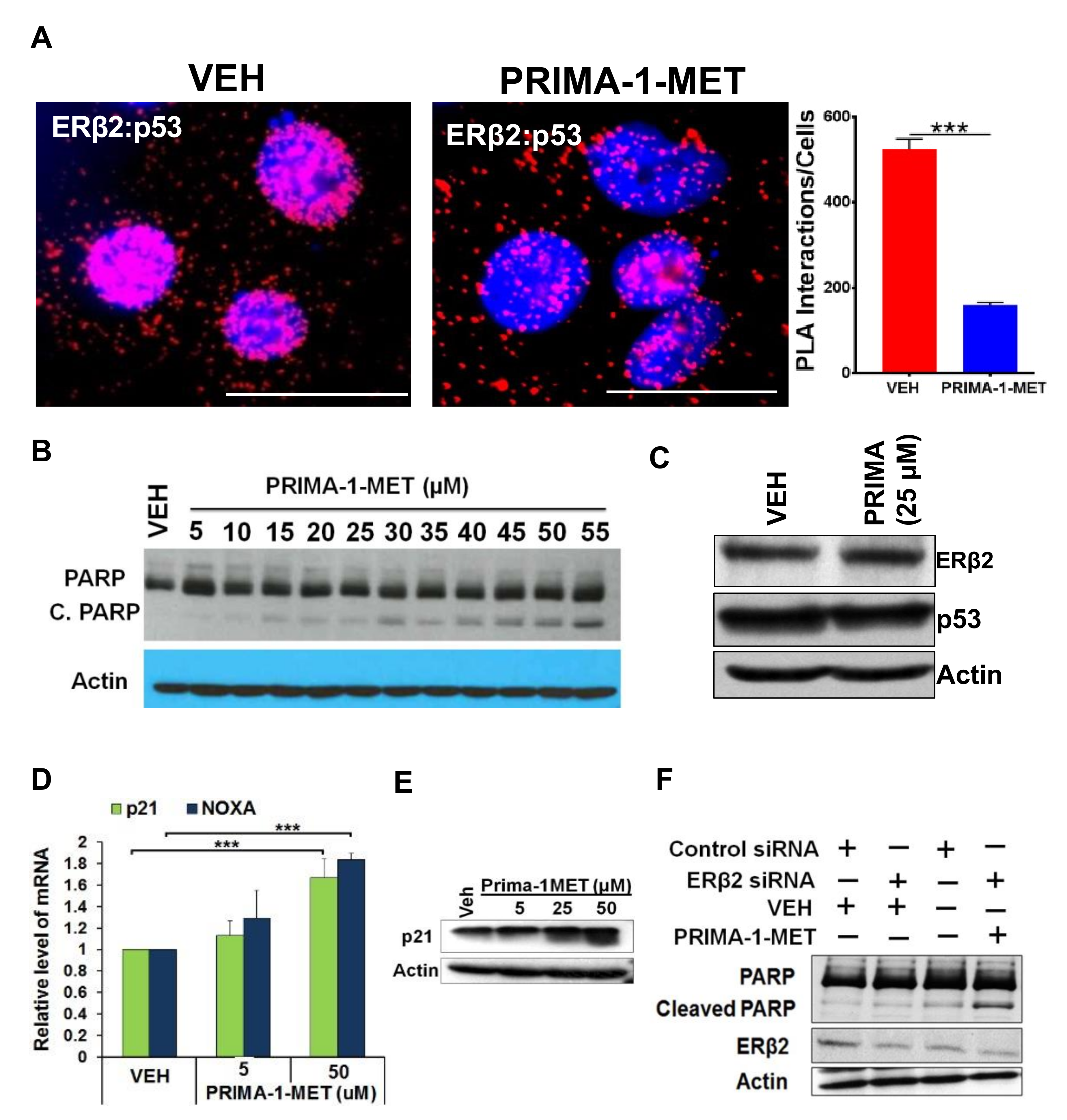
3.5. Disruption of ERβ2-Mutant p53 Crosstalk Leads to HGSOC Cells Apoptosis
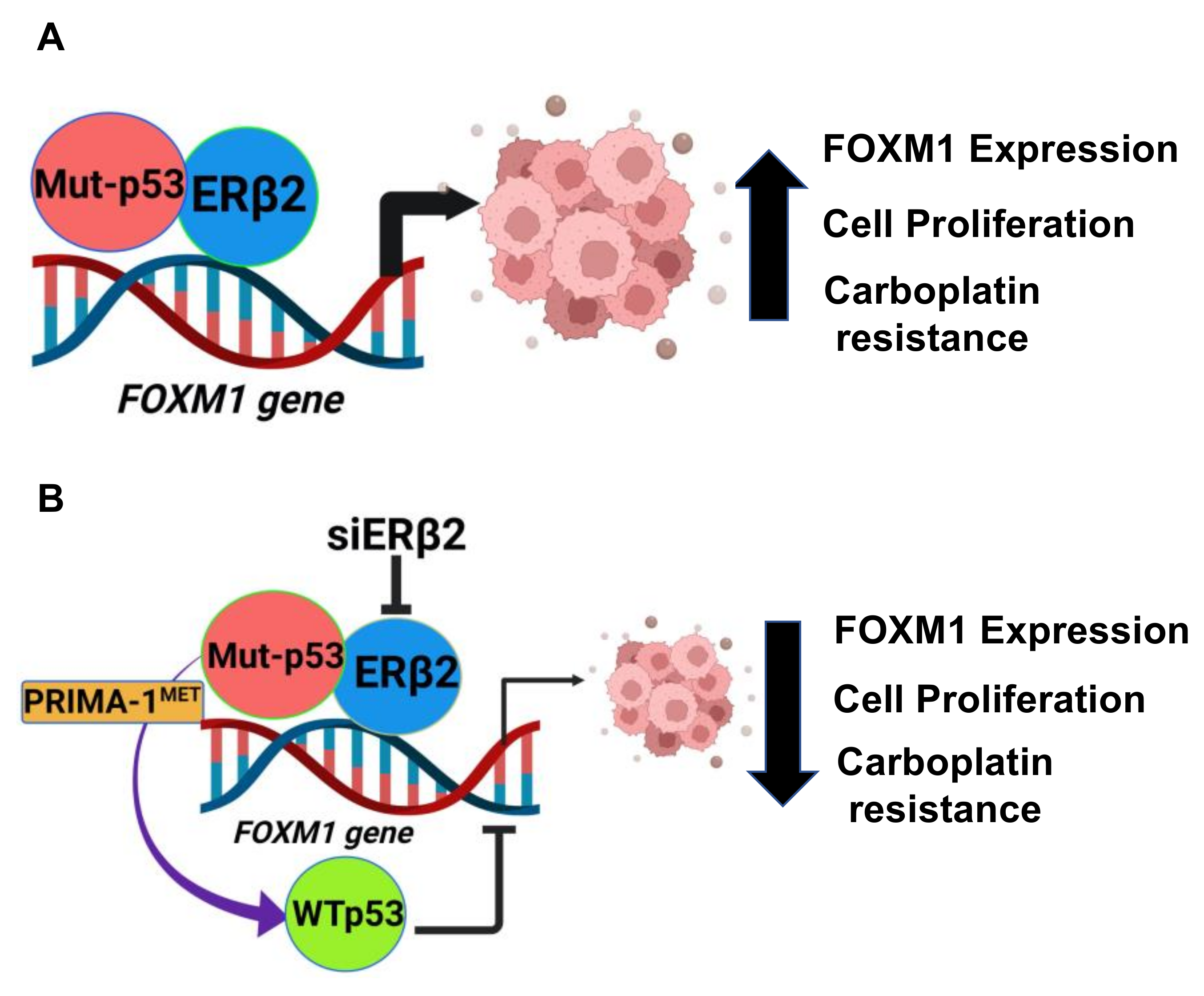
4. Discussion
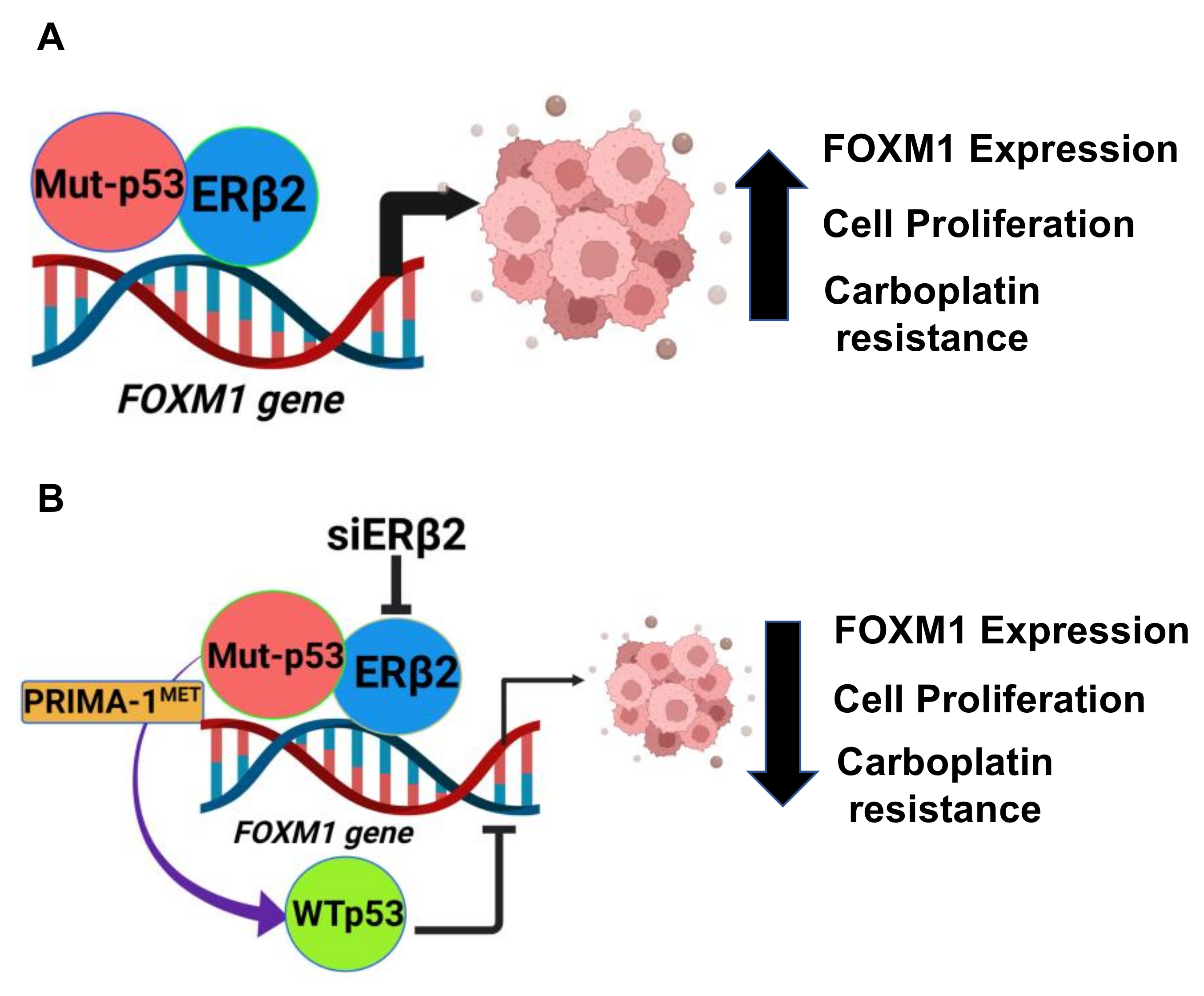
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.H.; Bast, R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meserve, E.E.K.; Brouwer, J.; Crum, C.P. Serous tubal intraepithelial neoplasia: The concept and its application. Mod. Pathol. 2017, 30, 710–721. [Google Scholar] [CrossRef]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 2019, 10, 5367. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Saleh, A.; Perets, R. Mutated p53 in HGSC-From a Common Mutation to a Target for Therapy. Cancers 2021, 13, 3465. [Google Scholar] [CrossRef]
- Tuna, M.; Ju, Z.; Yoshihara, K.; Amos, C.I.; Tanyi, J.L.; Mills, G.B. Clinical relevance of TP53 hotspot mutations in high-grade serous ovarian cancers. Br. J. Cancer 2020, 122, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.A.; Mullany, L.K.; Liu, Z.; Herron, A.J.; Wong, K.K.; Richards, J.S. Mutant p53 Promotes Epithelial Ovarian Cancer by Regulating Tumor Differentiation, Metastasis, and Responsiveness to Steroid Hormones. Cancer Res. 2016, 76, 2206–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum and taxanebased standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Chun, S.M.; Kim, K.R.; Sohn, I.; Sung, C.O. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS ONE 2013, 8, e72609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwanicki, M.P.; Chen, H.Y.; Iavarone, C.; Zervantonakis, I.K.; Muranen, T.; Novak, M.; Ince, T.A.; Drapkin, R.; Brugge, J.S. Mutant p53 regulates ovarian cancer transformed phenotypes through autocrine matrix deposition. JCI Insight 2016, 1, e86829. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen Signaling and Its Potential as a Target for Therapy in Ovarian Cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef]
