
Histological and Somatic Mutational Profiles of Mismatch Repair Deficient Endometrial Tumours of Different Aetiologies

 , , ,
, , ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
- Hereditary MMRd: ECs in individuals with a proven germline pathogenic variant in MLH1, MSH2, MSH6 or PMS2;
- Sporadic MMRd: MLH1 hypermethylated ECs from the Nature 2014 TCGA cBioportal.
2.1. Tumour Selection
2.2. Pathology Review
2.3. Immunohistochemistry
2.4. DNA Extraction and Next Generation Sequencing
2.5. Data Analysis
2.6. Statistical Analysis
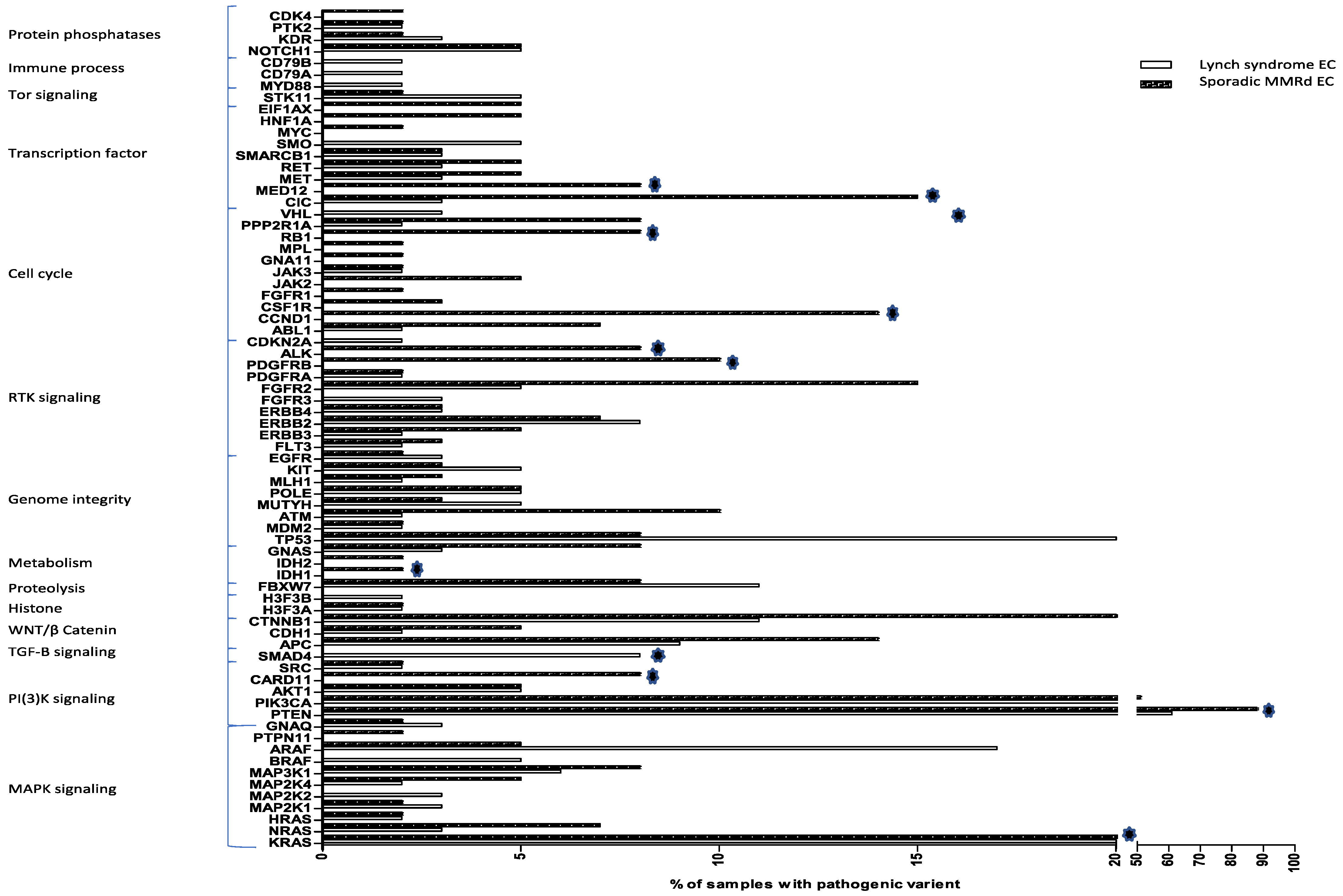
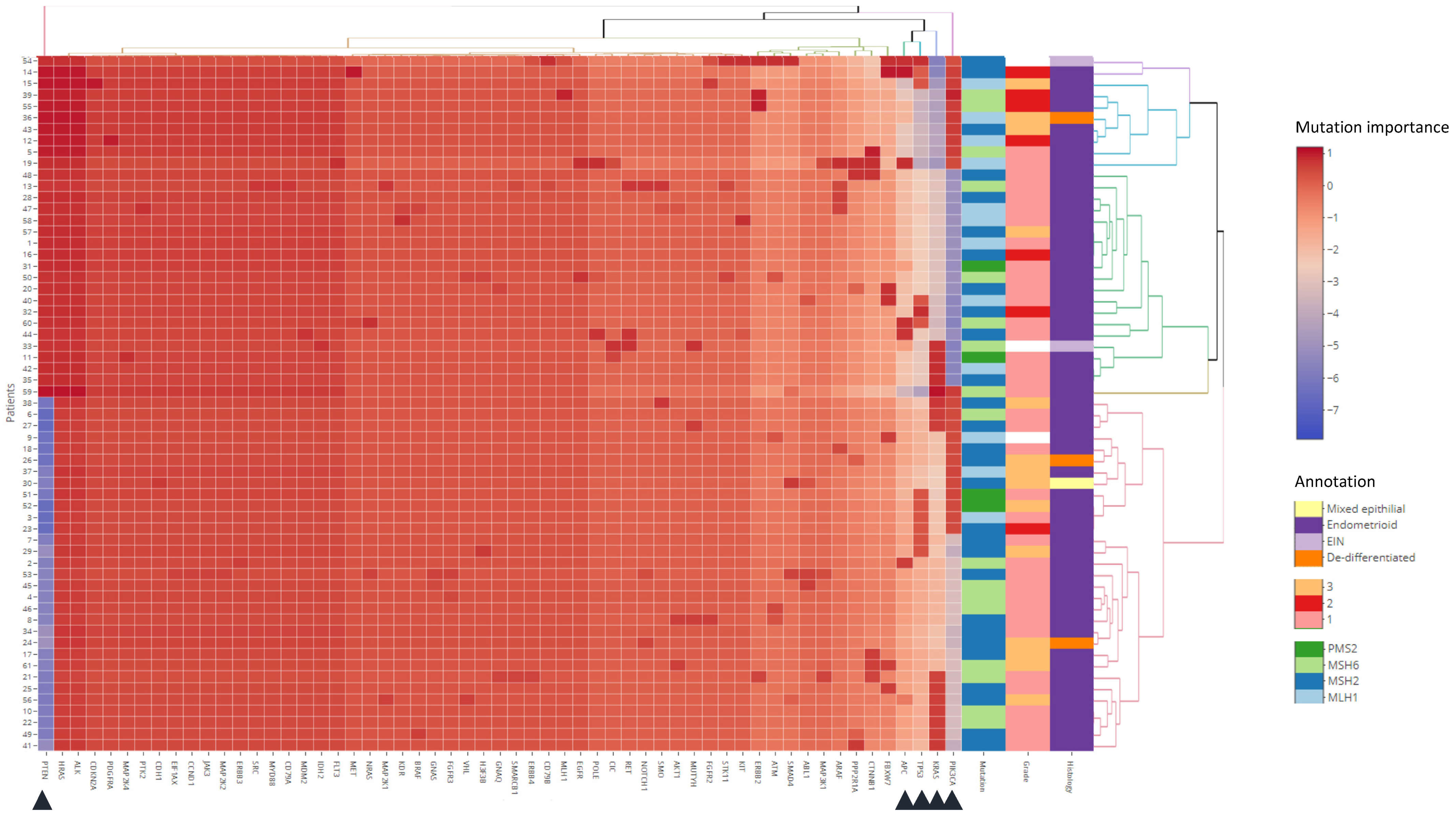
3. Results
3.1. Pathological Features
3.2. Somatic Mutations
3.3. Clustering Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levine, D.A.; Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; et al. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Ryan, N.A.J.; Glaire, M.A.; Blake, D.; Cabrera-Dandy, M.; Evans, D.G.; Crosbie, E.J. The Proportion of Endometrial Cancers Associated with Lynch Syndrome: A Systematic Review of the Literature and Meta-Analysis. Genet. Med. 2019, 21, 2167–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpkins, S.B.; Bocker, T.; Swisher, E.M.; Mutch, D.G.; Gersell, D.J.; Kovatich, A.J.; Palazzo, J.P.; Fishel, R.; Goodfellow, P.J. MLH1 Promoter Methylation and Gene Silencing Is the Primary Cause of Microsatellite Instability in Sporadic Endometrial Cancers. Hum. Mol. Genet. 1999, 8, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suter, C.M.; Martin, D.I.K.; Ward, R.L. Germline Epimutation of MLH1 in Individuals with Multiple Cancers. Nat. Genet. 2004, 36, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Piñol, V.; Xicola, R.M.; Bujanda, L.; Reñé, J.M.; et al. Mismatch Repair Status in the Prediction of Benefit from Adjuvant Fluorouracil Chemotherapy in Colorectal Cancer. Gut 2006, 55, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Sinicrope, F.A. DNA Mismatch Repair and Adjuvant Chemotherapy in Sporadic Colon Cancer. Nat. Rev. Clin. Oncol. 2010, 7, 174–177. