
Genomic Mechanisms Influencing Outcome in Chronic Myeloid Leukemia


Abstract
Simple Summary
Abstract
1. Introduction
2. BCR::ABL1 Kinase Domain Mutations and Their Role in Drug Resistance
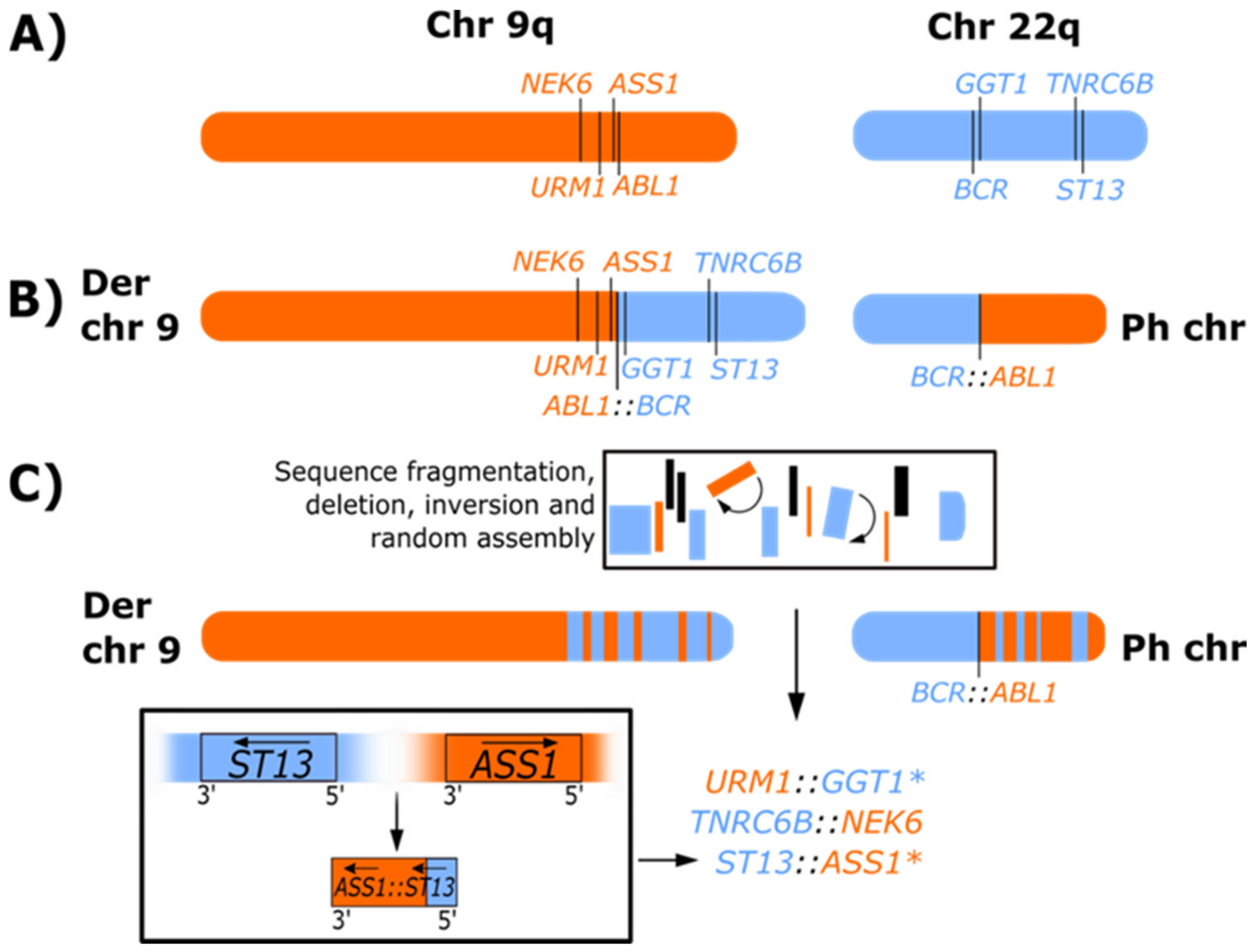
3. Rearrangements Associated with the Formation of the Ph Chromosome
3.1. The Influence of BCR::ABL1 Transcript Type and Treatment Response
3.2. Variant Translocations
3.3. Derivative Chromosome 9 Deletions

3.4. Ph-Associated Rearrangements
4. Role of Potentially Pathogenic Variants in Cancer Genes, Including Fusions and Deletions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bruford, E.A.; Antonescu, C.R.; Carroll, A.J.; Chinnaiyan, A.; Cree, I.A.; Cross, N.C.P.; Dalgleish, R.; Gale, R.P.; Harrison, C.J.; Hastings, R.J.; et al. HUGO Gene Nomenclature Committee (HGNC) recommendations for the designation of gene fusions. Leukemia 2021, 35, 3040–3043. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed]
- NCCN Clinical Practice Guidelines in Oncology: Chronic Myeloid Leukemia. Version 2.2022. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1427 (accessed on 2 January 2022).
