
Overview of Immune Checkpoint Inhibitors in Gynecological Cancer Treatment



Abstract
:Simple Summary
Abstract
1. Introduction
2. Methodology
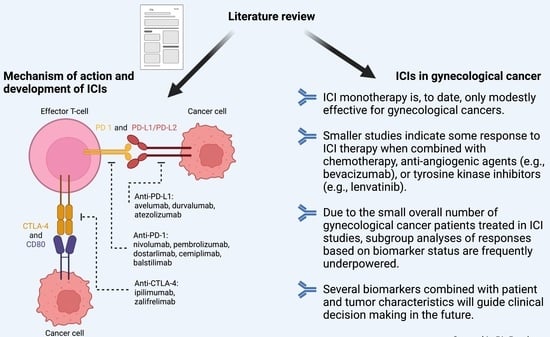
3. Mechanism of Action and Development of ICIs
3.1. Overview of Tumor Immunobiology
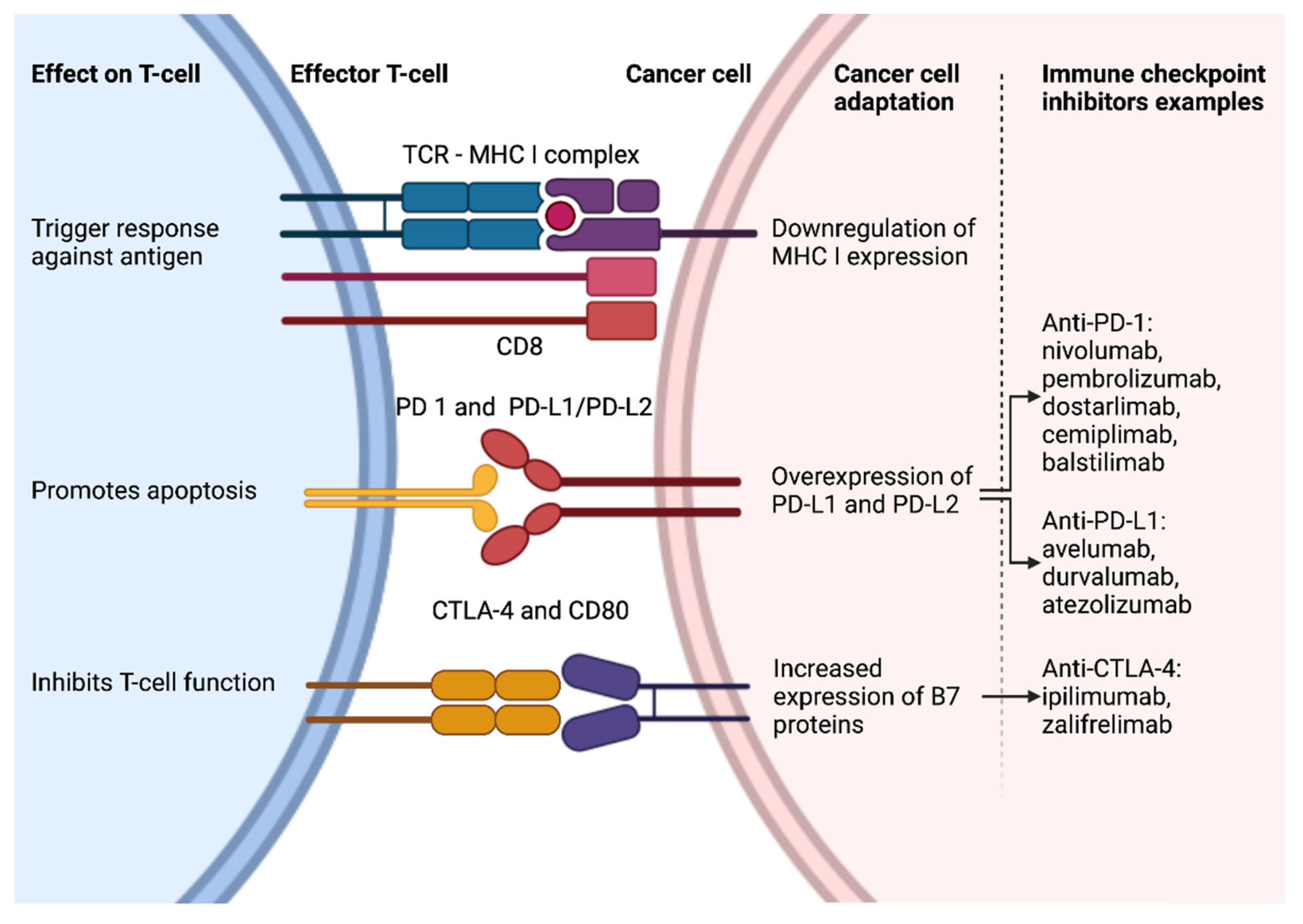
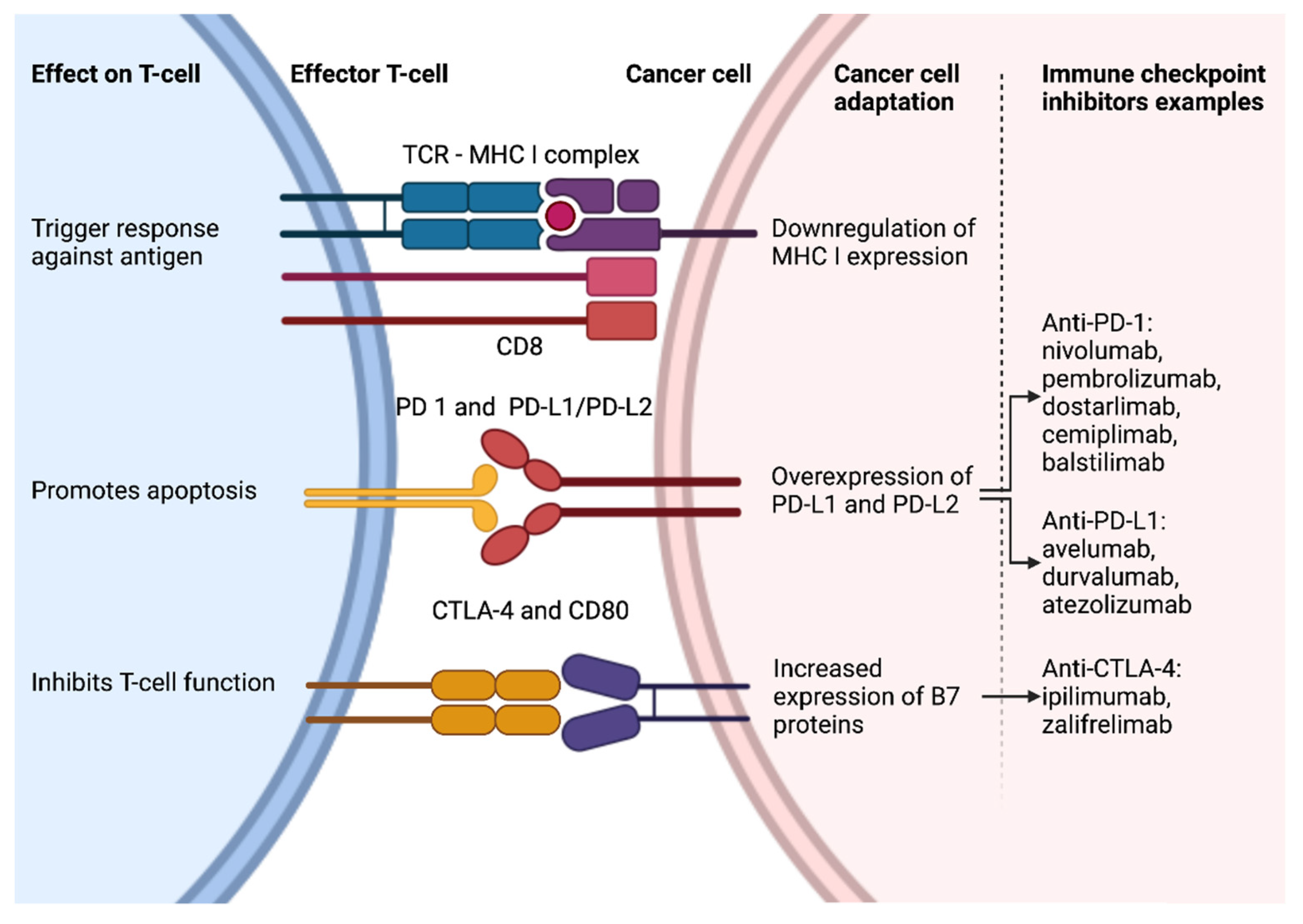
3.2. Overview of Immune Checkpoint Inhibitors
3.3. Immune Checkpoint Inhibitors—Related Adverse Events
4. Biomarkers That Predict the Response to ICIs
4.1. TILs
4.2. PD-L1 Expression
4.3. TME Gene Expression Profiles
4.4. Tumor Neoantigen Load and Mutational Burden
4.5. Microsatellite Instability (MSI) and Mismatch Repair (MMR) Deficiency
4.6. Microbiome
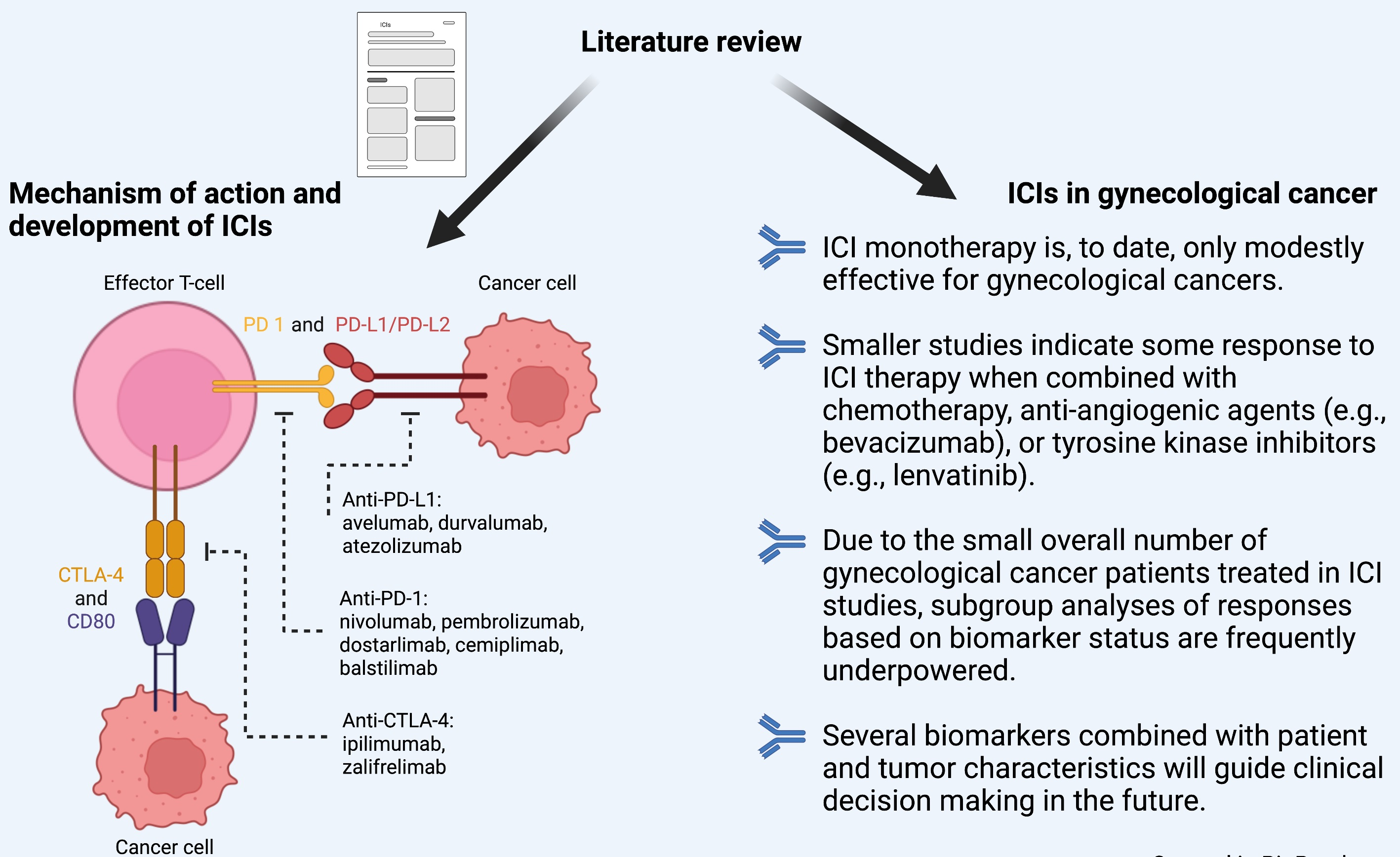
5. ICIs in Gynecological Cancer
5.1. Endometrial Cancer
5.2. Uterine Cervical Cancer
5.3. Ovarian Cancer
5.4. Vulvar and Vaginal Cancer
6. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer Immunotherapy Comes of Age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune Checkpoint Blockade Therapy for Cancer: An Overview of FDA-Approved Immune Checkpoint Inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of Action and Rationale for the Use of Checkpoint Inhibitors in Cancer. ESMO Open 2017, 2, e000213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on Tumor Cells in the Escape from Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. “Final Common Pathway” of Human Cancer Immunotherapy: Targeting Random Somatic Mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next Generation of Immune Checkpoint Inhibitors and Beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaniz, E.; Genestie, C.; Klein, C.; Salviat, F.; Ray-Coquard, I.L.; Joly, F.; Ferron, G.; Pujade-Lauraine, E.; Pautier, P.; Leary, A. Impact of Chemotherapy Alone or in Combination with an Anti-Angiogenic on the Immune Tumor Microenvironment (TME) of Ovarian Cancer: Data from the Randomized CHIVA Trial (a GINECO –GINEGEPS Study). J. Clin. Oncol. 2020, 38, 6011. [Google Scholar] [CrossRef]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of Anti-Angiogenic Therapy and Immune Checkpoint Blockade Normalizes Vascular-Immune Crosstalk to Potentiate Cancer Immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Ochoa de Olza, M.; Navarro Rodrigo, B.; Zimmermann, S.; Coukos, G. Turning up the Heat on Non-Immunoreactive Tumours: Opportunities for Clinical Development. Lancet Oncol. 2020, 21, e419–e430. [Google Scholar] [CrossRef]
- Lee, E.K.; Konstantinopoulos, P.A. Combined PARP and Immune Checkpoint Inhibition in Ovarian Cancer. Trends Cancer 2019, 5, 524–528. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the Mechanisms of Immune Checkpoint Therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-Related Adverse Events and Anti-Tumor Efficacy of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Weber, J.S. Immune-Related Toxicities of Checkpoint Inhibitors: Mechanisms and Mitigation Strategies. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse Effects of Immune-Checkpoint Inhibitors: Epidemiology, Management and Surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of Cancer Immunity and the Cancer–Immune Set Point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; van de Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv. Anat. Pathol. 2017, 24, 311–335. [Google Scholar] [PubMed]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive Biomarkers for Cancer Immunotherapy with Immune Checkpoint Inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.A.; Socinski, M.A.; Villaruz, L.C. PD-L1 Testing in Guiding Patient Selection for PD-1/PD-L1 Inhibitor Therapy in Lung Cancer. Mol. Diagn. Ther. 2018, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-Related MRNA Profile Predicts Clinical Response to PD-1 Blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.