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A.; Le, D.T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 373, 1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (N. Y.) 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F.; et al. ESMO Recommendations on Microsatellite Instability Testing for Immunotherapy in Cancer, and Its Relationship with PD-1/PD-L1 Expression and Tumour Mutational Burden: A Systematic Review-Based Approach. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Ramchander, N.C.; Ryan, N.A.J.; Walker, T.D.J.; Harries, L.; Bolton, J.; Bosse, T.; Evans, D.G.; Crosbie, E.J. Distinct Immunological Landscapes Characterize Inherited and Sporadic Mismatch Repair Deficient Endometrial Cancer. Front. Immunol. 2020, 10, 3023. [Google Scholar] [CrossRef] [Green Version]
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Ager, A.; Arends, M.J.; Frayling, I.M. Lynch Syndrome—Cancer Pathways, Heterogeneity and Immune Escape: Lynch Syndrome Cancer Pathways and Immune Escape. J. Pathol. 2018, 246, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.; Spurdle, A.; Plazzer, J.; Greenblatt, M.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; Dunnen, J.T.; et al. Application of a 5-Tiered Scheme for Standardized Classification of 2360 Unique Mismatch Repair Gene Variants in the InSiGHT Locus-Specific Database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.F. New Features in the 2014 WHO Classification of Uterine Neoplasms. Der Pathol. 2016, 37, 500–511. [Google Scholar]
- Quick, C.M.; May, T.; Horowitz, N.S.; Nucci, M.R. Low-Grade, Low-Stage Endometrioid Endometrial Adenocarcinoma: A Clinicopathologic Analysis of 324 Cases Focusing on Frequency and Pattern of Myoinvasion. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2012, 31, 337–343. [Google Scholar] [CrossRef]