- Andersen, C.L.; Sikora, M.J.; Boisen, M.M.; Ma, T.; Christie, A.; Tseng, G.; Park, Y.; Luthra, S.; Chandran, U.; Haluska, P.; et al. Active Estrogen Receptor-alpha Signaling in Ovarian Cancer Models and Clinical Specimens. Clin. Cancer Res. 2017, 23, 3802–3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, K.M.; Mok, S.C.; Ho, S.M. Expression of human estrogen receptor-alpha and -beta, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc. Natl. Acad. Sci. USA 1999, 96, 5722–5727. [Google Scholar] [CrossRef] [Green Version]
- Sieh, W.; Kobel, M.; Longacre, T.A.; Bowtell, D.D.; de Fazio, A.; Goodman, M.T.; Hogdall, E.; Deen, S.; Wentzensen, N.; Moysich, K.B.; et al. Hormone-receptor expression and ovarian cancer survival: An Ovarian Tumor Tissue Analysis consortium study. Lancet Oncol. 2013, 14, 853–862. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Sheridan, T.B.; Mabuchi, S.; Yoshino, K.; Hasegawa, K.; Studeman, K.D.; Im, D.D.; Rosenshein, N.B.; Roman, L.D.; Sood, A.K. Estrogen receptor expression and increased risk of lymphovascular space invasion in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2014, 133, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Ciucci, A.; Zannoni, G.F.; Travaglia, D.; Petrillo, M.; Scambia, G.; Gallo, D. Prognostic significance of the estrogen receptor beta (ERbeta) isoforms ERbeta1, ERbeta2, and ERbeta5 in advanced serous ovarian cancer. Gynecol. Oncol. 2014, 132, 351–359. [Google Scholar] [CrossRef]
- Drummond, A.E.; Fuller, P.J. Ovarian actions of estrogen receptor-beta: An update. Semin. Reprod. Med. 2012, 30, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Bossard, C.; Busson, M.; Vindrieux, D.; Gaudin, F.; Machelon, V.; Brigitte, M.; Jacquard, C.; Pillon, A.; Balaguer, P.; Balabanian, K.; et al. Potential role of estrogen receptor beta as a tumor suppressor of epithelial ovarian cancer. PLoS ONE 2012, 7, e44787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docquier, A.; Garcia, A.; Savatier, J.; Boulahtouf, A.; Bonnet, S.; Bellet, V.; Busson, M.; Margeat, E.; Jalaguier, S.; Royer, C.; et al. Negative regulation of estrogen signaling by ERbeta and RIP140 in ovarian cancer cells. Mol. Endocrinol. 