
Potential Involvement of NSD1, KRT24 and ACACA in the Genetic Predisposition to Colorectal Cancer

 ,
,  , , , , , , ,
, , , , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Germline Mutation Identification in Pooled Samples
2.3. Validation of the Obtained Results and Carrier Identification
2.4. In Silico Predictions
2.5. Co-Segregation and Second Hit Analyses
2.6. Gene Burden Test
2.7. Statistical Analysis
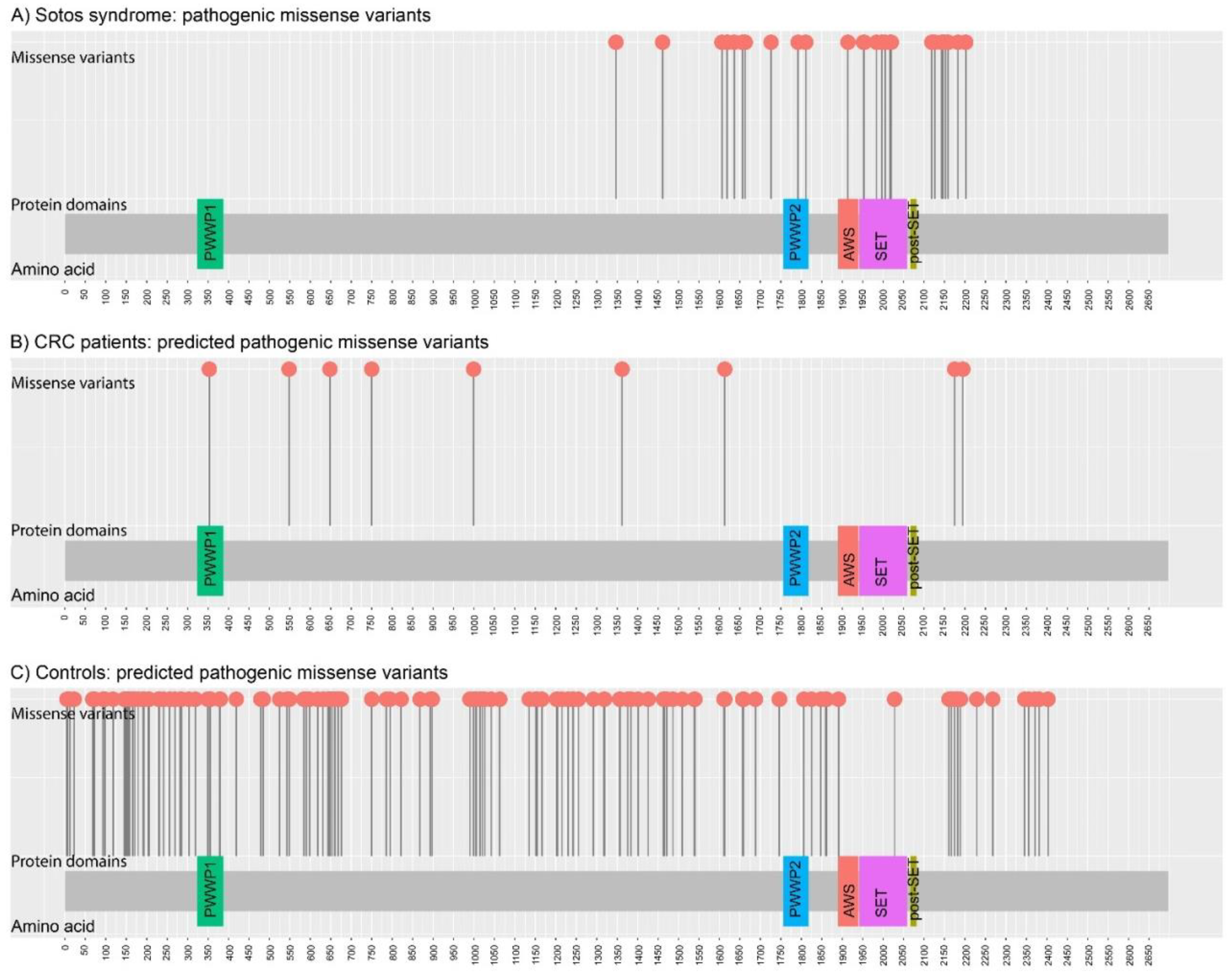
3. Results
3.1. Gene Selection
3.2. Gene Mutational Screening of Familial/Early-Onset CRC and Polyposis Patients

{kind=link}
{kind=link}
| Gene | Cohort or Study | Disruptive Alleles | Disruptive, Splice-Site, Start-Loss, Predicted Pathogenic Missense (REVEL > 0.4) | ||
|---|---|---|---|---|---|
| n/Total Alleles (%) | OR (95%CI); p-Value | n/Total Alleles (%) | OR (95%CI); p-Value | ||
| NSD1 | Controls (gnomAD non-cancer) | 15/268,374 (0.01%) | 868/268,374 (0.32%) | ||
| Familial/EOCRC | |||||
| Zhunussova et al. | 0/250 (0.00%) | 1/250 (0.40%) | |||
| Chubb et al. | 1/2012 (0.05%) | 8/2012 (0.40%) | |||
| Current study | 0/930 (0.00%) | 0/930 (0.00%) | |||
| Subtotal | 1/3192 (0.03%) | 5.61 (0.13–36.44); p = 0.17 | 9/3192 (0.28%) | 0.87 (0.40–1.66); p = 0.87 | |
| Polyposis (current study) | 0/542 (0.00%) | 0.00 (0.00–138.68); p = 1 | 1/542 (0.18%) | 0.57 (0.01–3.20); p = 1 | |
| TCGA CRC patients | 0/1086 (0.00%) | 0.00 (0.00–68.98); p = 1 | 2/1086 (0.18%) | 0.57 (0.07–2.07); p = 0.59 | |
| TOTAL patients | 1/4820 (0.02%) | 3.71 (0.09–24.15); p = 0.25 | 12/4820 (0.25%) | 0.77 (0.40–1.35); p = 0.44 | |
| HDAC10 | Controls (gnomAD non-cancer) | 303/268,374 (0.11%) | 1019/268,374 (0.38%) | ||
| Familial/EOCRC | |||||
| Chubb et al. | 3/2012 (0.15%) | 6/2012 (0.30%) | |||
| Current study | 0/930 (0.00%) | 0/930 (0.00%) | |||
| Subtotal | 3/2942 (0.10%) | 0.60 (0.07–2.20); p = 0.78 | 6/2942 (0.20%) | 0.54 (0.20–1.17); p = 0.17 | |
| Polyposis (current study) | 0/542 (0.00%) | 0.00 (0.00–6.08); p = 1 | 3/542 (0.55%) | 1.46 (0.30–4.30); p = 0.47 | |
| TCGA CRC patients | 0/1086 (0.00%) | 0.00 (0.00–3.03); p = 0.64 | 3/1086 (0.28%) | 0.72 (0.15–2.14); p = 0.80 | |
| TOTAL patients | 3/4570 (0.07%) | 0.58 (0.12–1.72); p = 0.50 | 12/4570 (0.26%) | 0.70 (0.36–1.21); p = 0.22 | |
| KRT24 | Controls (gnomAD non-cancer) | 186/268,374 (0.07%) | 1016/268,374 (0.38%) | ||
| Familial/EOCRC | |||||
| Chubb et al. | 2/2012 (0.10%) | 5/2012 (0.25%) | |||
| Current study | 1/930 (0.11%) | 2/930 (0.22%) | |||
| Subtotal | 3/2942 (0.10%) | 1.47 (0.30–4.37); p = 0.46 | 7/2942 (0.22%) | 0.63 (0.25–1.30); p = 0.29 | |
| Polyposis (current study) | 0/542 (0.00%) | 0.00 (0.00–9.94); p = 1 | 1/542 (0.18%) | 0.49 (0.01–2.73); p = 0.73 | |
| TCGA CRC patients | 2/1086 (0.18%) | 2.66 (0.32–9.77); p = 0.18 | 4/1086 (0.37%) | 0.97 (0.26–2.51); p = 1 | |
