
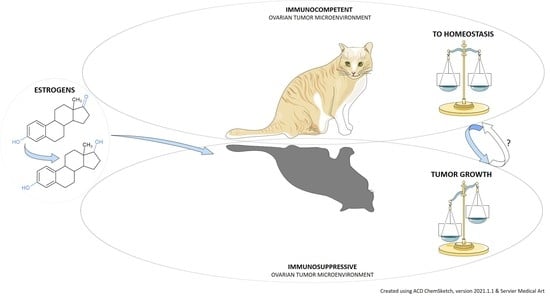
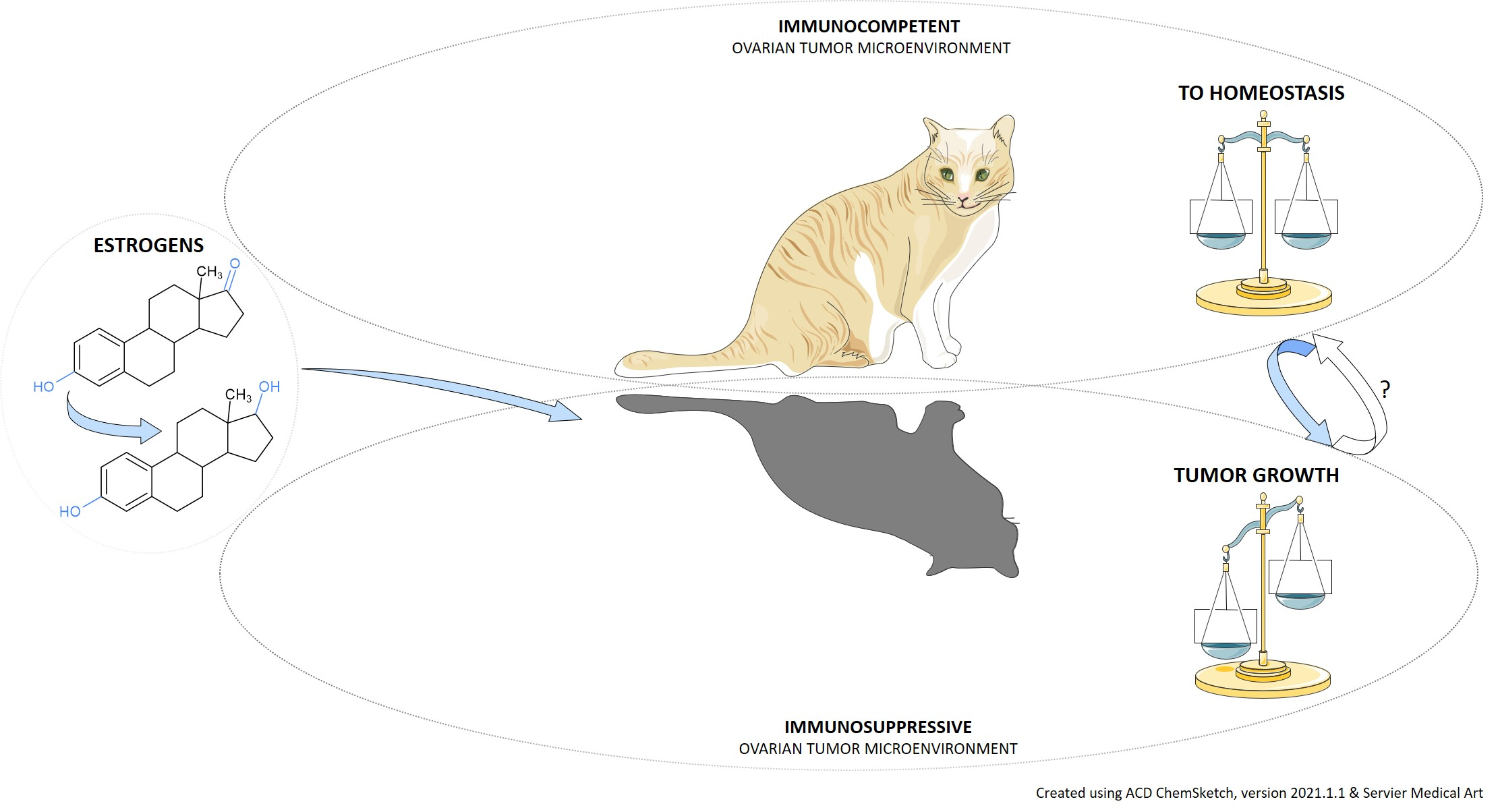
Estrogens and the Schrödinger’s Cat in the Ovarian Tumor Microenvironment



Simple Summary
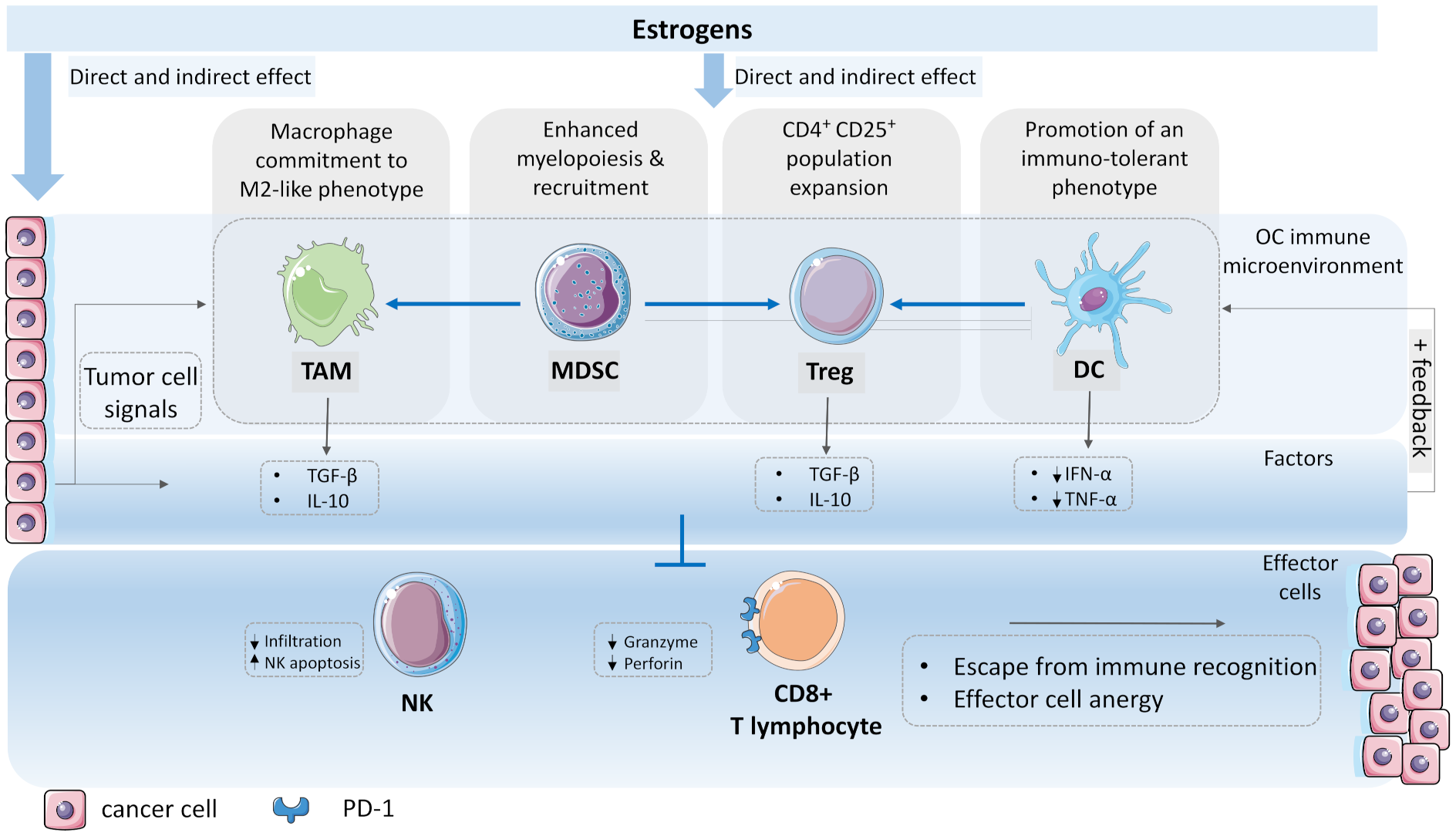
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Epithelial Ovarian Cancer Classification
3. OC and Estrogens
3.1. Estrogen Signaling in OC
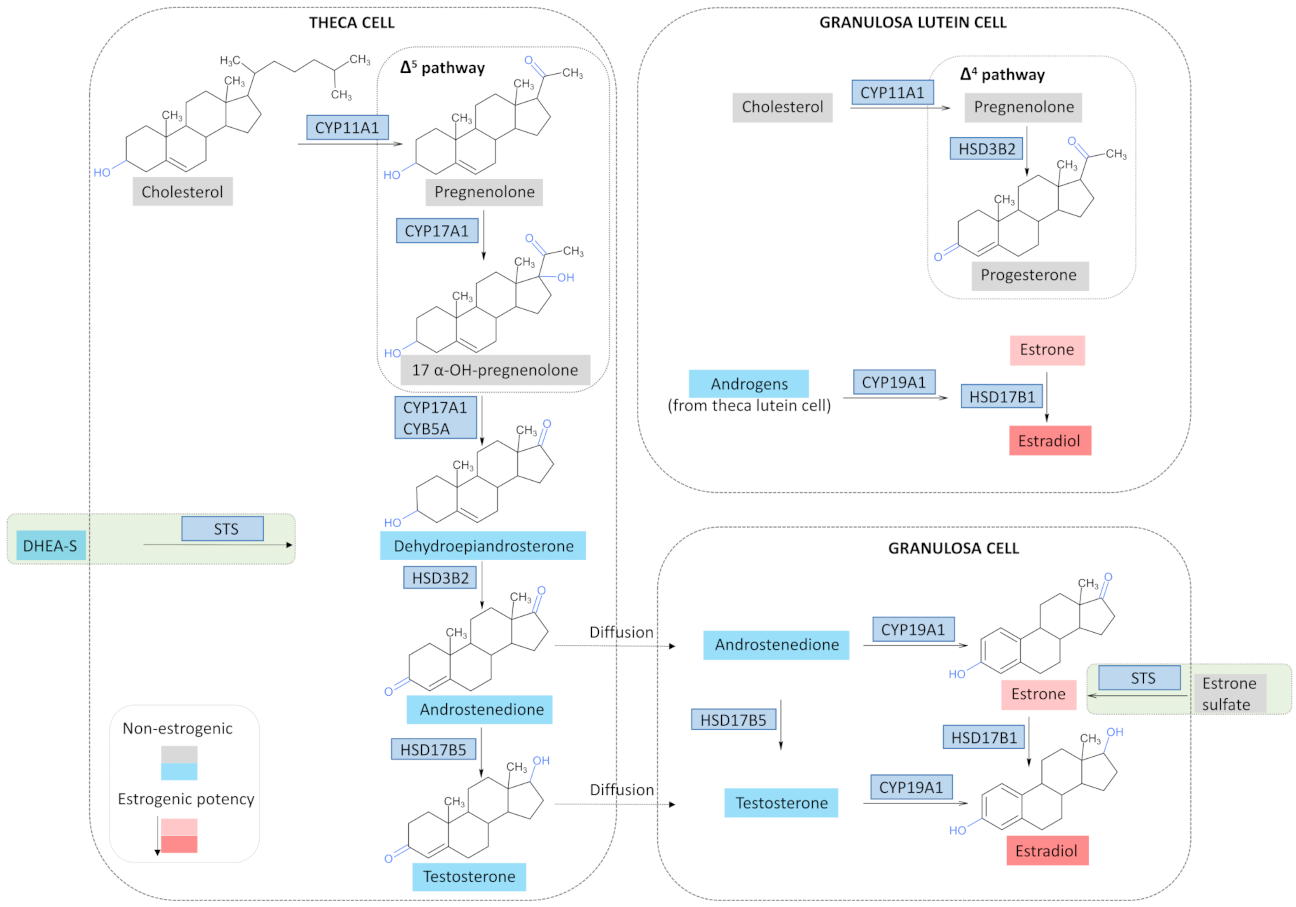
3.2. Estrogen Synthesis in a Physiological State
3.3. Estrogen Synthesis in OC
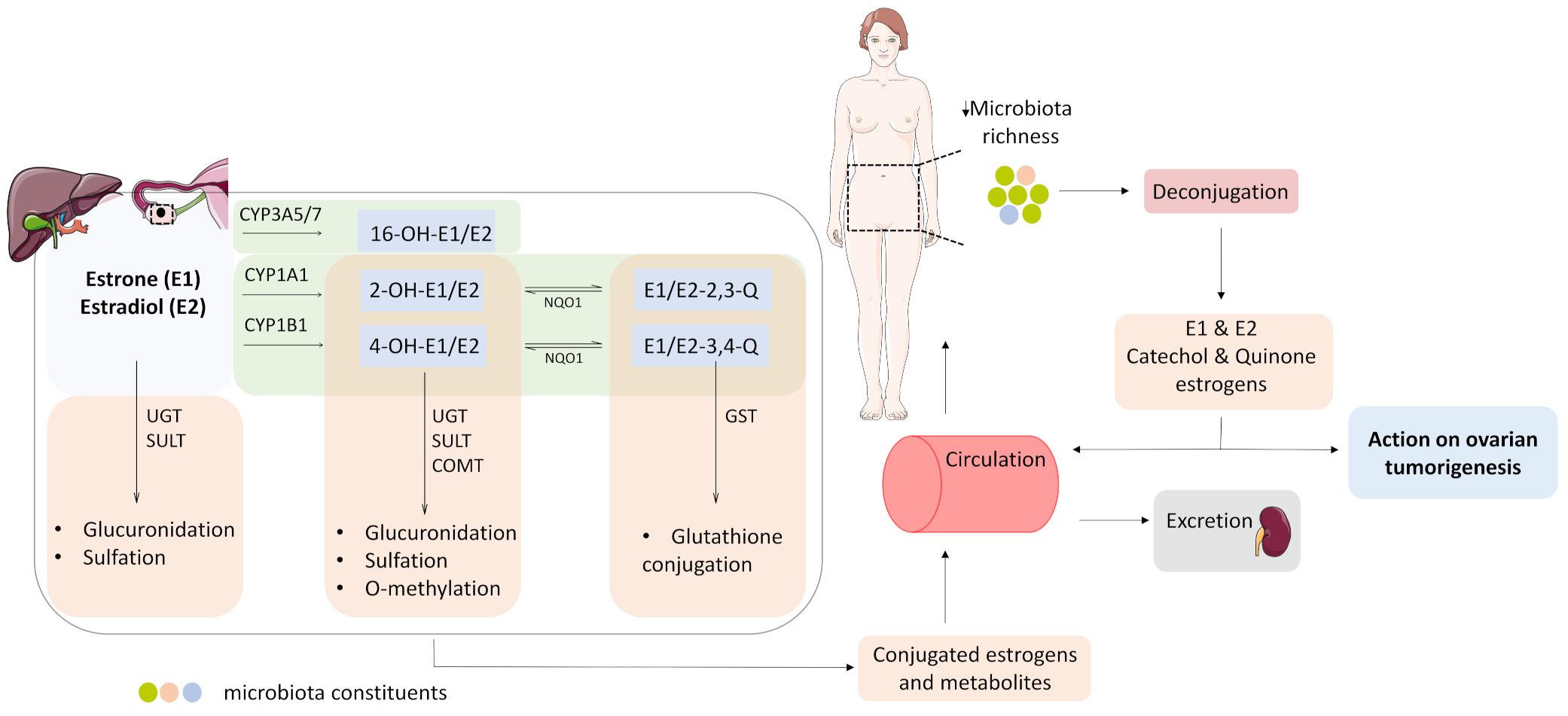
3.4. Estrogen Metabolism and the Microbiome Change in OC
4. The Emergence of a Tumor-Permissive Microenvironment and the Role of Estrogens
4.1. Macrophage Commitment to Tumor-Associated Type
4.2. Recruitment of Myeloid Derived Suppressor Cells
4.3. Expansion of Regulatory T Cell Population
4.4. The Dendritic Cell Immunotolerance
5. The Immune Override
6. Conclusions
7. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Matz, M.; Coleman, M.; Carreira, H.; Salmerón, D.; Chirlaque, M.D.; Allemani, C.; Bouzbid, S.; Hamdi-Chérif, M.; Zaidi, Z.; Bah, E.; et al. Worldwide comparison of ovarian cancer survival: Histological group and stage at diagnosis (CONCORD-2). Gynecol. Oncol. 2016, 144, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; I Weberpals, J.; Hendrickson, A.E.W.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Oza, A.M.; Estevez-Diz, M.D.P.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.M.; Uyar, D.S.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Du Bois, A.; Sehouli, J.; Vergote, I.; Ferron, G.; Reuss, A.; Meier, W.; Greggi, S.; Jensen, P.T.; Selle, F.; Guyon, F.; et al. Randomized phase III study to evaluate the impact of secondary cytoreductive surgery in recurrent ovarian cancer: Final analysis of AGO DESKTOP III/ENGOT-ov20. J. Clin. Oncol. 2020, 38, 6000. [Google Scholar] [CrossRef]
- Shi, T.; Zhu, J.; Feng, Y.; Tu, D.; Zhang, Y.; Zhang, P.; Jia, H.; Huang, X.; Cai, Y.; Yin, S.; et al. Secondary cytoreduction followed by chemotherapy versus chemotherapy alone in platinum-sensitive relapsed ovarian cancer (SOC-1): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 439–449. [Google Scholar] [CrossRef]
- Sieh, W.; Köbel, M.; A Longacre, T.; Bowtell, D.D.; Defazio, A.; Goodman, M.T.; Høgdall, E.; Deen, S.; Wentzensen, N.; Moysich, K.B.; et al. Hormone-receptor expression and ovarian cancer survival: An Ovarian Tumor Tissue Analysis consortium study. Lancet Oncol. 2013, 14, 853–862. [Google Scholar] [CrossRef]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer. Menopausal hormone use and ovarian cancer risk: Individual participant meta-analysis of 52 epidemiological studies. Lancet 2015, 385, 1835–1842. [Google Scholar] [CrossRef]
- Dallal, C.M.; for the B∼FIT Research Group; Lacey, J.V.; Pfeiffer, R.M.; Bauer, D.C.; Falk, R.T.; Buist, D.S.M.; A Cauley, J.; Hue, T.F.; Lacroix, A.Z.; et al. Estrogen Metabolism and Risk of Postmenopausal Endometrial and Ovarian Cancer: The B∼FIT Cohort. Horm. Cancer 2016, 7, 49–64. [Google Scholar] [CrossRef]
- Lindgren, P.R.; Backstrom, T.; Cajander, S.; Damber, M.-G.; Mahlck, C.-G.; Zhu, D.; Olofsson, J.I. The pattern of estradiol and progesterone differs in serum and tissue of benign and malignant ovarian tumors. Int. J. Oncol. 2002, 21, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, B.; Feigelson, H.S.; Flores, R.; Gail, M.H.; Xu, X.; Ravel, J.; Goedert, J.J. Associations of the Fecal Microbiome With Urinary Estrogens and Estrogen Metabolites in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2014, 99, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Sun, C.; Huang, J.; Xia, M.; Guo, E.; Li, N.; Lu, H.; Shan, W.; Wu, Y.; Li, Y.; et al. The biodiversity Composition of Microbiome in Ovarian Carcinoma Patients. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen Signaling and Its Potential as a Target for Therapy in Ovarian Cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef]