- Wedelin, C.; Björkholm, M.; Mellstedt, H.; Gahrton, G.; Holm, G. Clinical Findings and Prognostic Factors in Chronic Myeloid Leukemias. Acta Med. Scand. 2009, 220, 255–260. [Google Scholar] [CrossRef]
- Daley, G.Q.; Van Etten, R.A.; Baltimore, D. Induction of Chronic Myelogenous Leukemia in Mice by the P210 bcr/abl Gene of the Philadelphia Chromosome. Science 1990, 247, 824–830. [Google Scholar] [CrossRef]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.M.; Ghorab, A.; Sasaki, K.; Jabbour, E.J.; Gonzalez, G.N.; Kanagal-Shamanna, R.; Issa, G.C.; Garcia-Manero, G.; Kc, D.; et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: Cohort study of 477 patients. Cancer 2017, 123, 4391–4402. [Google Scholar] [CrossRef]
- Hughes, T.; Ross, D. Moving treatment-free remission into mainstream clinical practice in CML. Blood 2016, 128, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Martinelli, G.; Rosti, G.; Bassi, S.; Amabile, M.; Poerio, A.; Giannini, B.; Trabacchi, E.; Castagnetti, F.; Testoni, N.; et al. ABL Mutations in Late Chronic Phase Chronic Myeloid Leukemia Patients with Up-Front Cytogenetic Resistance to Imatinib Are Associated with a Greater Likelihood of Progression to Blast Crisis and Shorter Survival: A Study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J. Clin. Oncol. 2005, 23, 4100–4109. [Google Scholar] [CrossRef]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical Resistance to STI-571 Cancer Therapy Caused by BCR-ABL Gene Mutation or Amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Nicoll, J.M.; Nagar, B.; Gorre, M.E.; Paquette, R.L.; Kuriyan, J.; Sawyers, C.L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [Google Scholar] [CrossRef]
- Branford, S.; Rudzki, Z.; Walsh, S.; Grigg, A.; Arthur, C.; Taylor, K.; Herrmann, R.; Lynch, K.P.; Hughes, T.P. High frequency of point mutations clustered within the adenosine triphosphate–binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002, 99, 3472–3475. [Google Scholar] [CrossRef] [PubMed]
- Von Bubnoff, N.; Schneller, F.; Peschel, C.; Duyster, J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: A prospective study. Lancet 2002, 359, 487–491. [Google Scholar] [CrossRef]
- Roche-Lestienne, C.; Soenen, V.; Grardel, N.; Laï, J.-L.; Philippe, N.; Facon, T.; Fenaux, P.; Preudhomme, C. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood 2002, 100, 1014–1018. [Google Scholar] [CrossRef]
- Branford, S.; Rudzki, Z.; Walsh, S.; Parkinson, I.; Grigg, A.; Szer, J.; Taylor, K.; Herrmann, R.; Seymour, J.F.; Arthur, C.; et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 2003, 102, 276–283. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H.M.; Jones, D.T.L.; Talpaz, M.; Bekele, N.; Obrien, S.J.; Zhou, X.; Luthra, R.; Garciamanero, G.; Giles, F.J.; et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia 2006, 20, 1767–1773. [Google Scholar] [CrossRef]