2013, 27, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Viswanadhapalli, S.; Garcia, L.; Zhou, M.; Nair, B.C.; Kost, E.; Rao Tekmal, R.; Li, R.; Rao, M.K.; Curiel, T.; et al. Therapeutic utility of natural estrogen receptor beta agonists on ovarian cancer. Oncotarget 2017, 8, 50002–50014. [Google Scholar] [CrossRef] [Green Version]
- Pinton, G.; Nilsson, S.; Moro, L. Targeting estrogen receptor beta (ERbeta) for treatment of ovarian cancer: Importance of KDM6B and SIRT1 for ERbeta expression and functionality. Oncogenesis 2018, 7, 15. [Google Scholar] [CrossRef]
- Schuler-Toprak, S.; Moehle, C.; Skrzypczak, M.; Ortmann, O.; Treeck, O. Effect of estrogen receptor beta agonists on proliferation and gene expression of ovarian cancer cells. BMC Cancer 2017, 17, 319. [Google Scholar] [CrossRef]
- Aust, S.; Horak, P.; Pils, D.; Pils, S.; Grimm, C.; Horvat, R.; Tong, D.; Schmid, B.; Speiser, P.; Reinthaller, A.; et al. The prognostic value of estrogen receptor beta and proline-, glutamic acid- and leucine-rich protein 1 (PELP1) expression in ovarian cancer. BMC Cancer 2013, 13, 115. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.K.; Wei, N.; Liu, S.S.; Xiao-Yun, L.; Cheung, A.N.; Ngan, H.Y. Estrogen receptor subtypes in ovarian cancer: A clinical correlation. Obstet. Gynecol. 2008, 111, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Haring, J.; Schuler, S.; Lattrich, C.; Ortmann, O.; Treeck, O. Role of estrogen receptor beta in gynecological cancer. Gynecol. Oncol. 2012, 127, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Ciucci, A.; Zannoni, G.F.; Travaglia, D.; Scambia, G.; Gallo, D. Mitochondrial estrogen receptor beta2 drives antiapoptotic pathways in advanced serous ovarian cancer. Hum. Pathol. 2015, 46, 1138–1146. [Google Scholar] [CrossRef]