- Kaku, T.; Ogawa, S.; Kawano, Y.; Ohishi, Y.; Kobayashi, H.; Hirakawa, T.; Nakano, H. Histological classification of ovarian cancer. Med. Mol. Morphol. 2003, 36, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Mph, K.D.M.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Köbel, M.; Kalloger, S.E.; Huntsman, D.G.; Santos, J.L.; Swenerton, K.D.; Seidman, J.D.; Gilks, C.B. Differences in Tumor Type in Low-stage Versus High-stage Ovarian Carcinomas. Int. J. Gynecol. Pathol. 2010, 29, 203–211. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.-H.; Bast, R.C. Cell Origins of High-Grade Serous Ovar-ian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Sowamber, R.; Nelson, O.; Dodds, L.; DeCastro, V.; Paudel, I.; Milea, A.; Considine, M.; Cope, L.; Pinto, A.; Schlumbrecht, M.; et al. Integrative Transcriptome Analyses of the Human Fallopian Tube: Fimbria and Ampulla—Site of Origin of Serous Carcinoma of the Ovary. Cancers 2020, 12, 1090. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Cosio, S. Therapeutic Approach to Low-Grade Serous Ovarian Carcinoma: State of Art and Perspectives of Clinical Research. Cancers 2020, 12, 1336. [Google Scholar] [CrossRef]
- Singer, G.; Oldt, R.; Cohen, Y.; Wang, B.; Sidransky, D.; Kurman, R.J.; Shih, I.-M. Mutations in BRAF and KRAS Characterize the Development of Low-Grade Ovarian Serous Carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abu Shahin, N.; Pang, S.; Xiang, L.; Chambers, S.K.; Fadare, O.; Kong, B.; Zheng, W. Tubal origin of ‘ovarian’ low-grade serous carcinoma. Mod. Pathol. 2011, 24, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Savage, J.; Krishnan, J.; Vang, R.; Kurman, R.J. Intratumoral Heterogeneity Accounts for Apparent Progression of Noninvasive Serous Tumors to Invasive Low-grade Serous Carcinoma. Int. J. Gynecol. Pathol. 2020, 39, 43–54. [Google Scholar] [CrossRef]
- Perren, T.J. Mucinous epithelial ovarian carcinoma. Ann. Oncol. 2016, 27, i53–i57. [Google Scholar] [CrossRef]
- Cuatrecasas, M.; Villanueva, A.; Matias-Guiu, X.; Prat, J. K-ras mutations in mucinous ovarian tumors. Cancer 1997, 79, 1581–1586. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Luvero, D.; Shafer, A.; O’Connor, D.; Mangili, G.; Friedlander, M.; Pfisterer, J.; Mirza, M.R.; Kim, J.-W.; Alexandre, J.; et al. Gynecologic Cancer InterGroup (GCIG) Consensus Review for Mucinous Ovarian Carcinoma. Int. J. Gynecol. Cancer 2014, 24, S14–S19. [Google Scholar] [CrossRef]
- Cheasley, D.; Wakefield, M.J.; Ryland, G.L.; Allan, P.E.; Alsop, K.; Amarasinghe, K.C.; Ananda, S.; Anglesio, M.S.; Au-Yeung, G.; Böhm, M.; et al. The molecular origin and taxonomy of mucinous ovarian carcinoma. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kotani, Y.; Nakai, H.; Matsumura, N. Endometriosis-Associated Ovarian Cancer: The Origin and Targeted Therapy. Cancers 2020, 12, 1676. [Google Scholar] [CrossRef]