-J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.-C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell–Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated with Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non–Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Simoni, Y.; Zhuang, S.; Gabel, A.; Ma, S.; Chee, J.; Islas, L.; Cessna, A.; Creaney, J.; Bradley, R.K.; et al. Characterization of Neoantigen-Specific T Cells in Cancer Resistant to Immune Checkpoint Therapies. Proc. Natl. Acad. Sci. USA 2021, 118, e2025570118. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Y.; Wang, C. Beyond Tumor Mutation Burden: Tumor Neoantigen Burden as a Biomarker for Immunotherapy and Other Types of Therapy. Front. Oncol. 2021, 11, 672677. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of Tumor Mutation Burden as an Immunotherapy Biomarker: Utility for the Oncology Clinic. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB Measurement in Clinical Practice: Considerations on Assay Requirements. ESMO Open 2019, 4, e000442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F.; et al. ESMO Recommendations on Microsatellite Instability Testing for Immunotherapy in Cancer, and Its Relationship with PD-1/PD-L1 Expression and Tumour Mutational Burden: A Systematic Review-Based Approach. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration Center for Drug Evaluation and Research. FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication (accessed on 1 November 2021).
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High Tumor Mutation Burden Fails to Predict Immune Checkpoint Blockade Response across All Cancer Types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e–Mutated and Microsatellite-Instable Endometrial Cancers with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319. [Google Scholar] [CrossRef]
- Yamamoto, H.; Imai, K. An Updated Review of Microsatellite Instability in the Era of Next-Generation Sequencing and Precision Medicine. Semin. Oncol. 2019, 46, 261–270. [Google Scholar] [CrossRef]
- Bhangoo, M.S.; Boasberg, P.; Mehta, P.; Elvin, J.A.; Ali, S.M.; Wu, W.; Klempner, S.J. Tumor Mutational Burden Guides Therapy in a Treatment Refractory POLE-Mutant Uterine Carcinosarcoma. Oncologist 2018, 23, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Pierrard, J.; Seront, E. Impact of the Gut Microbiome on Immune Checkpoint Inhibitor Efficacy—A Systematic Review. Curr. Oncol. 2019, 26, 395–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Ahn, H.; Park, H. A Review on the Role of Gut Microbiota in Immune Checkpoint Blockade Therapy for Cancer. Mamm. Genome 2021, 32, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal Microbiota Transplant Promotes Response in Immunotherapy-Refractory Melanoma Patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal Microbiota Transplant Overcomes Resistance to Anti–PD-1 Therapy in Melanoma Patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Concin, N.; Matias-Guiu, X.; Vergote, I.; Cibula, D.; Mirza, M.R.; Marnitz, S.; Ledermann, J.; Bosse, T.; Chargari, C.; Fagotti, A.; et al. ESGO/ESTRO/ESP Guidelines for the Management of Patients with Endometrial Carcinoma. Int. J. Gynecol. Cancer 2021, 31, 12–39. [Google Scholar] [CrossRef]
- Miller, D.S.; Filiaci, V.L.; Mannel, R.S.; Cohn, D.E.; Matsumoto, T.; Tewari, K.S.; DiSilvestro, P.; Pearl, M.L.; Argenta, P.A.; Powell, M.A.; et al. Carboplatin and Paclitaxel for Advanced Endometrial Cancer: Final Overall Survival and Adverse Event Analysis of a Phase III Trial (NRG Oncology/GOG0209). J. Clin. Oncol. 2020, 38, 3841–3850. [Google Scholar] [CrossRef]