- van Kruchten, M.; van der Marel, P.; de Munck, L.; Hollema, H.; Arts, H.; Timmer-Bosscha, H.; de Vries, E.; Hospers, G.; Reyners, A. Hormone receptors as a marker of poor survival in epithelial ovarian cancer. Gynecol. Oncol. 2015, 138, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, A.M.; Prives, C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene 2009, 28, 4295–4305. [Google Scholar] [CrossRef] [Green Version]
- Pandit, B.; Halasi, M.; Gartel, A.L. p53 negatively regulates expression of FoxM1. Cell Cycle 2009, 8, 3425–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Barger, C.J.; Karpf, A.R. FOXM1: A Multifunctional Oncoprotein and Emerging Therapeutic Target in Ovarian Cancer. Cancers 2021, 13, 3065. [Google Scholar] [CrossRef]
- Mukhopadhyay, U.K.; Oturkar, C.C.; Adams, C.; Wickramasekera, N.; Bansal, S.; Medisetty, R.; Miller, A.; Swetzig, W.M.; Silwal-Pandit, L.; Borresen-Dale, A.L.; et al. TP53 Status as a Determinant of Pro- vs Anti-Tumorigenic Effects of Estrogen Receptor-Beta in Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 1202–1215. [Google Scholar] [CrossRef]
- Das, G.M.; Mukhopadhyay, U.K.; Bansal, S.; Medisetty, R.; Swetzig, W.; Wickramasekera, N. Tumor suppressor p53 status as a determinant of estrogen receptor beta signaling in breast cancer. FASEB J. 2013, 27, 1031.21. [Google Scholar] [CrossRef]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Gustafsson, J.A.; Thomas, C. ERbeta decreases the invasiveness of triple-negative breast cancer cells by regulating mutant p53 oncogenic function. Oncotarget 2016, 7, 13599–13611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oturkar, C.C.; Miller, A.; Zsiros, E.; Das, G.M. Estrogen receptor beta 2 (ERβ2)- p53- FOXM1 signaling axis in high-grade serous ovarian cancer (HGSOC): Underlying mechanisms and implications for resistance to therapy. Clin. Cancer Res. 2018, 24, 111–112. [Google Scholar]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Contal, C.; O’Quigly, J. An application of changepoint methods in studying the effect of age on survival in breast cancer. Comput. Stat. Data Anal. 1999, 30, 253–270. [Google Scholar] [CrossRef]