- Iida, Y.; Okamoto, A.; Hollis, R.L.; Gourley, C.; Herrington, C.S. Clear cell carcinoma of the ovary: A clinical and molecular perspective. Int. J. Gynecol. Cancer 2020, 31, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.-T.; Mao, T.-L.; Jones, S.; Veras, E.; Ayhan, A.; Wang, T.-L.; Glas, R.; Slamon, D.; Velculescu, V.E.; Kuman, R.J.; et al. Frequent Activating Mutations of PIK3CA in Ovarian Clear Cell Carcinoma. Am. J. Pathol. 2009, 174, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Pierson, W.E.; Peters, P.N.; Chang, M.T.; Chen, L.-M.; Quigley, D.A.; Ashworth, A.; Chapman, J.S. An integrated molecular profile of endometrioid ovarian cancer. Gynecol. Oncol. 2020, 157, 55–61. [Google Scholar] [CrossRef]
- Garavaglia, E.; Sigismondi, C.; Ferrari, S.; Candiani, M. The origin of endometriosis-associated ovarian cancer from uterine neoplastic lesions. Med. Hypotheses 2018, 110, 80–82. [Google Scholar] [CrossRef]
- Wang, Y.; Mang, M.; Wang, Y.; Wang, L.; Klein, R.; Kong, B.; Zheng, W. Tubal origin of ovarian endometriosis and clear cell and endometrioid carcinoma. Am. J. Cancer Res. 2015, 5, 869–879. [Google Scholar]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357. [Google Scholar] [CrossRef]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, L.; Yang, X.; Bie, J.; Li, D.; Sun, C.; Zhang, J.; Meng, Y.; Lin, J. Menopausal Hormone Replacement Therapy and the Risk of Ovarian Cancer: A Meta-Analysis. Front. Endocrinol. 2019, 10, 801. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Brinton, L.A.; Anderson, G.L.; Pfeiffer, R.M.; Falk, R.T.; Strickler, H.D.; Sliesoraitis, S.; Kuller, L.H.; Gass, M.L.; Fuhrman, B.; et al. Circulating Estrogens and Postmenopausal Ovarian Cancer Risk in the Women’s Health Initiative Observational Study. Cancer Epidemiol. Biomark. Prev. 2016, 25, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Schock, H.; Surcel, H.-M.; Zeleniuch-Jacquotte, A.; Grankvist, K.; Lakso, H.; Fortner, R.; Kaaks, R.; Pukkala, E.; Lehtinen, M.; Toniolo, P.; et al. Early pregnancy sex steroids and maternal risk of epithelial ovarian cancer. Endocr. Relat. Cancer 2014, 21, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gustafsson, J. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Arterburn, J.B.; Smith, H.O.; Oprea, T.; Sklar, L.A.; Hathaway, H.J. Estrogen Signaling through the Transmembrane G Protein–Coupled Receptor GPR30. Annu. Rev. Physiol. 2008, 70, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Pujol, P.; Rey, J.-M.; Nirde, P.; Roger, P.; Gastaldi, M.; Laffargue, F.; Rochefort, H.; Maudelonde, T. Differential Expres-sion of Estrogen Receptor-α and -β Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis. Cancer Res. 1998, 58, 5367. [Google Scholar] [PubMed]
- Schüler-Toprak, S.; Weber, F.; Skrzypczak, M.; Ortmann, O.; Treeck, O. Estrogen receptor β is associated with expression of cancer associated genes and survival in ovarian cancer. BMC Cancer 2018, 18, 981. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Cheung, L.W.T.; Wong, A.S.T.; Leung, P.C.K. Estrogen Regulates Snail and Slug in the Down-Regulation of E-Cadherin and Induces Metastatic Potential of Ovarian Cancer Cells through Estrogen Receptor α. Mol. Endocrinol. 2008, 22, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.L.; Siu, M.K.Y.; Jiang, Y.-X.; Wang, J.-J.; Wang, Y.; Leung, T.H.Y.; Liu, S.S.; Cheung, A.N.Y.; Ngan, H.Y.S. Differential expression of estrogen receptor subtypes and variants in ovarian cancer: Effects on cell invasion, proliferation and prognosis. BMC Cancer 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Chan, K.K.-L.; Leung, T.H.-Y.; Chan, D.; Wei, N.; Lau, G.T.-Y.; Liu, S.S.; Siu, M.K.-Y.; Ngan, H.Y.S. Targeting estrogen receptor subtypes (ERα and ERβ) with selective ER modulators in ovarian cancer. J. Endocrinol. 