| TOTAL patients | 5/4570 (0.11%) | 1.58 (0.51–3.75); p = 0.26 | 12/4570 (0.26%) | 0.69 (0.36–1.22); p = 0.27 | |
| ACACA | Controls (gnomAD non-cancer) | 43/268,374 (0.02%) | 988/268,374 (0.37%) | ||
| Familial/EOCRC | |||||
| Thutkawkorapin et al. | 0/102 (0.00%) | 1/102 (0.98%) | |||
| Chubb et al. | 1/2012 (0.05%) | 4/2012 (0.20%) | |||
| Current study | 0/930 (0.00%) | 0/930 (0.00%) | |||
| Subtotal | 1/3044 (0.03%) | 2.05 (0.05–12.06); p = 0.39 | 5/3044 (0.16%) | 0.45 (0.144–1.04); p = 0.07 | |
| Polyposis (current study) | 0/542 (0.00%) | 0.00 (0.00–44.53); p = 1 | 0/542 (0.00%) | 0.00 (0.00–1.85); p = 0.28 | |
| TCGA CRC patients | 1/1086 (0.09%) | 5.75 (0.14–33.85); p = 0.16 | 5/1086 (0.46%) | 1.25 (0.40–2.94); p = 0.61 | |
| TOTAL patients | 2/4672 (0.04%) | 2.67 (0.31–10.26); p = 0.18 | 10/4672 (0.21%) | 0.06 (0.03–0.11); p < 2.2 × 10–16 | |
| TP63 | Controls (gnomAD non-cancer) | 4/268,374 (0.001%) | 929/268,374 (0.35%) | ||
| Familial/EOCRC | |||||
| Chubb et al. | 0/2012 (0.00%) | 4/2012 (0.20%) | |||
| Current study | 0/930 (0.00%) | 3/930 (0.32%) | |||
| Subtotal | 0/2942 (0.00%) | 0.00 (0.00–137.82); p = 1 | 7/2942 (0.24%) | 0.69 (0.28–1.42); p = 0.43 | |
| Polyposis (current study) | 0/542 (0.00%) | 0.00 (0.00–736.86); p = 1 | 1/542 (0.18%) | 0.53 (0.01–2.98); p = 1 | |
| TCGA CRC patients | 0/1086 (0.00%) | 0.00 (0.00–374.35); p = 1 | 0/1086 (0.00%) | 0.00 (0.00–0.98); p = 0.06 | |
| TOTAL patients | 0/4570 (0.00%) | 0.00 (0.00–88.76); p = 1 | 8/4570 (0.18%) | 0.50 (0.22–1.00); p = 0.05 | |
3.2.1. NSD1
3.2.2. HDAC10
3.2.3. KRT24
3.2.4. TP63
3.3. Gene Burden Analysis: Assessment of the Association of the Selected Genes with CRC Predisposition
3.4. Somatic Second Hits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frank, C.; Sundquist, J.; Yu, H.; Hemminki, A.; Hemminki, K. Concordant and discordant familial cancer: Familial risks, proportions and population impact. Int. J. Cancer 2017, 140, 1510–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terradas, M.; Capellá, G.; Valle, L. Dominantly Inherited Hereditary Nonpolyposis Colorectal Cancer Not Caused by MMR Genes. J. Clin. Med. 2020, 9, 1954. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2019, 247, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; de Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Asp. Med. 2019, 69, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Supek, F.; Lehner, B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belhadj, S.; Mur, P.; Navarro, M.; González, S.; Moreno, V.; Capella, G.; Valle, L. Delineating the Phenotypic Spectrum of the NTHL1-Associated Polyposis. Clin. Gastroenterol. Hepatol. 2017, 15, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Belhadj, S.; Quintana, I.; Mur, P.; Munoz-Torres, P.M.; Alonso, M.H.; Navarro, M.; Terradas, M.; Piñol, V.; Brunet, J.; Moreno, V.; et al. NTHL1 biallelic mutations seldom cause colorectal cancer, serrated polyposis or a multi-tumor phenotype, in absence of colorectal adenomas. Sci. Rep. 2019, 9, 1–5. [Google Scholar] [CrossRef]