- Kyriakidis, I.; Papaioannidou, P. Estrogen receptor beta and ovarian cancer: A key to pathogenesis and response to therapy. Arch. Gynecol. Obstet. 2016, 293, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.R.; Freiman, R.N. Estrogen signaling crosstalk: Implications for endocrine resistance in ovarian cancer. J. Steroid Biochem. Mol. Biol. 2014, 143, 160–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefano, I.; Zannoni, G.F.; Prisco, M.G.; Fagotti, A.; Tortorella, L.; Vizzielli, G.; Mencaglia, L.; Scambia, G.; Gallo, D. Cytoplasmic expression of estrogen receptor beta (ERbeta) predicts poor clinical outcome in advanced serous ovarian cancer. Gynecol. Oncol. 2011, 122, 573–579. [Google Scholar] [CrossRef]
- Mei, J.; Tian, H.; Huang, H.S.; Hsu, C.F.; Liou, Y.; Wu, N.; Zhang, W.; Chu, T.Y. Cellular models of development of ovarian high-grade serous carcinoma: A review of cell of origin and mechanisms of carcinogenesis. Cell Prolif. 2021, 54, e13029. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanova, A.; Vang, R.; Kshirsagar, M.; Lu, D.; Marks, M.A.; Shih Ie, M.; Kurman, R.J. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: An immunohistochemical and nucleotide sequencing analysis. Mod. Pathol. 2011, 24, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Alkema, W.; Engstrom, P.; Wasserman, W.W.; Lenhard, B. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004, 32, D91–D94. [Google Scholar] [CrossRef] [Green Version]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim. Biophys. Acta 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Bjorklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [Green Version]
- Millour, J.; de Olano, N.; Horimoto, Y.; Monteiro, L.J.; Langer, J.K.; Aligue, R.; Hajji, N.; Lam, E.W. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011, 10, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of Estrogen Receptor Beta on Gene Networks Regulated by Estrogen Receptor Alpha in Breast Cancer Cells. Endocrinology 2006, 147, 4831–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.; Edvardsson, K.; Lewandowski, S.A.; Strom, A.; Gustafsson, J.A. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene 2008, 27, 1019–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [Green Version]