2014, 221, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Heublein, S.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; et al. The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro. Cells 2021, 10, 619. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, X.; Zhao, Y.; Wen, H.; Liu, G. Role of GPER on proliferation, migration and invasion in ligand-independent manner in human ovarian cancer cell line SKOV3. Cell Biochem. Funct. 2015, 33, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Molecular Biology of Steroid Hormone Synthesis. Endocr. Rev. 1988, 9, 295–318. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Minireview: Regulation of Steroidogenesis by Electron Transfer. Endocrinology 2005, 146, 2544–2550. [Google Scholar] [CrossRef]
- Simard, J.; Ricketts, M.-L.; Gingras, S.; Soucy, P.; Feltus, F.; Melner, M.H. Molecular Biology of the 3β-Hydroxysteroid Dehydrogenase/Δ5-Δ4 Isomerase Gene Family. Endocr. Rev. 2005, 26, 525–582. [Google Scholar] [CrossRef]
- Fogle, R.H.; Stanczyk, F.Z.; Zhang, X.; Paulson, R.J. Ovarian Androgen Production in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2007, 92, 3040–3043. [Google Scholar] [CrossRef]
- Simpson, E. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230. [Google Scholar] [CrossRef]
- Yamagata, S.; Yamamoto, K.; Yamamoto, K.; Yamamoto, A.; Kawamura, N.; Nakamura, T.; Shimizu, S.; Sugawa, T. Es-trogen production in epithelial tumors of the ovary--identification of estrogen-synthesizing cells. Nihon Sanka Fujinka Gakkai Zasshi 1989, 41, 1791–1796. [Google Scholar]
- Aiman, J.; Forney, J.P.; Parker, C.R., Jr. Secretion of androgens and estrogens by normal and neoplastic ovaries in post-menopausal women. Obstet. Gynecol. 1986, 68, 1–5. [Google Scholar] [CrossRef]
- Matsumoto, Y. Study on the estrogen production in parenchymatous cells of epithelial ovarian tumor. Osaka City Med. J. 1992, 38. [Google Scholar]
- Blanco, L.Z., Jr.; Kuhn, E.; Morrison, J.C.; Bahadirli-Talbott, A.; Smith-Sehdev, A.; Kurman, R.J. Steroid hormone synthesis by the ovarian stroma surrounding epithelial ovarian tumors: A potential mechanism in ovarian tumorigenesis. Mod. Pathol. 2017, 30, 563–576. [Google Scholar] [CrossRef]
- Rižner, T.L.; Thalhammer, T.; Özvegy-Laczka, C. The Importance of Steroid Uptake and Intracrine Action in Endometrial and Ovarian Cancers. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wu, X.; Hillier, S.G.; Fegan, K.S.; Critchley, H.O.; Mason, J.I.; Sarvi, S.; Harlow, C.R. Local estrogen metabolism in epithelial ovarian cancer suggests novel targets for therapy. J. Steroid Biochem. Mol. Biol. 2015, 150, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Raftogianis, R.; Creveling, C.; Weinshilboum, R.; Weisz, J. Chapter 6: Estrogen Metabolism by Conjugation. J. Natl. Cancer Inst. Monogr. 2000, 2000, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Downie, D.; McFadyen, M.; Rooney, P.H.; Cruickshank, M.E.; Parkin, D.E.; Miller, I.D.; Telfer, C.; Melvin, W.T.; Murray, G.I. Profiling Cytochrome P450 Expression in Ovarian Cancer: Identification of Prognostic Markers. Clin. Cancer Res. 2005, 11, 7369–7375. [Google Scholar] [CrossRef]
- Cui, X.; Li, L.; Yan, G.; Meng, K.; Lin, Z.; Nan, Y.; Jin, G.; Li, C. High expression of NQO1 is associated with poor prognosis in serous ovarian carcinoma. BMC Cancer 2015, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sawers, L.; Ferguson, M.J.; Ihrig, B.R.; Young, H.C.; Chakravarty, P.; Wolf, C.R.; Smith, G.D.W. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br. J. Cancer 2014, 111, 1150–1158. [Google Scholar] [CrossRef]