- Nicolini, F.; Corm, S.; Lê, Q.-H.; Sorel, N.; Hayette, S.; Bories, D.; Leguay, T.; Roy, L.; Giraudier, S.; Tulliez, M.; et al. Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: A retrospective analysis from the French intergroup of CML (Fi(ϕ)-LMC GROUP). Leukemia 2006, 20, 1061–1066. [Google Scholar] [CrossRef]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Soverini, S.; Hochhaus, A.; Nicolini, F.E.; Gruber, F.; Lange, T.; Saglio, G.; Pane, F.; Müller, M.C.; Ernst, T.; Rosti, G.; et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: Recommendations from an expert panel on behalf of European LeukemiaNet. Blood 2011, 118, 1208–1215. [Google Scholar] [CrossRef]
- Thomas, E.; Philipp, E.; Martin, C.M.; Peter, P.; Thomas, S.; Jana, H.; Sebastian, K.; Paul, R.; Rüdiger, H.; Andreas, H. Dynamics of BCR-ABL mutated clones prior to hematologic or cytogenetic resistance to imatinib. Haematologica 2008, 93, 186–192. [Google Scholar] [CrossRef][Green Version]
- Hughes, T.; Saglio, G.; Branford, S.; Soverini, S.; Kim, D.-W.; Müller, M.C.; Martinelli, G.; Cortes, J.; Beppu, L.; Gottardi, E.; et al. Impact of Baseline BCR-ABL Mutations on Response to Nilotinib in Patients with Chronic Myeloid Leukemia in Chronic Phase. J. Clin. Oncol. 2009, 27, 4204–4210. [Google Scholar] [CrossRef]
- Müller, M.C.; Cortes, J.; Kim, D.-W.; Druker, B.; Erben, P.; Pasquini, R.; Branford, S.; Hughes, T.; Radich, J.P.; Ploughman, L.; et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: Analysis of responses according to preexisting BCR-ABL mutations. Blood 2009, 114, 4944–4953. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Melo, J.V.; Hughes, T.P. Selecting optimal second-line tyrosine kinase inhibitor therapy for chronic myeloid leukemia patients after imatinib failure: Does the BCR-ABL mutation status really matter? Blood 2009, 114, 5426–5435. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.; Bornmann, W.; Pellicena, P.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Structural Mechanism for STI-571 Inhibition of Abelson Tyrosine Kinase. Science 2000, 289, 1938–1942. [Google Scholar] [CrossRef]
- Nicolini, F.E.; Mauro, M.J.; Martinelli, G.; Kim, D.-W.; Soverini, S.; Müller, M.C.; Hochhaus, A.; Cortes, J.; Chuah, C.; Dufva, I.H.; et al. Epidemiologic study on survival of chronic myeloid leukemia and Ph(+) acute lymphoblastic leukemia patients with BCR-ABL T315I mutation. Blood 2009, 114, 5271–5278. [Google Scholar] [CrossRef]
- Nicolini, F.E.; Ibrahim, A.R.; Soverini, S.; Martinelli, G.; Müller, M.C.; Hochhaus, A.; Dufva, I.H.; Kim, D.-W.; Cortes, J.; Mauro, M.J.; et al. The BCR-ABLT315I mutation compromises survival in chronic phase chronic myelogenous leukemia patients resistant to tyrosine kinase inhibitors, in a matched pair analysis. Haematologica 2013, 98, 1510–1516. [Google Scholar] [CrossRef]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.-S.; Xu, Q.; et al. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef]
- Soverini, S.; Colarossi, S.; Gnani, A.; Rosti, G.; Castagnetti, F.; Poerio, A.; Iacobucci, I.; Amabile, M.; Abruzzese, E.; Orlandi, E.; et al. Contribution of ABL Kinase Domain Mutations to Imatinib Resistance in Different Subsets of Philadelphia-Positive Patients: By the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin. Cancer Res. 2006, 12, 7374–7379. [Google Scholar] [CrossRef]