- Decruze, S.B.; Green, J.A. Hormone Therapy in Advanced and Recurrent Endometrial Cancer: A Systematic Review. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2007, 17, 964–978. [Google Scholar] [CrossRef]
- Ueda, Y.; Miyake, T.; Egawa-Takata, T.; Miyatake, T.; Matsuzaki, S.; Yokoyama, T.; Yoshino, K.; Fujita, M.; Enomoto, T.; Kimura, T. Second-Line Chemotherapy for Advanced or Recurrent Endometrial Carcinoma Previously Treated with Paclitaxel and Carboplatin, with or without Epirubicin. Cancer Chemother. Pharmacol. 2011, 67, 829–835. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network; Levine, D.A. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.S.; et al. Confirmation of ProMisE: A Simple, Genomics-Based Clinical Classifier for Endometrial Cancer. Cancer 2017, 123, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; González-Martín, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.M.; Mutch, D.G. Endometrial Tumor Immune Response: Predictive Biomarker of Response to Immunotherapy. Clin. Cancer Res. 2019, 25, 2366–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oaknin, A.; Gilbert, L.; Tinker, A.V.; Sabatier, R.; Boni, V.; O’Malley, D.M.; Ghamande, S.; Duska, L.; Ghatage, P.; Guo, W.; et al. LBA36 Safety and Antitumor Activity of Dostarlimab in Patients (Pts) with Advanced or Recurrent DNA Mismatch Repair Deficient (DMMR) or Proficient (MMRp) Endometrial Cancer (EC): Results from GARNET. Ann. Oncol. 2020, 31, S1166. [Google Scholar] [CrossRef]
- Antill, Y.C.; Kok, P.S.; Robledo, K.; Barnes, E.; Friedlander, M.; Baron-Hay, S.E.; Shannon, C.M.; Coward, J.; Beale, P.J.; Goss, G.; et al. Activity of Durvalumab in Advanced Endometrial Cancer (AEC) According to Mismatch Repair (MMR) Status: The Phase II PHAEDRA Trial (ANZGOG1601). J. Clin. Oncol. 2019, 37, 5501. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Luo, W.; Liu, J.F.; Gulhan, D.C.; Krasner, C.; Ishizuka, J.J.; Gockley, A.A.; Buss, M.; Growdon, W.B.; Crowe, H.; et al. Phase II Study of Avelumab in Patients with Mismatch Repair Deficient and Mismatch Repair Proficient Recurrent/Persistent Endometrial Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 2786–2794. [Google Scholar] [CrossRef]
- Oaknin, A.; Gilbert, L.; Tinker, A.; Brown, J.; Mathews, C.; Press, J.Z.; Sabatier, R.; O’Malley, D.; Samouelian, V.; Boni, V.; et al. 76P Analysis of Antitumor Activity of Dostarlimab by Tumor Mutational Burden (TMB) in Patients (Pts) with Endometrial Cancer (EC) in the GARNET Trial. Ann. Oncol. 2021, 32, S388–S389. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Korkaya, H.; Orsulic, S. Editorial: The Tumor Microenvironment: Recent Advances and Novel Therapeutic Approaches. Front. Cell Dev. Biol. 2020, 8, 586176. [Google Scholar] [CrossRef]
- Colombo, N.; Lorusso, D.; Herráez, A.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Floquet, A.; Monk, B.J.; et al. 726MO Outcomes by Histology and Prior Therapy with Lenvatinib plus Pembrolizumab vs. Treatment of Physician’s Choice in Patients with Advanced Endometrial Cancer (Study 309/KEYNOTE-775). Ann. Oncol. 2021, 32, S729–S730. [Google Scholar] [CrossRef]
- Lheureux, S.; Matei, D.; Konstantinopoulos, P.A.; Block, M.S.; Jewell, A.; Gaillard, S.; McHale, M.S.; McCourt, C.K.; Temkin, S.; Girda, E.; et al. A Randomized Phase II Study of Cabozantinib and Nivolumab versus Nivolumab in Recurrent Endometrial Cancer. J. Clin. Oncol. 2020, 38, 6010. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Center for Drug Evaluation and Research. FDA Grants Accelerated Approval to Dostarlimab-Gxly for DMMR Endometrial Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-endometrial-cancer (accessed on 1 November 2021).