- Bosse, T.; Peters, E.E.M.; Creutzberg, C.L.; Jürgenliemk-Schulz, I.M.; Jobsen, J.J.; Mens, J.W.M.; Lutgens, L.C.H.W.; van der Steen-Banasik, E.M.; Smit, V.T.H.B.M.; Nout, R.A. Substantial Lymph-Vascular Space Invasion (LVSI) Is a Significant Risk Factor for Recurrence in Endometrial Cancer—A Pooled Analysis of PORTEC 1 and 2 Trials. Eur. J. Cancer Oxf. Engl. 1990 2015, 51, 1742–1750. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; den Eynden, G.V.; Baehner, F.L.; Penault-Llorca, F.; et al. The Evaluation of Tumor-Infiltrating Lymphocytes (TILs) in Breast Cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2014, 26, 259–271. [Google Scholar] [CrossRef]
- Barrow, E.; Jagger, E.; Brierley, J.; Wallace, A.; Evans, G.; Hill, J.; McMahon, R. Semiquantitative Assessment of Immunohistochemistry for Mismatch Repair Proteins in Lynch Syndrome. Histopathology 2010, 56, 331–344. [Google Scholar] [CrossRef]
- Köbel, M.; Ronnett, B.M.; Singh, N.; Soslow, R.A.; Gilks, C.B.; McCluggage, W.G. Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int. J. Gynecol. Pathol. 2019, 38, S123–S131. [Google Scholar] [CrossRef]
- Van Eijk, R.; Stevens, L.; Morreau, H.; van Wezel, T. Assessment of a Fully Automated High-Throughput DNA Extraction Method from Formalin-Fixed, Paraffin-Embedded Tissue for KRAS, and BRAF Somatic Mutation Analysis. Exp. Mol. Pathol. 2013, 94, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Richardson, J.T.E. The Analysis of 2 × 2 Contingency Tables—Yet Again. Stat. Med. 2011, 30, 890. [Google Scholar] [CrossRef]
- Roux, B.L.; Rouanet, H. Geometric Data Analysis; Springer: Dordrecht, The Netherlands, 2005; ISBN 978-1-4020-2235-7. [Google Scholar]
- Fader, A.N.; Diaz, L.A.; Armstrong, D.K.; Tanner, E.J.; Uram, J.; Eyring, A.; Wang, H.; Fisher, G.; Greten, T.; Le, D. Preliminary Results of a Phase II Study: PD-1 Blockade in Mismatch Repair–Deficient, Recurrent or Persistent Endometrial Cancer. Gynecol. Oncol. 2016, 141, 206–207. [Google Scholar] [CrossRef]
- An, H.J.; Kim, K.I.; Kim, J.Y.; Shim, J.Y.; Kang, H.; Kim, T.H.; Kim, J.K.; Jeong, J.K.; Lee, S.Y.; Kim, S.J. Microsatellite Instability in Endometrioid Type Endometrial Adenocarcinoma Is Associated with Poor Prognostic Indicators. Am. J. Surg. Pathol. 