- Khorashad, J.S.; de Lavallade, H.; Apperley, J.F.; Milojkovic, D.; Reid, A.G.; Bua, M.; Szydlo, R.; Olavarria, E.; Kaeda, J.; Goldman, J.M.; et al. Finding of Kinase Domain Mutations in Patients with Chronic Phase Chronic Myeloid Leukemia Responding to Imatinib May Identify Those at High Risk of Disease Progression. J. Clin. Oncol. 2008, 26, 4806–4813. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Kantarjian, H.; O’Brien, S.; Jabbour, E.; Borthakur, G.; Ravandi, F.; Verstovsek, S.; Shan, J.; Cortes, J. Outcome of patients with chronic myeloid leukemia with multiple ABL1 kinase domain mutations receiving tyrosine kinase inhibitor therapy. Haematologica 2011, 96, 918–924. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kizilors, A.; Crisà, E.; Lea, N.; Passera, R.; Mian, S.; Anwar, J.; Best, S.; Nicolini, F.; Ireland, R.; Aldouri, M.; et al. Effect of low-level BCR-ABL1 kinase domain mutations identified by next-generation sequencing in patients with chronic myeloid leukaemia: A population-based study. Lancet Haematol. 2019, 6, e276–e284. [Google Scholar] [CrossRef]
- Parker, W.T.; Ho, M.; Scott, H.S.; Hughes, T.P.; Branford, S. Poor response to second-line kinase inhibitors in chronic myeloid leukemia patients with multiple low-level mutations, irrespective of their resistance profile. Blood 2012, 119, 2234–2238. [Google Scholar] [CrossRef] [PubMed]
- Zabriskie, M.S.; Eide, C.A.; Tantravahi, S.K.; Vellore, N.A.; Estrada, J.; Nicolini, F.E.; Khoury, H.J.; Larson, R.; Konopleva, M.; Cortes, J.; et al. BCR-ABL1 Compound Mutations Combining Key Kinase Domain Positions Confer Clinical Resistance to Ponatinib in Ph Chromosome-Positive Leukemia. Cancer Cell 2014, 26, 428–442. [Google Scholar] [CrossRef]
- Parker, W.T.; Yeung, D.T.O.; Yeoman, A.L.; Altamura, H.K.; Jamison, B.A.; Field, C.R.; Hodgson, J.G.; Lustgarten, S.; Rivera, V.M.; Hughes, T.; et al. The impact of multiple low-level BCR-ABL1 mutations on response to ponatinib. Blood 2016, 127, 1870–1880. [Google Scholar] [CrossRef]
- Parker, W.T.; Lawrence, R.M.; Ho, M.; Irwin, D.L.; Scott, H.S.; Hughes, T.P.; Branford, S. Sensitive Detection of BCR-ABL1 Mutations in Patients with Chronic Myeloid Leukemia After Imatinib Resistance Is Predictive of Outcome During Subsequent Therapy. J. Clin. Oncol. 2011, 29, 4250–4259. [Google Scholar] [CrossRef]
- Soverini, S.; Bavaro, L.; De Benedittis, C.; Martelli, M.; Iurlo, A.; Orofino, N.; Sica, S.; Sorà, F.; Lunghi, F.; Ciceri, F.; et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: The NEXT-in-CML study. Blood 2020, 135, 534–541. [Google Scholar] [CrossRef]
- Soverini, S.; De Benedittis, C.; Polakova, K.M.; Linhartova, J.; Castagnetti, F.; Gugliotta, G.; Papayannidis, C.; Mancini, M.; Klamová, H.; Salvucci, M.; et al. Next-generation sequencing for sensitive detection of BCR-ABL1 mutations relevant to tyrosine kinase inhibitor choice in imatinib-resistant patients. Oncotarget 2016, 7, 21982–21990. [Google Scholar] [CrossRef]
- Polakova, K.M.; Kulvait, V.; Benesova, A.; Linhartova, J.; Klamova, H.; Jaruskova, M.; De Benedittis, C.; Haferlach, T.; Baccarani, M.; Martinelli, G.; et al. Next-generation deep sequencing improves detection of BCR-ABL1 kinase domain mutations emerging under tyrosine kinase inhibitor treatment of chronic myeloid leukemia patients in chronic phase. J. Cancer Res. Clin. Oncol. 2014, 141, 887–899. [Google Scholar] [CrossRef]