- Terradas, M.; Munoz-Torres, P.M.; Belhadj, S.; Aiza, G.; Navarro, M.; Brunet, J.; Capellá, G.; Valle, L. Contribution to colonic polyposis of recently proposed predisposing genes and assessment of the prevalence of NTHL1- and MSH3- associated polyposes. Hum. Mutat. 2019, 40, 1910–1923. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Belhadj, S.; Terradas, M.; Munoz-Torres, P.M.; Aiza, G.; Navarro, M.; Capellá, G.; Valle, L. Candidate genes for hereditary colorectal cancer: Mutational screening and systematic review. Hum. Mutat. 2020, 41, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rozadilla, C.; Álvarez-Barona, M.; Quintana, I.; López-Novo, A.; Amigo, J.; Cameselle-Teijeiro, J.M.; Roman, E.; Gonzalez, D.; Llor, X.; Bujanda, L.; et al. Exome sequencing of early-onset patients supports genetic heterogeneity in colorectal cancer. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Houlston, R.S. CanVar: A resource for sharing germline variation in cancer patients. F1000Research 2016, 5, 2813. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- D’Afonseca, V.; Gónzalez, G.; Salazar, M.; Arencibia, A.D. Computational analyses on genetic alterations in the NSD genes family and the implications for colorectal cancer development. Ecancermedicalscience 2020, 14, 1001. [Google Scholar] [CrossRef]
- Zhunussova, G.; Afonin, G.; Abdikerim, S.; Jumanov, A.; Perfilyeva, A.; Kaidarova, D.; Djansugurova, L. Mutation Spectrum of Cancer-Associated Genes in Patients With Early Onset of Colorectal Cancer. Front. Oncol. 2019, 9, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götze, S.; Coersmeyer, M.; Müller, O.; Sievers, S. Histone deacetylase inhibitors induce attenuation of Wnt signaling and TCF7L2 depletion in colorectal carcinoma cells. Int. J. Oncol. 2014, 45, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Yan, Y.; Lu, L.; Chen, B. HDAC10 expression is associated with DNA mismatch repair gene and is a predictor of good prognosis in colon carcinoma. Oncol. Lett. 2017, 14, 4923–4929. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, R.; Li, Y.; Xiang, S.; Yuan, F.; Yuan, Z.; Telles, E.; Fang, J.; Coppola, D.; Shibata, D.; Lane, W.S.; et al. Histone Deacetylase 10 Regulates DNA Mismatch Repair and May Involve the Deacetylation of MutS Homolog. J. Biol. Chem. 2015, 290, 22795–22804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Ho, K.S.; Eu, K.W.; Cheah, P.Y. A Susceptibility Gene Set for Early Onset Colorectal Cancer That Integrates Diverse Signaling Pathways: Implication for Tumorigenesis. Clin. Cancer Res. 2007, 13, 1107–1114. [Google Scholar] [CrossRef] [Green Version]