- European Medicines Agency. Jemperli. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/jemperli (accessed on 1 November 2021).
- U.S. Food and Drug Administration Center for Drug Evaluation and Research. FDA Grants Regular Approval to Pembrolizumab and Lenvatinib for Advanced Endometrial Carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-pembrolizumab-and-lenvatinib-advanced-endometrial-carcinoma (accessed on 1 November 2021).
- European Commission Approves KEYTRUDA® (Pembrolizumab) Plus LENVIMA® (Lenvatinib) as First-Line Treatment for Adult Patients with Advanced Renal Cell Carcinoma. Available online: https://www.merck.com/news/european-commission-approves-keytruda-pembrolizumab-plus-lenvima-lenvatinib-as-first-line-treatment-for-adult-patients-with-advanced-renal-cell-carcinoma/ (accessed on 5 January 2022).
- ESMO. ESMO Clinical Practice Guidelines: Gynaecological Cancers. Available online: https://www.esmo.org/guidelines/gynaecological-cancers (accessed on 7 November 2021).
- National Comprehensive Cancer Network. Uterine Neoplasms (Version 1.2022). Available online: https://www.nccn.org/professionals/physician_gls/pdf/uterine.pdf (accessed on 5 November 2021).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.; Babb, P.; Jones, J.; Allen, E. Effect of Screening on Incidence of and Mortality from Cancer of Cervix in England: Evaluation Based on Routinely Collected Statistics. BMJ 1999, 318, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.D.; Matsuo, K.; Huang, Y.; Tergas, A.I.; Hou, J.Y.; Khoury-Collado, F.; St. Clair, C.M.; Ananth, C.V.; Neugut, A.I.; Hershman, D.L. Prognostic Performance of the 2018 International Federation of Gynecology and Obstetrics Cervical Cancer Staging Guidelines. Obstet. Gynecol. 2019, 134, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Cibula, D.; Pötter, R.; Planchamp, F.; Avall-Lundqvist, E.; Fischerova, D.; Haie Meder, C.; Köhler, C.; Landoni, F.; Lax, S.; Lindegaard, J.C.; et al. The European Society of Gynaecological Oncology/European Society for Radiotherapy and Oncology/European Society of Pathology Guidelines for the Management of Patients with Cervical Cancer. Int. J. Gynecol. Cancer 2018, 28, 641–655. [Google Scholar] [CrossRef]
- Moore, D.H.; Tian, C.; Monk, B.J.; Long, H.J.; Omura, G.A.; Bloss, J.D. Prognostic Factors for Response to Cisplatin-Based Chemotherapy in Advanced Cervical Carcinoma: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2010, 116, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Tewari, K.S.; Sill, M.W.; Long, H.J.; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; et al. Improved Survival with Bevacizumab in Advanced Cervical Cancer. N. Engl. J. Med. 2014, 370, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Heeren, A.M.; Punt, S.; Bleeker, M.C.; Gaarenstroom, K.N.; van der Velden, J.; Kenter, G.G.; de Gruijl, T.D.; Jordanova, E.S. Prognostic Effect of Different PD-L1 Expression Patterns in Squamous Cell Carcinoma and Adenocarcinoma of the Cervix. Mod. Pathol. 2016, 29, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.C.; Ros, W.; Delord, J.-P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Hasegawa, K.; Tamura, K.; Katsumata, N.; Matsumoto, K.; Takahashi, S.; Mukai, H.; Nomura, H.; Minami, H. Efficacy and Safety of Nivolumab (Nivo) in Patients (Pts) with Advanced or Recurrent Uterine Cervical or Corpus Cancers. J. Clin. Oncol. 2018, 36, 5594. [Google Scholar] [CrossRef]
- Naumann, R.W.; Hollebecque, A.; Meyer, T.; Devlin, M.-J.; Oaknin, A.; Kerger, J.; López-Picazo, J.M.; Machiels, J.-P.; Delord, J.-P.; Evans, T.R.J.; et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results From the Phase I/II CheckMate 358 Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Monk, B.J.; Vergote, I.; Miller, A.; de Melo, A.C.; Kim, H.S.; Kim, Y.M.; Lisyanskaya, A.; Samouëlian, V.; Lorusso, D.; et al. VP4-2021: EMPOWER-Cervical 1/GOG-3016/ENGOT-Cx9: Interim Analysis of Phase III Trial of Cemiplimab vs. Investigator’s Choice (IC) Chemotherapy (Chemo) in Recurrent/Metastatic (R/M) Cervical Carcinoma. Ann. Oncol. 2021, 32, 940–941. [Google Scholar] [CrossRef]
- VP4_2021—EMPOWER-Cervical 1/GOG-3016/ENGOT-Cx9: Interim Analysis of Phase III Trial of Cemiplimab vs. Investigator’s Choice (IC) Chemotherapy (Che... | OncologyPRO. Available online: https://oncologypro.esmo.org/meeting-resources/esmo-congress-2021/vp4_2021-empower-cervical-1-gog-3016-engot-cx9-interim-analysis-of-phase-iii-trial-of-cemiplimab-vs-investigator-s-choice-ic-chemotherapy-che (accessed on 3 November 2021).