- Mungenast, F.; Aust, S.; Vergote, I.; Vanderstichele, A.; Sehouli, J.; Braicu, E.; Mahner, S.; Castillo-Tong, D.C.; Zeillinger, R.; Thalhammer, T. Clinical significance of the estrogen-modifying enzymes steroid sulfatase and estrogen sulfotransferase in epithelial ovarian cancer. Oncol. Lett. 2017, 13, 4047–4054. [Google Scholar] [CrossRef]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef]
- Pollet, R.M.; D’Agostino, E.H.; Walton, W.G.; Xu, Y.; Little, M.S.; Biernat, K.A.; Pellock, S.J.; Patterson, L.M.; Creekmore, B.; Isenberg, H.N.; et al. An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25, 967–977.e5. [Google Scholar] [CrossRef] [PubMed]
- Ervin, S.M.; Simpson, J.B.; Gibbs, M.E.; Creekmore, B.C.; Lim, L.; Walton, W.G.; Gharaibeh, R.Z.; Redinbo, M.R. Structural Insights into Endobiotic Reactivation by Human Gut Microbiome-Encoded Sulfatases. Biochemistry 2020, 59, 3939–3950. [Google Scholar] [CrossRef]
- Tong, J.; Zhang, X.; Fan, Y.; Chen, L.; Ma, X.; Yu, H.; Li, J.; Guan, X.; Zhao, P.; Yang, J. Changes of Intestinal Microbiota in Ovarian Cancer Patients Treated with Surgery and Chemotherapy. Cancer Manag. Res. 2020, 12, 8125–8135. [Google Scholar] [CrossRef]
- Nené, N.R.; Reisel, D.; Leimbach, A.; Franchi, D.; Jones, A.; Evans, I.; Knapp, S.; Ryan, A.; Ghazali, S.; Timms, J.; et al. Association between the cervicovaginal microbiome, BRCA1 mutation status, and risk of ovarian cancer: A case-control study. Lancet Oncol. 2019, 20, 1171–1182. [Google Scholar] [CrossRef]
- Miao, R.; Badger, T.C.; Groesch, K.; Diaz-Sylvester, P.L.; Wilson, T.; Ghareeb, A.; Martin, J.A.; Cregger, M.; Welge, M.; Bushell, C.; et al. Assessment of peritoneal microbial features and tumor marker levels as potential diagnostic tools for ovarian cancer. PLoS ONE 2020, 15, e0227707. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Yoshihara, M.; Douagi, I.; Damdimopoulos, A.; Panula, S.; Petropoulos, S.; Lu, H.; Pettersson, K.; Palm, K.; Katayama, S.; et al. Single-cell analysis of human ovarian cortex identifies distinct cell populations but no oogonial stem cells. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Zhang, Z.; Schlamp, F.; Huang, L.; Clark, H.; Brayboy, L.M. Inflammaging is associated with shifted macrophage ontogeny and polarization in the aging mouse ovary. Reproduction 2020, 159, 325–337. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef]
- Quail, D.F.; A Joyce, J. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Coukos, G. Deciphering and Reversing Tumor Immune Suppression. Immunity 2013, 39, 61–73. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Chen, D.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Winterhoff, B.J.; Maile, M.; Mitra, A.; Sebe, A.; Bazzaro, M.; Geller, M.A.; Abrahante, J.; Klein, M.; Hellweg, R.; Mullany, S.A.; et al. Single cell sequencing reveals heterogeneity within ovarian cancer epithelium and cancer associated stromal cells. Gynecol. Oncol. 2017, 144, 598–606. [Google Scholar] [CrossRef]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.-J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Pepe, G.; Locati, M.; Della Torre, S.; Mornata, F.; Cignarella, A.; Maggi, A.; Vegeto, E. The estrogen–macrophage interplay in the homeostasis of the female reproductive tract. Hum. Reprod. Update 2018, 24, 652–672. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Liu, J.-H.; Zheng, L.-M.; Cai, M.-Y.; Ding, T. Prognostic significance of tumor-associated macrophage infiltration in advanced epithelial ovarian carcinoma. Ai Zheng Aizheng Chin. J. Cancer 2009, 28, 323–327. [Google Scholar]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of M2-Polarized Macrophages is Associated with Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technol. Cancer Res. Treat. 2013, 12, 259–267. [Google Scholar] [CrossRef]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer 2013, 134, 32–42. [Google Scholar] [CrossRef]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef]