- Erbilgin, Y.; Eskazan, A.E.; Ng, O.H.; Salihoglu, A.; Elverdi, T.; Firtina, S.; Tasar, O.; Mercan, S.; Sisko, S.; Khodzhaev, K.; et al. Deep sequencing of BCR-ABL1 kinase domain mutations in chronic myeloid leukemia patients with resistance to tyrosine kinase inhibitors. Leuk. Lymphoma 2018, 60, 200–207. [Google Scholar] [CrossRef]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutiérrez, V.; Luna, A.; Alonso-Dominguez, J.M.; Estrada, N.; Boque, C.; Xicoy, B.; Giraldo, P.; Angona, A.; Alvarez-Larrán, A.; Sanchez-Guijo, F.; et al. Safety and efficacy of asciminib treatment in chronic myeloid leukemia patients in real-life clinical practice. Blood Cancer J. 2021, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Réa, D.; Mauro, M.J.; Boquimpani, C.; Minami, Y.; Lomaia, E.; Voloshin, S.; Turkina, A.G.; Kim, D.-W.; Apperley, J.F.; Abdo, A.; et al. A phase 3, open-label, randomized study of asciminib, a STAMP inhibitor, vs bosutinib in CML after 2 or more prior TKIs. Blood 2021, 138, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Adrián, F.J.; Jahnke, W.; Cowan-Jacob, S.W.; Li, A.G.; Iacob, R.E.; Sim, T.; Powers, J.; Dierks, C.; Sun, F.; et al. Targeting Bcr–Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010, 463, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.; Minami, H.; Rea, D.; DeAngelo, D.; Breccia, M.; Goh, Y.-T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef]
- Eide, C.A.; Zabriskie, M.S.; Stevens, S.L.S.; Antelope, O.; Vellore, N.A.; Than, H.; Schultz, A.R.; Clair, P.; Bowler, A.D.; Pomicter, A.; et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 2019, 36, 431–443.e5. [Google Scholar] [CrossRef]
- Lindström, H.J.G.; Friedman, R. The effects of combination treatments on drug resistance in chronic myeloid leukaemia: An evaluation of the tyrosine kinase inhibitors axitinib and asciminib. BMC Cancer 2020, 20, 397. [Google Scholar] [CrossRef]
- Luskin, M.R.; Stevenson, K.E.; Mendez, L.M.; Wang, E.S.; Wadleigh, M.; Garcia, J.S.; Stone, R.M.; An, H.H.; Hagopian, E.; Galinsky, I.; et al. A Phase I Study of Asciminib (ABL001) in Combination with Dasatinib and Prednisone for BCR-ABL1-Positive ALL in Adults. Blood 2021, 138, 2305. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Filik, Y.; Berger, D.; Schewzik, C.; Stefanzl, G.; Sadovnik, I.; Degenfeld-Schonburg, L.; Eisenwort, G.; Schneeweiss-Gleixner, M.; Byrgazov, K.; et al. Asciminib and ponatinib exert synergistic anti-neoplastic effects on CML cells expressing BCR-ABL1 (T315I)-compound mutations. Am. J. Cancer Res. 2021, 11, 4470–4484. [Google Scholar]
- Lee, B.J.; Shah, N.P. Identification and characterization of activating ABL1 1b kinase mutations: Impact on sensitivity to ATP-competitive and allosteric ABL1 inhibitors. Leukemia 2016, 31, 1096–1107. [Google Scholar] [CrossRef]