- Lorusso, D. Role of Immunotherapy in Advanced or Recurrent Cervical Cancer. In Proceedings of the 22nd European Congress on Gynaecological Oncology, Prague, Czech Republic, 23–25 October 2021. [Google Scholar]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos Usta, E.; Yañez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Oaknin, A.; Gladieff, L.; Colombo, N.; Villacampa, G.; Mirza, M.R.; De Giorgi, U.; Randall, L.M.; Takekuma, M.; González-Martín, A. BEATcc (ENGOT-Cx10/GEICO 68-C/GOG3030/JGOG1084): A Randomized, Open Label, Phase III Study of Cisplatin and Paclitaxel Chemotherapy with Bevacizumab (CTx plus B) with or without Atezolizumab (Atz) as First-Line Treatment for Metastatic, Persistent, or Recurrent (m/r) Carcinoma of the Cervix (CCx). J. Clin. Oncol. 2019, 37, TPS5594. [Google Scholar] [CrossRef]
- Monk, B.J.; Mayadev, J.; Nunes, A.T.; Dabrowska Brown, A.; Marcovitz, M.; Lanasa, M.C. CALLA: Efficacy and Safety of Durvalumab with and Following Concurrent Chemoradiotherapy (CCRT) versus CCRT Alone in Women with Locally Advanced Cervical Cancer: A Phase III, Randomized, Double-Blind, Multicenter Study. J. Clin. Oncol. 2019, 37, TPS5597. [Google Scholar] [CrossRef]
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 Co-Inhibitory Molecules Collaborate to Limit CD8 + T Cell Signaling and Dampen Antitumor Immunity in a Murine Ovarian Cancer Model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef] [PubMed]
- Naumann, R.W.; Oaknin, A.; Meyer, T.; Lopez-Picazo, J.M.; Lao, C.; Bang, Y.-J.; Boni, V.; Sharfman, W.H.; Park, J.C.; Devriese, L.A.; et al. LBA62—Efficacy and Safety of Nivolumab (Nivo) + Ipilimumab (Ipi) in Patients (Pts) with Recurrent/Metastatic (R/M) Cervical Cancer: Results from CheckMate 358. Ann. Oncol. 2019, 30, v898–v899. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Randall, L.M.; Jackson, C.G.; Coleman, R.L.; Hays, J.L.; Moore, K.N.; Naumann, R.W.; Rocconi, R.P.; Slomovitz, B.M.; Tewari, K.S.; et al. RaPiDS (GOG-3028): Randomized Phase II Study of Balstilimab Alone or in Combination with Zalifrelimab in Cervical Cancer. Future Oncol. 2021, 17, 3433–3443. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Oaknin, A.; Monk, B.J.; Leary, A.; Selle, F.; Alexandre, J.; Randall, L.M.; Rojas, C.; Neffa, M.; Kryzhanivska, A.; et al. LBA34 Single-Agent Anti-PD-1 Balstilimab or in Combination with Anti-CTLA-4 Zalifrelimab for Recurrent/Metastatic (R/M) Cervical Cancer (CC): Preliminary Results of Two Independent Phase II Trials. Ann. Oncol. 2020, 31, S1164–S1165. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Center for Drug Evaluation and Research. FDA Approves Pembrolizumab for Advanced Cervical Cancer with Disease Progression during or after Chemotherapy. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-advanced-cervical-cancer-disease-progression-during-or-after-chemotherapy (accessed on 3 November 2021).
- U.S. Food and Drug Administration Center for Drug Evaluation and Research. FDA Approves Pembrolizumab Combination for the First-Line Treatment of Cervical Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-combination-first-line-treatment-cervical-cancer (accessed on 7 November 2021).
- National Comprehensive Cancer Network. Cervical Cancer (Version 1.2022). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cervical.pdf (accessed on 5 November 2021).
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO Consensus Conference Recommendations on Ovarian Cancer: Pathology and Molecular Biology, Early and Advanced Stages, Borderline Tumours and Recurrent Disease. Int. J. Gynecol. Cancer 2019, 29, 728–760. [Google Scholar] [CrossRef] [Green Version]
- Hanby, A.M.; Walker, C.; Tavassoli, F.A.; Devilee, P. Pathology and Genetics: Tumours of the Breast and Female Genital Organs. WHO Classification of Tumours Series—Volume IV. Lyon, France: IARC Press. Breast Cancer Res. 2004, 6, 133. [Google Scholar] [CrossRef] [Green Version]
- Pignata, S.; Cecere, S.C.; Bois, A.D.; Harter, P.; Heitz, F. Treatment of Recurrent Ovarian Cancer. Ann. Oncol. 2017, 28, viii51–viii56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.D.; Fares, C.M.; Campos, M.; Chen, H.-W.; Shintaku, I.P.; Konecny, G.E.; Rao, J. Association of PD-L1 Expression by Immunohistochemistry and Gene Microarray with Molecular Subtypes of Ovarian Tumors. Mod. Pathol. 2020, 33, 2001–2010. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor Activity and Safety of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer: Results from the Phase II KEYNOTE-100 Study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Ryan, N.A.J.; McVey, R.J.; Lalloo, F.; Bowers, N.; Green, K.; Woodward, E.R.; Clancy, T.; Bolton, J.; Wallace, A.J.; et al. Assessment of Mismatch Repair Deficiency in Ovarian Cancer. J. Med. Genet. 2021, 58, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.A.; Wentzensen, N. Frequency of Mismatch Repair Deficiency in Ovarian Cancer: A Systematic Review This Article Is a US Government Work and, as Such, Is in the Public Domain of the United States of America. Int. J. Cancer 2011, 129, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Fan, W.; Ye, M.; Tian, C.; Zhao, L.; Wang, J.; Han, W.; Yang, W.; Gu, C.; Li, M.; et al. Molecular Profiles and Tumor Mutational Burden Analysis in Chinese Patients with Gynecologic Cancers. Sci. Rep. 2018, 8, 8990. [Google Scholar] [CrossRef]
- Fan, S.; Gao, X.; Qin, Q.; Li, H.; Yuan, Z.; Zhao, S. Association between Tumor Mutation Burden and Immune Infiltration in Ovarian Cancer. Int. Immunopharmacol. 2020, 89, 107126. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Van Wilpe, S.; Tolmeijer, S.H.; Koornstra, R.H.T.; de Vries, I.J.M.; Gerritsen, W.R.; Ligtenberg, M.; Mehra, N. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers 2021, 13, 2249. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Sun, C.; Feng, Y.; Jia, Q.; Zhu, B. Potent Immunogenicity in BRCA1-mutated Patients with High-grade Serous Ovarian Carcinoma. J. Cell. Mol. Med. 2018, 22, 3979–3986. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, M. Evaluation of BRCA1 and BRCA2 as Indicators of Response to Immune Checkpoint Inhibitors. JAMA Netw. Open 2021, 4, e217728. [Google Scholar] [CrossRef]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA Somatic Mutations and Epigenetic BRCA Modifications in Serous Ovarian Cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO Recommendations on Predictive Biomarker Testing for Homologous Recombination Deficiency and PARP Inhibitor Benefit in Ovarian Cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Disis, M.L.; Patel, M.R.; Pant, S.; Hamilton, E.P.; Lockhart, A.C.; Kelly, K.; Beck, J.T.; Gordon, M.S.; Weiss, G.J.; Taylor, M.H.; et al. Avelumab (MSB0010718C; Anti-PD-L1) in Patients with Recurrent/Refractory Ovarian Cancer from the JAVELIN Solid Tumor Phase Ib Trial: Safety and Clinical Activity. J. Clin. Oncol. 2016, 34, 5533. [Google Scholar] [CrossRef]