- Thutkawkorapin, J.; Lindblom, A.; Tham, E. Exome sequencing in 51 early onset non-familial CRC cases. Mol. Genet. Genom. Med. 2019, 7, e605. [Google Scholar] [CrossRef]
- Lin, C.W.; Li, X.R.; Zhang, Y.; Hu, G.; Guo, Y.H.; Zhou, J.Y.; Du, J.; Lv, L.; Gao, K.; Deng, H. TAp63 suppress metastasis via miR-133b in colon cancer cells. Br. J. Cancer 2014, 110, 2310–2320. [Google Scholar] [CrossRef] [Green Version]
- Mo, S.; Dai, W.; Xiang, W.; Li, Y.; Feng, Y.; Zhang, L.; Li, Q.; Cai, G. Prognostic and predictive value of an autophagy-related signature for early relapse in stages I–III colon cancer. Carcinogenesis 2019, 40, 861–870. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Tauchmann, S.; Schwaller, J. NSD1: A Lysine Methyltransferase between Developmental Disorders and Cancer. Life 2021, 11, 877. [Google Scholar] [CrossRef]
- Saugier-Veber, P.; Bonnet, C.; Afenjar, A.; Drouin-Garraud, V.; Coubes, C.; Fehrenbach, S.; Holder-Espinasse, M.; Roume, J.; Malan, V.; Portnoi, M.-F.; et al. Heterogeneity ofNSD1alterations in 116 patients with Sotos syndrome. Hum. Mutat. 2007, 28, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Hanks, S.; Temple, I.K.; Davies, S.; Murray, A.; Upadhyaya, M.; Tomkins, S.; Hughes, H.E.; Cole, R.T.; Rahman, N. NSD1 Mutations Are the Major Cause of Sotos Syndrome and Occur in Some Cases of Weaver Syndrome but Are Rare in Other Overgrowth Phenotypes. Am. J. Hum. Genet. 2003, 72, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, C.; Zhang, X. Mutplot: An easy-to-use online tool for plotting complex mutation data with flexibility. PLoS ONE 2019, 14, e0215838. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Yadav, V.; Kumar, S.; Saini, N. MicroRNA-195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci. Rep. 2015, 5, 17454. [Google Scholar] [CrossRef]
- Keenan, M.M.; Liu, B.; Tang, X.; Wu, J.; Cyr, D.; Stevens, R.D.; Ilkayeva, O.; Huang, Z.; Tollini, L.A.; Murphy, S.K.; et al. ACLY and ACC1 Regulate Hypoxia-Induced Apoptosis by Modulating ETV4 via α-ketoglutarate. PLoS Genet. 2015, 11, e1005599. [Google Scholar] [CrossRef] [Green Version]
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Müller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855.e5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Freedman, J.A.; Liu, H.; Moorman, P.G.; Hyslop, T.; George, D.J.; Lee, N.H.; Patierno, S.R.; Wei, Q. Associations between RNA splicing regulatory variants of stemness-related genes and racial disparities in susceptibility to prostate cancer. Int. J. Cancer 2017, 141, 731–743. [Google Scholar] [CrossRef]
- Kiemeney, L.A.; Thorlacius, S.; Sulem, P.; Geller, F.; Aben, K.K.H.; Stacey, S.N.; Gudmundsson, J.; Jakobsdottir, M.; Bergthorsson, J.T.; Sigurdsson, A.; et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat. Genet. 2008, 40, 1307–1312. [Google Scholar] [CrossRef]