- Liu, R.; Hu, R.; Zeng, Y.; Zhang, W.; Zhou, H.-H. Tumour immune cell infiltration and survival after platinum-based chemotherapy in high-grade serous ovarian cancer subtypes: A gene expression-based computational study. EBioMedicine 2020, 51, 102602. [Google Scholar] [CrossRef] [PubMed]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Villa, A.; Rizzi, N.; Vegeto, E.; Ciana, P.; Maggi, A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci. Rep. 2015, 5, 15224. [Google Scholar] [CrossRef]
- Pepe, G.; Braga, D.; Renzi, T.A.; Villa, A.; Bolego, C.; D’Avila, F.; Barlassina, C.; Maggi, A.; Locati, M.; Vegeto, E. Self-renewal and phenotypic conversion are the main physiological responses of macrophages to the endogenous estrogen surge. Sci. Rep. 2017, 7, 44270. [Google Scholar] [CrossRef]
- Ciucci, A.; Zannoni, G.F.; Buttarelli, M.; Lisi, L.; Travaglia, D.; Martinelli, E.; Scambia, G.; Gallo, D. Multiple direct and indirect mechanisms drive estrogen-induced tumor growth in high grade serous ovarian cancers. Oncotarget 2016, 7, 8155–8171. [Google Scholar] [CrossRef]
- Thibault, B.; Castells, M.; Delord, J.-P.; Couderc, B. Ovarian cancer microenvironment: Implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 2013, 33, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.C.; Goedemans, R.; Jha, V.; Nortier, J.W.R.; Welters, M.J.P.; Kroep, J.; Van Der Burg, S.H. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor Microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef]
- Mlynska, A.; Povilaityte, E.; Zemleckaite, I.; Zilionyte, K.; Strioga, M.; Krasko, J.; Dobrovolskiene, N.; Peng, M.-W.; Intaite, B.; Pasukoniene, V. Platinum sensitivity of ovarian cancer cells does not influence their ability to induce M2-type macrophage polarization. Am. J. Reprod. Immunol. 2018, 80, e12996. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.A.M.; De Groot, A.F.; Dijkgraaf, E.M.; Simões, A.M.C.; Van Der Noord, V.E.; Van Ham, J.J.; Welters, M.J.P.; Kroep, J.; Van Der Burg, S.H. The blood mMDSC to DC ratio is a sensitive and easy to assess independent predictive factor for epithelial ovarian cancer survival. OncoImmunology 2018, 7, e1465166. [Google Scholar] [CrossRef]
- Okła, K.; Czerwonka, A.; Wawruszak, A.; Bobiński, M.; Bilska, M.; Tarkowski, R.; Bednarek, W.; Wertel, I.; Kotarski, J. Clinical Relevance and Immunosuppressive Pattern of Circulating and Infiltrating Subsets of Myeloid-Derived Suppressor Cells (MDSCs) in Epithelial Ovarian Cancer. Front. Immunol. 2019, 10, 691. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Yamanoi, K.; Horikawa, N.; Hosoe, Y.; Nakamura, E.; et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Coosemans, A.; Baert, T.; Ceusters, J.; Busschaert, P.; Landolfo, C.; Verschuere, T.; Van Rompuy, A.