2007, 31, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; von Knebel Doeberitz, M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, C.; Stelloo, E.; Smit, V.; Ruano, D.; Tops, C.; Vermij, L.; Rutten, T.; Jürgenliemk-Schulz, I.; Lutgens, L.; Jobsen, J.; et al. 28 Prevalence and Prognosis of Lynch Syndrome and Sporadic Mismatch Repair Deficiency in the Combined PORTEC-1,-2 and -3 Endometrial Cancer Trials. Int. J. Gynecol. Cancer 2020, 30, A20. [Google Scholar] [CrossRef]
- Libera, L.; Craparotta, I.; Sahnane, N.; Chiaravalli, A.M.; Mannarino, L.; Cerutti, R.; Riva, C.; Marchini, S.; Furlan, D. Targeted Gene Sequencing of Lynch Syndrome–Related and Sporadic Endometrial Carcinomas. Hum. Pathol. 2018, 81, 235–244. [Google Scholar] [CrossRef]
- Bussaglia, E.; Delrio, E.; Matiasguiu, X.; Prat, J. PTEN Mutations in Endometrial Carcinomas: A Molecular and Clinicopathologic Analysis of 38 Cases. Hum. Pathol. 2000, 31, 312–317. [Google Scholar] [CrossRef]
- Zöchbauer-Müller, S.; Fong, K.M.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant Promoter Methylation of Multiple Genes in Non-Small Cell Lung Cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar] [PubMed]
- Ignatov, A.; Bischoff, J.; Ignatov, T.; Schwarzenau, C.; Krebs, T.; Kuester, D.; Costa, S.D.; Roessner, A.; Semczuk, A.; Schneider-Stock, R. APC Promoter Hypermethylation Is an Early Event in Endometrial Tumorigenesis. Cancer Sci. 2010, 101, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Cohort, T.C.; Wong, S.Q.; Li, J.; Tan, A.Y.-C.; Vedururu, R.; Pang, J.-M.B.; Do, H.; Ellul, J.; Doig, K.; Bell, A.; et al. Sequence Artefacts in a Prospective Series of Formalin-Fixed Tumours Tested for Mutations in Hotspot Regions by Massively Parallel Sequencing. BMC Med. Genom. 2014, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Carrick, D.M.; Mehaffey, M.G.; Sachs, M.C.; Altekruse, S.; Camalier, C.; Chuaqui, R.; Cozen, W.; Das, B.; Hernandez, B.Y.; Lih, C.-J.; et al. Robustness of Next Generation Sequencing on Older Formalin-Fixed Paraffin-Embedded Tissue. PLoS ONE 2015, 10, e0127353. [Google Scholar] [CrossRef] [Green Version]