- Liu, J.F.; Gordon, M.; Veneris, J.; Braiteh, F.; Balmanoukian, A.; Eder, J.P.; Oaknin, A.; Hamilton, E.; Wang, Y.; Sarkar, I.; et al. Safety, Clinical Activity and Biomarker Assessments of Atezolizumab from a Phase I Study in Advanced/Recurrent Ovarian and Uterine Cancers. Gynecol. Oncol. 2019, 154, 314–322. [Google Scholar] [CrossRef]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in Patients with Programmed Death Ligand 1-Positive Advanced Ovarian Cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef]
- Hamanishi, J.; Takeshima, N.; Katsumata, N.; Ushijima, K.; Kimura, T.; Takeuchi, S.; Matsumoto, K.; Ito, K.; Mandai, M.; Nakai, H.; et al. Nivolumab Versus Gemcitabine or Pegylated Liposomal Doxorubicin for Patients with Platinum-Resistant Ovarian Cancer: Open-Label, Randomized Trial in Japan (NINJA). J. Clin. Oncol. 2021, 39, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.; Ray-Coquard, I.-L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab Alone or in Combination with Chemotherapy versus Chemotherapy Alone in Platinum-Resistant or Platinum-Refractory Ovarian Cancer (JAVELIN Ovarian 200): An Open-Label, Three-Arm, Randomised, Phase 3 Study. Lancet Oncol. 2021, 22, 1034–1046. [Google Scholar] [CrossRef]
- Monk, B.J.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. Chemotherapy with or without Avelumab Followed by Avelumab Maintenance versus Chemotherapy Alone in Patients with Previously Untreated Epithelial Ovarian Cancer (JAVELIN Ovarian 100): An Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2021, 22, 1275–1289. [Google Scholar] [CrossRef]
- Ledermann, J.; Colombo, N.; Oza, A.; Fujiwara, K.; Birrer, M.J.; Randall, L.; Poddubskaya, E.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. 1 Avelumab in Combination with and/or Following Chemotherapy vs. Chemotherapy in Treatment-Naive Patients with Ovarian Cancer: Biomarker Analyses from the Phase 3 JAVELIN Ovarian 100 Trial. Int. J. Gynecol. Cancer 2020, 30. [Google Scholar] [CrossRef]
- Zsiros, E.; Lynam, S.; Attwood, K.M.; Wang, C.; Chilakapati, S.; Gomez, E.C.; Liu, S.; Akers, S.; Lele, S.; Frederick, P.J.; et al. Efficacy and Safety of Pembrolizumab in Combination with Bevacizumab and Oral Metronomic Cyclophosphamide in the Treatment of Recurrent Ovarian Cancer: A Phase 2 Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, 78–85. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Mäenpää, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1842–1855. [Google Scholar] [CrossRef]
- González-Martín, A.; Chung, H.; Saada-Bouzid, E.; Yanez, E.; Senellart, H.; Cassier, P.A.; Basu, B.; Ghori, R.; Kubiak, P.; Smith, A.; et al. 2 Efficacy and Safety of Lenvatinib plus Pembrolizumab in Patients with Previously Treated Ovarian Cancer in the Multicohort Phase 2 LEAP-005 Study. Int. J. Gynecol. Cancer 2020, 30. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.-W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II Study of Olaparib (O) plus Durvalumab (D) and Bevacizumab (B) (MEDIOLA): Initial Results in Patients (Pts) with Non-Germline BRCA-Mutated (Non-GBRCAm) Platinum Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2020, 31, S615–S616. [Google Scholar] [CrossRef]
- Drew, Y.; de Jonge, M.; Hong, S.H.; Park, Y.H.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.E.; Alvarez, R.H.; Gresty, C.; et al. An Open-Label, Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Germline BRCA -Mutated ( GBRCA m) Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J., Jr.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized Phase II Trial of Nivolumab Versus Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- ARCAGY/ GINECO GROUP A Multicentre Feasibility Randomized Study of Anti-PD-L1 Durvalumab (MEDI4736) With or Without Anti-CTLA-4 Tremelimumab in Patients With Ovarian, Fallopian Tube or Primary Peritoneal Adenocarcinoma, Treated With a First-Line Neo-Adjuvant Strategy. Available online: https://clinicaltrials.gov/ct2/show/NCT03249142 (accessed on 10 November 2021).
- Randall, L.M.; O’Malley, D.M.; Monk, B.J.; Coleman, R.L.; O’Cearbhaill, R.E.; Gaillard, S.; Adams, S.; Cappuccini, F.; Huang, M.; Chon, H.S.; et al. 883TiP MOONSTONE/GOG-3032: A Phase II, Open-Label, Single-Arm Study to Evaluate the Efficacy and Safety of Niraparib + Dostarlimab in Patients with Platinum-Resistant Ovarian Cancer. Ann. Oncol. 2020, 31, S646–S647. [Google Scholar] [CrossRef]
- González-Martín, A.; Colombo, N.; Heitz, F.; dePont Christensen, R.; Selle, F.; Vergote, I.; Oaknin, A. ENGOT-Ov41/GEICO-69-O/ANITA Trial: A Phase III Randomized, Double-Blinded Trial of Platinum-Based Chemotherapy (CT) with or without Atezolizumab (ATZ) Followed by Niraparib Maintenance with or without ATZ in Patients with Recurrent Ovarian, Tubal or Peritoneal Cancer (OC) and Platinum Treatment-Free Interval (TFIp) >6 Months. J. Clin. Oncol. 2019, 37, TPS5599. [Google Scholar] [CrossRef]
- Nordic Society of Gynaecological Oncology - Clinical Trials Unit ENGOT-OV42 / NSGO-AVATAR: A Three-Arm Randomized Study to Evaluate the Efficacy of Niraparib-Bevacizumab-Dostarlimab Triplet Combination Against Niraparib-Bevacizumab Doublet Combination and Against Standard of Care Therapy in Women With Relapsed Ovarian Cancer Where Platinum Combination Therapy Is an Option. Available online: https://clinicaltrials.gov/ct2/show/NCT03806049 (accessed on 10 November 2021).
- Barber, E.; Matei, D. Immunotherapy in Ovarian Cancer: We Are Not There Yet. Lancet Oncol. 2021, 22, 903–905. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Ovarian Cancer/Fallopian Tube Cancer/Primary Peritoneal Cancer (Version 3.2021). Available online: https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf (accessed on 5 November 2021).