-S.; Vanderstichele, A.; Froyman, W.; Neven, P.; et al. Myeloid-derived suppressor cells at diagnosis may discriminate between benign and malignant ovarian tumors. Int. J. Gynecol. Cancer 2019, 29, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell–Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2016, 7, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; You, M.; Fan, H.; Ji, J.; Ding, L.; Li, P.; Hou, Y. 17β-estradiol contributes to the accumulation of myeloid-derived suppressor cells in blood by promoting TNF-α secretion. Acta Biochim. Biophys. Sin. 2015, 47, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Milette, S.; Hashimoto, M.; Perrino, S.; Qi, S.; Chen, M.; Ham, B.; Wang, N.; Istomine, R.; Lowy, A.M.; Piccirillo, C.A.; et al. Sexual dimorphism and the role of estrogen in the immune microenvironment of liver metastases. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.; Zhang, H.-G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Shevach, E.M. Regulatory T Cells in Autoimmmunity. Annu. Rev. Immunol. 2000, 18, 423–449. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Barnett, J.C.; Bean, S.M.; Whitaker, R.S.; Kondoh, E.; Baba, T.; Fujii, S.; Marks, J.R.; Dressman, H.K.; Murphy, S.K.; Berchuck, A. Ovarian cancer tumor infiltrating T-regulatory (Treg) cells are associated with a metastatic phenotype. Gynecol. Oncol. 2010, 116, 556–562. [Google Scholar] [CrossRef]
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of regulatory T cells by physiological level estrogen. J. Cell. Physiol. 2007, 214, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Adurthi, S.; Kumar, M.M.; Vinodkumar, H.S.; Mukherjee, G.; Krishnamurthy, H.; Acharya, K.K.; Bafna, U.D.; Uma, D.K.; Abhishekh, B.; Krishna, S.; et al. Oestrogen Receptor-α binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci. Rep. 2017, 7, 17289. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.A.; Rosenstein, Y. Oestradiol potentiates the suppressive function of human CD4+ CD25+ regulatory T cells by promoting their proliferation. Immunology 2006, 118, 58–65. [Google Scholar] [CrossRef]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- Valor, L.; Teijeiro, R.; Aristimuño, C.; Faure, F.; Alonso, B.; De Andrés, C.; Tejera, M.; López-Lazareno, N.; Fernández-Cruz, E.; Sánchez-Ramón, S. Estradiol-dependent perforin expression by human regulatory T-cells. Eur. J. Clin. Investig. 2010, 41, 357–364. [Google Scholar] [CrossRef]
- Wculek, S.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2019, 20, 7–24. [Google Scholar] [CrossRef]
- Sichien, D.; Lambrecht, B.; Guilliams, M.; Scott, C. Development of conventional dendritic cells: From common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. 2017, 10, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Patidar, A.; Selvaraj, S.; Sarode, A.; Chauhan, P.; Chattopadhyay, D.; Saha, B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 2018, 104, 114–123. [Google Scholar] [CrossRef]
- Harimoto, H.; Shimizu, M.; Nakagawa, Y.; Nakatsuka, K.; Wakabayashi, A.; Sakamoto, C.; Takahashi, H. Inactivation of tumor-specific CD8+ CTLs by tumor-infiltrating tolerogenic dendritic cells. Immunol. Cell Biol. 2013, 91, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Kovats, S. Estrogen receptors regulate an inflammatory pathway of dendritic cell differentiation: Mechanisms and implications for immunity. Horm. Behav. 2012, 62, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Papenfuss, T.L.; Powell, N.D.; McClain, M.A.; Bedarf, A.; Singh, A.; Gienapp, I.E.; Shawler, T.; Whitacre, C.C. Estriol Generates Tolerogenic Dendritic Cells In Vivo That Protect against Autoimmunity. J. Immunol. 2011, 186, 3346–3355. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Sisirak, V.; Meeus, P.; Gobert, M.; Treilleux, I.; Bajard, A.; Combes, J.