- Ryan, N.A.J.; McMahon, R.; Tobi, S.; Snowsill, T.; Esquibel, S.; Wallace, A.J.; Bunstone, S.; Bowers, N.; Mosneag, I.E.; Kitson, S.J.; et al. The Proportion of Endometrial Tumours Associated with Lynch Syndrome (PETALS): A Prospective Cross-Sectional Study. PLoS Med. 2020, 17, e1003263. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-Genome Sequencing and Comprehensive Molecular Profiling Identify New Driver Mutations in Gastric Cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LS-EC Total Cohort (n = 135) | LS-EC NGS Cohort (n = 64) | Sporadic MMRd EC (n = 59) | LS-EC (Total Cohort) vs. Sporadic MMRd EC p-Value | |
|---|---|---|---|---|
| Age at diagnosis in years (SEM) | 53 (3.1) | 53 (2.1) | 62 (1.2) | <0.0001 |
| Path_MMR variant | ||||
| path_MLH1 | 29 (21.8%) | 14 (21.9%) | NA | NA |
| path_MSH2 | 50 (36.1%) | 29 (45.3%) | NA | NA |
| path_MSH6 | 43 (32.3%) | 17 (26.6%) | NA | NA |
| path_PMS2 | 13 (9.8%) | 4 (6.3%) | NA | NA |
| Histology | ||||
| Endometrioid | 125 (92%) | 58 (91%) | 59 (100%) | <0.0001 |
| Clear cell | 1 (0.8%) | 0 | 0 | NA |
| Undifferentiated | 4 (3%) | 3 (4.7%) | 0 | NA |
| Mixed epithelial | 1 (0.8%) | 1 (1.5%) | 0 | NA |
| EIN | 4 (3%) | 2 (3%) | 0 | NA |
| Grade | ||||
| 1 | 87 (64.7%) | 39 (60.9%) | 17 (28.8%) | <0.0001 |
| 2 | 19 (13.5%) | 7 (10.9%) | 19 (32.2%) | 0.0025 |
| 3 | 25 (18.8%) | 16 (25%) | 23 (39%) | 0.0029 |
| EIN | 4 (3%) | 2 (3%) | 0 | NA |
| Stage | ||||
| I | 47 (34.8%) | 26 (40.6%) | 41 (69.5%) | 0.0006 |
| II | 3 (2.3%) | 1 (1.5%) | 4 (6.8%) | 0.13 |
| III | 11 (8.2%) | 7 (11%) | 11 (18.6%) | 0.19 |
| IV | 0 | 0 | 3 (5.1%) | NA |
| EIN | 4 (3%) | 2 (3%) | 0 | NA |
| Not known | 70 (51.9%) | 28 (43.7%) | 0 | NA |
| Myometrial invasion | ||||
| Broad front | 66 (48.1%) | 31 (48.4%) | 15 (25.4%) | 0.003 |
| Infiltrating gland | 19 (14.3%) | 13 (20.3%) | 12 (20.3%) | 0.3 |
| MELF | 4 (3%) | 3 (4.7%) | 0 | NA |
| Adenomyosis invasion | 3 (2.3%) | 2 (3%) | 0 | NA |
| Non-specific invasion | 16 (12%) | 4 (6%) | 0 | NA |
| No invasion/superficial | 21 (15.8) | 9 (14.1%) | 28 (47.5%) | <0.0001 |
| Not known/not applicable * | 6 (4.5%) | 2 (3%) | 4 (6.8%) | 0.51 |
| LVSI | ||||