- Rogers, L.J.; Cuello, M.A. Cancer of the Vulva. Int. J. Gynecol. Obstet. 2018, 143, 4–13. [Google Scholar] [CrossRef]
- Oonk, M.H.M.; Planchamp, F.; Baldwin, P.; Bidzinski, M.; Brännström, M.; Landoni, F.; Mahner, S.; Mahantshetty, U.; Mirza, M.; Petersen, C.; et al. European Society of Gynaecological Oncology Guidelines for the Management of Patients with Vulvar Cancer. Int. J. Gynecol. Cancer 2017, 27, 832–837. [Google Scholar] [CrossRef]
- Dolled-Filhart, M.; Locke, D.; Murphy, T.; Lynch, F.; Yearley, J.H.; Frisman, D.; Pierce, R.; Weiner, R.; Wu, D.; Emancipator, K. Development of a Prototype Immunohistochemistry Assay to Measure Programmed Death Ligand-1 Expression in Tumor Tissue. Arch. Pathol. Lab. Med. 2016, 140, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.; Russo, A.L.; Lee, H.; Spriggs, D. A Phase 2 Study of Combined Chemo-Immunotherapy with Cisplatin-Pembrolizumab and Radiation for Unresectable Vulvar Squamous Cell Carcinoma. J. Transl. Med. 2020, 18, 350. [Google Scholar] [CrossRef]
- Shields, L.B.E.; Gordinier, M.E. Pembrolizumab in Recurrent Squamous Cell Carcinoma of the Vulva: Case Report and Review of the Literature. Gynecol. Obstet. Investig. 2019, 84, 94–98. [Google Scholar] [CrossRef]
- How, J.A.; Jazaeri, A.A.; Soliman, P.T.; Fleming, N.D.; Gong, J.; Piha-Paul, S.A.; Janku, F.; Stephen, B.; Naing, A. Pembrolizumab in Vaginal and Vulvar Squamous Cell Carcinoma: A Case Series from a Phase II Basket Trial. Sci. Rep. 2021, 11, 3667. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Vulvar Cancer (Version 1.2022). Available online: https://www.nccn.org/professionals/physician_gls/pdf/vulvar.pdf (accessed on 5 November 2021).
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II Study of Olaparib + Durvalumab (MEDIOLA): Updated Results in Germline BRCA-Mutated Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2019, 30, v485–v486. [Google Scholar] [CrossRef]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Yoo, S.-K.; Valero, C.; Pastore, A.; Krishna, C.; Lee, M.; Hoen, D.; Shi, H.; Kelly, D.W.; Patel, N.; et al. Improved Prediction of Immune Checkpoint Blockade Efficacy across Multiple Cancer Types. Nat. Biotechnol. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Biomarker | Rationale | Method of Detection | Use in Gynecological Cancer | Discriminating Result |
|---|---|---|---|---|
| Tumor-infiltrating lymphocytes (TILs) | Presence of TILs is indicator of intrinsic immune response | Histopathology of TT, semi-quantitatively, spatial pattern (1. between tumor cells, 2. in stroma, 3. absence) | No | 1. Immune-hot tumors 2. Immune-excluded tumors 3. Immune-deserted tumors |
| PD-1/PD-L1 expression | PD-L1 on tumor cells indicate activated immune escape mechanisms, PD-1 on TILs indicates T-cell exhaustion | Histopathology of TT -> IHC staining, semi-quantitatively; 1. Expression on tumor cells—tumor proportion score (TPS) 2. Expression in TT—combined proportion score (CPS) 3. Pattern of IHC staining; | 1. Uterine cervical cancer 2. Ovarian cancer 3. Vulvar cancer | Score expressed as number or percent; cut-offs varied from 1%-50% in studies, depending on tumor histotype |
| TME gene expression profiles | Immune-related gene expression is a marker of immune response | mRNA isolation from TT -> gene expression quantification -> model-based score | No | Not clearly defined |
| Tumor neoantigen load | Tumor neoantigens initiate immune response | DNA isolation from TT -> NGS -> in silico neoantigen prediction | No | Not clearly defined |
| Tumor mutational burden (TMB) | High TMB = hypermutated genome -> high tumor neoantigen load | DNA isolation from TT ->NGS(WES)/targeted assays (FoundationOne, MSK-IMPACT) | Potentially all (tissue FDA approval of pembrolizumab) | TMB-high -> 10–20 mut/Mb (varies) |
| Microsatellite instability (MSI), mismatch repair deficiency (MMR) | Defective DNA repair mechanism -> hypermutated genome -> high tumor neoantigen load | 1. MMR—histopathology of TT with IHC for 4 proteins involved in MMR 2. MSI—TT DNA isolation -> PCR with 5 probes | 1. Endometrial cancer 2. Ovarian cancer | 1. Non-expression in ≥ 1 protein -> MMRd (otherwise pMMR) 2. Altered repeat length in ≥ probes—MSI-H (otherwise MSS) |
| Homologous recombination deficiency (HRD) | Defective DNA repair mechanism -> hypermutated genome -> high tumor neoantigen load | 1. Germline BRCA mutation (genetic testing of normal tissue) 2. Tumor BRCA mutation (genetic testing of TT) 3. Genomic scar assay (Myriad myChoice, FoundationFocusCDxBRCA) 4. Other, less clinically validated assays | Ovarian cancer | 1. gBRCA mut/wt 2. tBRCA mut/wt 3. HRD +/− |
| Trial | Phase | Intervention | Study Population and Biomarker Status (Number of Patients) | Outcomes | |||
|---|---|---|---|---|---|---|---|
| Endometrial Cancer—ICI Monotherapy | |||||||
| KEYNOTE—158 [37] | 2 | Pembro | A/R/M disease, ≥1 PST | MSI-H/dMMR (49) | ORR 57%, mPFS 25.7 mo | ||
| TMB-H (15) a | ORR 46% | ||||||
| Non-TMB-H (67) a | ORR 6% | ||||||
| GARNET [54] | 1 | Dostarlimab | A/R/M disease, ≥1 PST | MSI-H/dMMR (103) | ORR 46% (CR in 10.7%), DCR 59% | ||
| MSS/MMRp (156) | TMB-H a (141) | ORR 13% | ORR 45.5% | ||||
| Non TMB-H a (13) | DCR 35% | ORR 12.1% | |||||
| Endometrial Cancer—ICI Combined with Other Agents | |||||||
| KEYNOTE-775 [60] | 3 | Pembro + Lenvatinib vs. Chemo | A/R/M disease, ≥ 1 PST | All patients (827) | ORR 31.9% vs. 14.7%; mPFS 7.2 vs. 3.8 mo; mOS 18.3 vs. 11.4 mo | ||
| MSI-H/dMMR (130) | ORR 40% vs. 12.3%; mPFS 10.7 vs. 3.7 mo; mOS NR vs. 8.6 mo | ||||||