-D.; Faget, J.; Mithieux, F.; Cassignol, A.; et al. Quantitative and Functional Alterations of Plasmacytoid Dendritic Cells Contribute to Immune Tolerance in Ovarian Cancer. Cancer Res. 2011, 71, 5423–5434. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, S.; Herfs, M.; Delvenne, P.; Hubert, P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: Insight into the molecular mechanisms. J. Leukoc. Biol. 2012, 93, 343–352. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.S.; Ohashi, P.S. The Roles of CD8+ T Cell Subsets in Antitumor Immunity. Trends Cell Biol. 2020, 30, 695–704. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Henriksen, J.R.; Donskov, F.; Waldstrøm, M.; Jakobsen, A.; Hjortkjaer, M.; Petersen, C.B.; Steffensen, K.D. Favorable prognostic impact of Natural Killer cells and T cells in high-grade serous ovarian carcinoma. Acta Oncol. 2020, 59, 652–659. [Google Scholar] [CrossRef]
- Brummelman, J.; Mazza, E.M.C.; Alvisi, G.; Colombo, F.S.; Grilli, A.; Mikulak, J.; Mavilio, D.; Alloisio, M.; Ferrari, F.; Lopci, E.; et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med. 2018, 215, 2520–2535. [Google Scholar] [CrossRef]
- Pesce, S.; Tabellini, G.; Cantoni, C.; Patrizi, O.; Coltrini, D.; Rampinelli, F.; Matta, J.; Vivier, E.; Moretta, A.; Parolini, S.; et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. OncoImmunology 2015, 4, e1001224. [Google Scholar] [CrossRef]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Prognostic effect of programmed death-ligand 1 (PD-L1) in ovarian cancer: A systematic review, meta-analysis and bioinformatics study. J. Ovarian Res. 2019, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bekos, C.; Pils, D.; Dekan, S.; Hofstetter, G.; Horak, P.; Reinthaller, A.; Polterauer, S.; Schwameis, R.; Aust, S. PD-1 and PD-L1 expression on TILs in peritoneal metastases compared to ovarian tumor tissues and its associations with clinical outcome. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Domagala, J.; Lachota, M.; Klopotowska, M.; Graczyk-Jarzynka, A.; Domagala, A.; Zhylko, A.; Soroczynska, K.; Winiarska, M. The Tumor Microenvironment—A Metabolic Obstacle to NK Cells’ Activity. Cancers 2020, 12, 3542. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients With Recurrent or Refractory Ovarian Cancer. JAMA Oncol. 2019, 5, 393–401. [Google Scholar] [CrossRef]
- Matulonis, U.; Shapira-Frommer, R.; Santin, A.; Lisyanskaya, A.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.; Birrer, M.; Provencher, D.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Fujiwara, K.; Dychter, S.S.; Devgan, G.; Monk, B.J. Avelumab (anti-PD-L1) in platinum-resistant/refractory ovarian cancer: JAVELIN Ovarian 200 Phase III study design. Futur. Oncol. 2018, 14, 2103–2113. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gjorgoska, M.; Rižner, T.L. Estrogens and the Schrödinger’s Cat in the Ovarian Tumor Microenvironment. Cancers 2021, 13, 5011. https://doi.org/10.3390/cancers13195011
Gjorgoska M, Rižner TL. Estrogens and the Schrödinger’s Cat in the Ovarian Tumor Microenvironment. Cancers. 2021; 13(19):5011. https://doi.org/10.3390/cancers13195011
Chicago/Turabian StyleGjorgoska, Marija, and Tea Lanišnik Rižner. 2021. "Estrogens and the Schrödinger’s Cat in the Ovarian Tumor Microenvironment" Cancers 13, no. 19: 5011. https://doi.org/10.3390/cancers13195011
APA StyleGjorgoska, M., & Rižner, T. L. (2021). Estrogens and the Schrödinger’s Cat in the Ovarian Tumor Microenvironment. Cancers, 13(19), 5011. https://doi.org/10.3390/cancers13195011