- Branford, S.; Hughes, T.P.; Rudzki, Z. Dual transcription of b2a2 and b3a2 BCR-ABL transcripts in chronic myeloid leukaemia is confined to patients with a linked polymorphism within the BCR gene. Br. J. Haematol. 2002, 117, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Castagnetti, F.; Gugliotta, G.; Rosti, G.; Soverini, S.; Albeer, A.; Pfirrmann, M. The proportion of different BCR-ABL1 transcript types in chronic myeloid leukemia. An international overview. Leukemia 2019, 33, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Saglio, G.; Guerrasio, A.; Rosso, C.; Zaccaria, A.; Tassinari, A.; Serra, A.; Cambrin, G.R.; Mazza, U.; Gavosto, F. New type of Bcr/Abl junction in Philadelphia chromosome-positive chronic myelogenous leukemia. Blood 1990, 76, 1819–1824. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, L.; Mohanty, S.; Kochupillai, V. Response to Imatinib mesylate in chronic myeloid leukemia patients with variant BCR-ABL fusion transcripts. Ann. Hematol. 2009, 89, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Abruzzese, E.; Breccia, M. Prognostic Significance of Transcript-Type BCR-ABL1 in Chronic Myeloid Leukemia. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020062. [Google Scholar] [CrossRef]
- Marum, J.E.; Branford, S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther. Adv. Hematol. 2016, 7, 237–251. [Google Scholar] [CrossRef]
- Shepherd, P.; Suffolk, R.; Halsey, J.; Allan, N. Analysis of molecular breakpoint and m-RNA transcripts in a prospective randomized trial of interferon in chronic myeloid leukaemia: No correlation with clinical features, cytogenetic response, duration of chronic phase, or survival. Br. J. Haematol. 1995, 89, 546–554. [Google Scholar] [CrossRef]
- Schaefer-Rego, K.; Dudek, H.; Popenoe, D.; Arlin, Z.; Mears, J.; Bank, A.; Leibowitz, D. CML patients in blast crisis have breakpoints localized to a specific region of the BCR. Blood 1987, 70, 448–455. [Google Scholar] [CrossRef]
- Baccarani, M.; Tura, S.; Russo, D.; Baccarani, M.; Zaccaria, A.; Saglio, G.; Guerrasio, A.; Martinelli, G.; Zuffa, E.; Testoni, N. Chronic myeloid-leukemia, BCR/ABL transcript, response to alpha-interferon and survival. Leukemia 1995, 9, 1648–1652. [Google Scholar]
- Hanfstein, B.; Lauseker, M.; Hehlmann, R.; Saussele, S.; Erben, P.; Dietz, C.; Fabarius, A.; Proetel, U.; Schnittger, S.; Haferlach, C.; et al. Distinct characteristics of e13a2 versus e14a2 BCR-ABL1 driven chronic myeloid leukemia under first-line therapy with imatinib. Haematologica 2014, 99, 1441–1447. [Google Scholar] [CrossRef]
- Castagnetti, F.; Gugliotta, G.; Breccia, M.; Iurlo, A.; Levato, L.; Albano, F.; Vigneri, P.; Abruzzese, E.; Rossi, G.; Rupoli, S.; et al. The BCR-ABL1 transcript type influences response and outcome in Philadelphia chromosome-positive chronic myeloid leukemia patients treated frontline with imatinib. Am. J. Hematol. 2017, 92, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Harris, R.J.; Giannoudis, A.; Davies, A.; Knight, K.; Watmough, S.J.; Wang, L.; Clark, R.E. Chronic myeloid leukemia patients with the e13a2 BCR-ABL fusion transcript have inferior responses to imatinib compared to patients with the e14a2 transcript. Haematologica 2009, 94, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Pagnano, K.B.B.; Miranda, E.C.; Delamain, M.T.; Duarte, G.O.; de Paula, E.V.; Lorand-Metze, I.; de Souza, C.A. Influence of BCR-ABL Transcript Type on Outcome in Patients with Chronic-Phase Chronic Myeloid Leukemia Treated with Imatinib. Clin. Lymphoma