- Shafrir, A.L.; Babic, A.; Gates Kuliszewski, M.; Rice, M.S.; Townsend, M.K.; Hecht, J.L.; Tworoger, S.S. Estrogen Receptor-beta Expression of Ovarian Tumors and Its Association with Ovarian Cancer Risk Factors. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2211–2219. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oturkar, C.C.; Gandhi, N.; Rao, P.; Eng, K.H.; Miller, A.; Singh, P.K.; Zsiros, E.; Odunsi, K.O.; Das, G.M. Estrogen Receptor-Beta2 (ERβ2)–Mutant p53–FOXM1 Axis: A Novel Driver of Proliferation, Chemoresistance, and Disease Progression in High Grade Serous Ovarian Cancer (HGSOC). Cancers 2022, 14, 1120. https://doi.org/10.3390/cancers14051120
Oturkar CC, Gandhi N, Rao P, Eng KH, Miller A, Singh PK, Zsiros E, Odunsi KO, Das GM. Estrogen Receptor-Beta2 (ERβ2)–Mutant p53–FOXM1 Axis: A Novel Driver of Proliferation, Chemoresistance, and Disease Progression in High Grade Serous Ovarian Cancer (HGSOC). Cancers. 2022; 14(5):1120. https://doi.org/10.3390/cancers14051120
Chicago/Turabian StyleOturkar, Chetan C., Nishant Gandhi, Pramod Rao, Kevin H. Eng, Austin Miller, Prashant K. Singh, Emese Zsiros, Kunle O. Odunsi, and Gokul M. Das. 2022. "Estrogen Receptor-Beta2 (ERβ2)–Mutant p53–FOXM1 Axis: A Novel Driver of Proliferation, Chemoresistance, and Disease Progression in High Grade Serous Ovarian Cancer (HGSOC)" Cancers 14, no. 5: 1120. https://doi.org/10.3390/cancers14051120
APA StyleOturkar, C. C., Gandhi, N., Rao, P., Eng, K. H., Miller, A., Singh, P. K., Zsiros, E., Odunsi, K. O., & Das, G. M. (2022). Estrogen Receptor-Beta2 (ERβ2)–Mutant p53–FOXM1 Axis: A Novel Driver of Proliferation, Chemoresistance, and Disease Progression in High Grade Serous Ovarian Cancer (HGSOC). Cancers, 14(5), 1120. https://doi.org/10.3390/cancers14051120