- Pineda, S.; Milne, R.L.; Calle, M.; Rothman, N.; de Maturana, E.L.; Herranz, J.; Kogevinas, M.; Chanock, S.J.; Tardon, A.; Márquez, M.; et al. Genetic Variation in the TP53 Pathway and Bladder Cancer Risk. A Comprehensive Analysis. PLoS ONE 2014, 9, e89952. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, Y.; Ma, S. A functional Variant (Rs35592567) in TP63 at 3q28 is Associated with Gastric Cancer Risk via Modifying its Regulation by MicroRNA-140. Cell. Physiol. Biochem. 2018, 47, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wu, W.; Zhu, M.; Wang, C.; Shen, W.; Cheng, Y.; Geng, L.; Liguo, G.; Zhang, J.; Dai, J.; et al. Integrating expression-related SNPs into genome-wide gene- and pathway-based analyses identified novel lung cancer susceptibility genes. Int. J. Cancer 2018, 142, 1602–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Gene | Function | Role in CRC | Previous Association with Cancer Predisposition | a Expression in Normal Colon Mucosa | Significantly Enriched Cancer Type by ALFRED | b Cancer Driver Gene (Tumor Type) | c Observed vs. Expected LoF Variants (gnomAD) | d Disruptive Variants in Cases vs. Controls. (Chubb et al.) | Syndromes Caused by Germline Mutations (Mode of Inheritance) |
|---|---|---|---|---|---|---|---|---|---|
| NSD1 | Negative regulation of RNApol II transcription. Regulation of histone H3K36 methylation. | NSD1 expression is a marker of poor prognosis [30]. | Germline NSD1 c.1135G>A (p.A379T) identified in an early-onset CRC patient without family history of cancer [31]. | Yes | Pancancer (FDR 20%), BLCA, LIHC, LUSC, OV, STAC. | Yes (HNSCC, EC, LUSC, ESC, CSC, STAC, UCS) | 5/110.6 | Cases: 0 Controls: 0 | Sotos syndrome (AD) |
| HDAC10 | Chromatin organization, transcriptional regulation, cell cycle progression and DNA repair. | Wnt pathway regulator in CRC cell lines [32]. Possible tumor suppressor function in CRC [33]. Potential involvement in DNA mismatch repair [33,34]. | No | Yes | Pancancer (FDR 20–50%) | No | 43/36.8 | Cases: 1 Controls: 0 | None |
| KRT24 | Organization of membrane proteins. Apoptotic cellular response. | None reported. | Overexpressed in normal mucosa of early-onset CRC patients [35]. | Yes | Pancancer (FDR 50–60%), OV. | No | 24/23.3 | Cases: 2 Controls: 0 | None |
| ACACA | Cell energy maintenance (fatty acid biosynthesis). Cell proliferation control. | None reported. | Germline c.6623G>A (p.R2208Q) identified in an early-onset CRC patient without family history of cancer [36]. | Yes | Pancancer (FDR 50–60%) | No | 20/134.5 | Cases: 1 Controls: 0 | Acetyl-CoA carboxylase deficiency (AR) |