| Present | 13 (9.8%) | 4 (6%) | 12 (20.3%) | 0.047 |
| Significant | 8 (6%) | 7 (10.9%) | 3 (5.1%) | 0.81 |
| Absent | 108 (79.7%) | 51 (79.7%) | 41 (69.5%) | 0.13 |
| Not known/not applicable * | 6 (4.5%) | 2 (3%) | 3 (5.1%) | 0.86 |
| TILs ^ | ||||
| >80% | 33 (24.8%) | 19 (29.7%) | 1 (1.7%) | <0.0001 |
| 51–80% | 50 (36.1%) | 22 (34.4%) | 13 (22%) | 0.05 |
| 11–50% | 33 (24.8%) | 18 (28.1%) | 26 (44.1%) | 0.56 |
| 0–10% | 12 (9%) | 3 (4.5%) | 18 (30.5%) | <0.0001 |
| Not known/Not applicable * | 7 (5.3%) | 2 (3%) | 1 (1.7%) | 0.13 |
| Squamous differentiation | 25 (18.8%) | 13 (20.3%) | 19 (32.2%) | 0.042 |
| Mucinous differentiation | 22 (16.5%) | 12 (18.75%) | 17 (28.8%) | 0.051 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryan, N.A.J.; Walker, T.D.J.; Bolton, J.; ter Haar, N.; Van Wezel, T.; Glaire, M.A.; Church, D.N.; Evans, D.G.; Bosse, T.; Crosbie, E.J. Histological and Somatic Mutational Profiles of Mismatch Repair Deficient Endometrial Tumours of Different Aetiologies. Cancers 2021, 13, 4538. https://doi.org/10.3390/cancers13184538
Ryan NAJ, Walker TDJ, Bolton J, ter Haar N, Van Wezel T, Glaire MA, Church DN, Evans DG, Bosse T, Crosbie EJ. Histological and Somatic Mutational Profiles of Mismatch Repair Deficient Endometrial Tumours of Different Aetiologies. Cancers. 2021; 13(18):4538. https://doi.org/10.3390/cancers13184538
Chicago/Turabian StyleRyan, Neil A. J., Thomas D. J. Walker, James Bolton, Natalja ter Haar, Tom Van Wezel, Mark A. Glaire, David N. Church, D. Gareth Evans, Tjalling Bosse, and Emma J. Crosbie. 2021. "Histological and Somatic Mutational Profiles of Mismatch Repair Deficient Endometrial Tumours of Different Aetiologies" Cancers 13, no. 18: 4538. https://doi.org/10.3390/cancers13184538
APA StyleRyan, N. A. J., Walker, T. D. J., Bolton, J., ter Haar, N., Van Wezel, T., Glaire, M. A., Church, D. N., Evans, D. G., Bosse, T., & Crosbie, E. J. (2021). Histological and Somatic Mutational Profiles of Mismatch Repair Deficient Endometrial Tumours of Different Aetiologies. Cancers, 13(18), 4538. https://doi.org/10.3390/cancers13184538