| MSS/MMRp (697) | ORR 30.3% vs. 15.1%; mPFS 6.6 vs. 3.8 mo; mOS 17.4 vs. 12.0 mo | ||||||
| KEYNOTE-158 [76] | 2 | Pembro | A/R/M disease, 78% pts received ≥1 PSTs | All patients (98) | ORR 12.2%, DCR 30.6% | ||
| PD-L1+ b (82) | ORR 14.6%, DCR 32.9% | ||||||
| PD-L1– b (15) | ORR 0%, DCR 20% | ||||||
| CHECKMATE-358 [78] | 1/2 | Nivo | A/R/M disease, ≥1 PST | All patients (24), of those 10 PD-L1+, 6 PD-L1−, 3 NA c | ORR 26.3%, DCR 68.4%, mPFS 5.1 mo | ||
| EMPOWER-Cervical 1 [79] | 3 | Cemiplimab vs. Chemo | Recurrent/metastatic ≥1 prior syst. Th. | All patients (608) | mOS 12 v 8.5 mo | ||
| PD-L1+ d (162) | mOS ~ 14.5 vs. 9 mo | ||||||
| PD-L1– d (92) | mOS ~ 8 vs. 6 mo | ||||||
| Uterine Cervical Cancer—ICI Combined with Other Agents | |||||||
| KEYNOTE-826 [82] | 3 | SoC + pembro vs. SoC (Platinum-based doublet + bev) | A/R/M, no PST (first line) | All patients (619) | mPFS 10.4 vs. 8.2; HRPO 0.65 | ||
| PD-L1 CPS 1 (35, 11%) | HRPO 0.94 | ||||||
| PD-L1 CPS ≥ 1 (548, 88%) | mPFS 10.4 vs. 8.2 mo; HRPO 0.62 | ||||||
| PD-L1 CPS >10 (158, 51%) | mPFS 10.4 vs. 8.1 mo; HRPO 0.58 | ||||||
| Uterine Cervical Cancer—ICI Combined with Another ICI | |||||||
| CHECKMATE-358 [86] | 1/2 | Ipi + nivo | A/R/M, 0–2 PST | All patients(91) | ORR 46%, mPFS 8.5 mo, mOS 25.4 mo | ||
| RaPiDS [87] | 2 | Balstilimab+/−zalifrelimab | A/R/M, ≥1 PST | Combination group (143) | All (143) | ORR 22% | |
| PD-L1+ b (55%) | ORR 27% | ||||||
| PD-L1– b (25%) | ORR 11 % | ||||||
| PD-L1 NA (20) | ORR 21% | ||||||
| Ovarian Cancer—ICI Monotherapy | |||||||
| NINJA [113] | 3 | Nivo vs. single -agent chemo (GEM or PLD) | Relapsed, platinum resistant | All (316) | mPFS 2 vs. 3.8 mo; mOS 10.1 vs. 12.1 (favours chemo) | ||
| PD-L1+ (123) d | |||||||
| PD-L1− (189) d | |||||||
| Ovarian Cancer—ICI Combined with Other Agents | |||||||
| JAVELIN Ovarian 200 [114] | 3 | Ave (188) vs. Ave + PLD (188) vs. PLD (190) | Relapsed, platinum -resistant or refractory (no PST for platinum resistant disease) | All (566) | PD-L1+ e (288) | ORR 4% vs. 13% vs. 4%; DCR 33% vs. 49% vs. 57% | |
| PD-L1– e (220) | |||||||
| TIL+ f (228) | |||||||
| TIL– f (227) | |||||||
| JAVELIN Ovarian 100 [115] | 3 | PDC + maintenance Ave vs. PDC + Ave + maintenance Ave vs. PDC | First line—ACT or NACT | All (998) | PD-L1+ e (477) | mPFS 16.8 vs. 18.1 vs. NE Mo; HRP 1.43 vs. 1.14 vs. 1 (results favour PDC) | HRP 1.23 vs. 0.98 vs. 1 |
| PD-L1– e (326) | HRP 1.02 vs. 1.36 vs. 1 | ||||||
| gBRCA mut. (93) | HRP 1.98 vs. 2.51 vs. 1 | ||||||
| gBRCA wt (854) | HRP 1.32 vs. 1.14 vs. 1 | ||||||
| Kunde et.al. [117] | 2 | Pembro + metronomical CPA + bev | Relapsed, platinum—sensitive (25%) and platinum—resistant (75%) | 40 patients | BRCA g mut 35% | ORR 47.5%, DCR 95%, mPFS 10 mo | / |
| BRCA g wt 57.5% | / | ||||||
| PD-L1+ h 47.5% | ORR 52.6% | ||||||
| PD-L1– h 42.5% | ORR 35.3% | ||||||
| IMagyno-050 [118] | 3 | PDC + bev vs. PDC + bev + atezolizumab | First line—ACT or NACT | All patients (1301) | mPFS 18.4 vs. 19.5 (HRP 0.92) | ||
| PD-L1 < 1% IC i (517) | |||||||
| PD-L1 ≥ 1% IC i (784) | |||||||
| PD-L1 ≥ 5% IC i (260) | |||||||
| PD-L1 >= 1% TC i (73) | |||||||
| LEAP-005 [119] | 2 | Lenvatinib + pembro | Relapsed, 3 PST, (80% platinum resistant/refractory) | All patients (31) | ORR 32%, DCR 74%, mPFS 4.4 mo | ||
| TOPACIO/KEYNOTE-162 [120] | 1/2 | Niraparib + pembro | Relapsed, platinum sensitive and resistant disease, 1–5 PST; median number of PSTs was 3 | All patients (60) | ORR 18%, DCR 65% | ||
| tBRCAj | Mut(11, 18%) | ORR 18% | |||||
| Wt(49, 79%) | ORR 18% | ||||||
| PD-L1 k | +(35, 56%) | ORR 21% | |||||
| −(21, 34% | ORR 10% | ||||||
| HRDb | +(22, 35%) | ORR 14% | |||||
| −(33, 53% | ORR 19% | ||||||
| MEDIOLA [121,137] | 2 | Olaparib + durva | Relapsed, platinum sensitive, ≥ 1 PST | gBRCA mut (32) (doublet) | ORR 71.9%, mPFS 11.1 mo | ||
| sBRCA mut. (32) (doublet) | ORR 31 %, mPFS 5.5 mo | ||||||
| Olaparib + durva + bev | sBRCA mut. (31) (triplet) | ORR 77%, mPFS 14.7 mo | |||||
| Ovarian Cancer—ICI Combined with Another ICI | |||||||
| NRG-GY003 [123] | 2 | Nivo vs. Nivo + ipi | Relapsed, 1–3 PST, platinum resistant or platinum sensitive, PFI < 12 mo | All (100) | mPFS 2 vs. 3.9 mo; mOS 22 vs. 28 mo | ||
| Nivo or Nivo + Ipi (pooled data) | Any PD-L1 in TC l | +(5) | ORR 36%, mPFS 2.5 mo | ||||
| −(26) | ORR 23%, mPFS 4 mo | ||||||
| PD-L1 ≥ 1% in IC l | +(20) | ORR 31%, mPFS 4 mo | |||||
| −(11) | ORR 19%, mPFS 2.3 mo | ||||||
| Vulvar Cancer | |||||||
| KEYNOTE-028 [25] | 1 b | Pembro | Advanced, PD-L1 positive h | All (18) | ORR 6%, mPS 3.1 mo, mOS 3.8 mo | ||
| CHECKMATE-358 [78] | 1/2 | Nivo | Advanced | All (5), of those 4 PD-L1+ m, 1 pt NA | ORR 20%, DCR 80% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirš, B.; Škof, E.; Smrkolj, V.; Smrkolj, Š. Overview of Immune Checkpoint Inhibitors in Gynecological Cancer Treatment. Cancers 2022, 14, 631. https://doi.org/10.3390/cancers14030631
Pirš B, Škof E, Smrkolj V, Smrkolj Š. Overview of Immune Checkpoint Inhibitors in Gynecological Cancer Treatment. Cancers. 2022; 14(3):631. https://doi.org/10.3390/cancers14030631
Chicago/Turabian StylePirš, Boštjan, Erik Škof, Vladimir Smrkolj, and Špela Smrkolj. 2022. "Overview of Immune Checkpoint Inhibitors in Gynecological Cancer Treatment" Cancers 14, no. 3: 631. https://doi.org/10.3390/cancers14030631
APA StylePirš, B., Škof, E., Smrkolj, V., & Smrkolj, Š. (2022). Overview of Immune Checkpoint Inhibitors in Gynecological Cancer Treatment. Cancers, 14(3), 631. https://doi.org/10.3390/cancers14030631