| TP63 | Development, stem cell regulation, premature aging, and DNA damage response. WNT negative regulator. | TAp63 expression is downregulated in CRC [37]. Expression of TP63 is a prognostic marker [38]. | No | Yes (low) | Pancancer (FDR 50–60%) | Yes (EC, BLCA, HNSCC, NB) | 4/19.5 | Cases: 0 Controls: 0 | Various developmental syndromes with craniofacial and skeletal abnormalities (AD) [OMIM 603273] |
| Gene (Transcript) | Family ID | Variant | dbSNP | a Population MAF% | b In Silico Prediction (REVEL Score) | c Evolutionary Conservation (PhyloP/Phast-Cons Scores) |
|---|---|---|---|---|---|---|
| NSD1 (NM_022455) | F1 | c.3056G>A (p.R1019H) | rs750354456 | 0.00195 | 0.416 (D) | 3.876/1.000 |
| F2 | c.3089T>C (p.L1030S) | rs200856103 | 0.04579 | 0.365 | 2.905/1.000 | |
| F3 | c.3151G>A (p.E1051K) | rs141014337 | 0 | 0.329 | 3.287/1.000 | |
| HDAC10 (NM_001159286) | F4, F5 | c.308C>T (p.A103V) | rs143228101 | 0.03606 | 0.658 (D) | 9.957/1.000 |
| F6 | c.827G>A (p.R276G) | rs752737416 | 0.00186 | 0.6179 (D) | 1.6579/0.987 | |
| KRT24 (NM_019016) | F7 | c.130C>T (p.R44*) | rs148493418 | 0.02725 | - | - |
| F8 | c.449G>A (p.R150H) | rs146614779 | 0.00762 | 0.880 (D) | 6.124/1.000 | |
| F9, F10, F11, F12, F13 | c.1096C>T (p.R366C) | rs16966138 | 0.05585 | 0.514 (D) | 2.990/1.000 | |
| F14 | c.1143G>A (p.M381I) | rs375745897 | 0.01523 | 0.515 (D) | 7.501/1.000 | |
| TP63 (NM_003722) | F15, F16 | c.84T>G (p.H28Q) | rs370716448 | 0.00509 | 0.449 (D) | 1.792/1.000 |
| F17 | c.1127G>A (p.R376H) | rs143591434 | 0.00195 | 0.495 (D) | 7.106/1.000 | |
| F18 | c.1459C>T (p.R487C) | rs777306829 | 0.01696 | 0.636 (D) | 3.485/1.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintana, I.; Mur, P.; Terradas, M.; García-Mulero, S.; Aiza, G.; Navarro, M.; Piñol, V.; Brunet, J.; Moreno, V.; Sanz-Pamplona, R.; et al. Potential Involvement of NSD1, KRT24 and ACACA in the Genetic Predisposition to Colorectal Cancer. Cancers 2022, 14, 699. https://doi.org/10.3390/cancers14030699
Quintana I, Mur P, Terradas M, García-Mulero S, Aiza G, Navarro M, Piñol V, Brunet J, Moreno V, Sanz-Pamplona R, et al. Potential Involvement of NSD1, KRT24 and ACACA in the Genetic Predisposition to Colorectal Cancer. Cancers. 2022; 14(3):699. https://doi.org/10.3390/cancers14030699
Chicago/Turabian StyleQuintana, Isabel, Pilar Mur, Mariona Terradas, Sandra García-Mulero, Gemma Aiza, Matilde Navarro, Virginia Piñol, Joan Brunet, Victor Moreno, Rebeca Sanz-Pamplona, and et al. 2022. "Potential Involvement of NSD1, KRT24 and ACACA in the Genetic Predisposition to Colorectal Cancer" Cancers 14, no. 3: 699. https://doi.org/10.3390/cancers14030699
APA StyleQuintana, I., Mur, P., Terradas, M., García-Mulero, S., Aiza, G., Navarro, M., Piñol, V., Brunet, J., Moreno, V., Sanz-Pamplona, R., Capellá, G., & Valle, L. (2022). Potential Involvement of NSD1, KRT24 and ACACA in the Genetic Predisposition to Colorectal Cancer. Cancers, 14(3), 699. https://doi.org/10.3390/cancers14030699