Myeloma Leuk. 2017, 17, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Polampalli, S.; Choughule, A.; Negi, N.; Shinde, S.; Baisane, C.; Amre, P.; Subramanian, P.; Gujral, S.; Prabhash, K.; Parikh, P. Analysis and comparison of clinicohematological parameters and molecular and cytogenetic response of two Bcr/Abl fusion transcripts. Genet. Mol. Res. 2008, 7, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Kantarjian, H.; Patel, K.P.; Gonzalez, G.N.; Luthra, R.; Kanagal-Shamanna, R.; Sasaki, K.; Jabbour, E.; Romo, C.G.; Kadia, T.M.; et al. Impact of BCR-ABL transcript type on outcome in patients with chronic-phase CML treated with tyrosine kinase inhibitors. Blood 2016, 127, 1269–1275. [Google Scholar] [CrossRef]
- Breccia, M.; Molica, M.; Colafigli, G.; Massaro, F.; Quattrocchi, L.; Latagliata, R.; Mancini, M.; Diverio, D.; Guarini, A.; Alimena, G.; et al. Prognostic factors associated with a stable MR4.5 achievement in chronic myeloid leukemia patients treated with imatinib. Oncotarget 2017, 9, 7534–7540. [Google Scholar] [CrossRef]
- Shanmuganathan, N.; Pagani, I.S.; Ross, D.M.; Park, S.; Yong, A.S.M.; Braley, J.A.; Altamura, H.K.; Hiwase, D.K.; Yeung, D.T.; Kim, D.-W.; et al. Early BCR-ABL1 kinetics are predictive of subsequent achievement of treatment-free remission in chronic myeloid leukemia. Blood 2021, 137, 1196–1207. [Google Scholar] [CrossRef]
- D’Adda, M.; Farina, M.; Schieppati, F.; Borlenghi, E.; Bottelli, C.; Cerqui, E.; Ferrari, S.; Gramegna, D.; Pagani, C.; Passi, A.; et al. The e13a2 BCR-ABL transcript negatively affects sustained deep molecular response and the achievement of treatment-free remission in patients with chronic myeloid leukemia who receive tyrosine kinase inhibitors. Cancer 2019, 125, 1674–1682. [Google Scholar] [CrossRef]
- Claudiani, S.; Apperley, J.F.; Gale, R.P.; Clark, R.; Szydlo, R.; Deplano, S.; Palanicawandar, R.; Khorashad, J.; Foroni, L.; Milojkovic, D. E14a2 BCR-ABL1 transcript is associated with a higher rate of treatment-free remission in individuals with chronic myeloid leukemia after stopping tyrosine kinase inhibitor therapy. Haematologica 2017, 102, e297–e299. [Google Scholar] [CrossRef]
- Rojas, J.M.; Knight, K.; Wang, L.; Clark, R. Clinical evaluation of BCR-ABL peptide immunisation in chronic myeloid leukaemia: Results of the EPIC study. Leukemia 2007, 21, 2287–2295. [Google Scholar] [CrossRef]
- Greiner, J.; Schmitt, M. Leukemia-associated antigens as target structures for a specific immunotherapy in chronic myeloid leukemia. Eur. J. Haematol. 2008, 80, 461–468. [Google Scholar] [CrossRef]
- Dragani, M.; Petiti, J.; Rege-Cambrin, G.; Gottardi, E.; Daraio, F.; Caocci, G.; Aguzzi, C.; Crisà, E.; Andreani, G.; Caciolli, F.; et al. Treatment-free remission in Chronic Myeloid Leukemia harboring atypical BCR-ABL1 transcripts. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020066. [Google Scholar] [CrossRef]
- Pagani, I.S.; Dang, P.; Saunders, V.A.; Braley, J.; Thieleke, A.; Branford, S.; Hughes, T.P.; Ross, D.M. Clinical utility of genomic DNA Q-PCR for the monitoring of a patient with atypical e19a2 BCR-ABL1 transcripts in chronic myeloid leukemia. Leuk. Lymphoma 2020, 61, 2527–2529. [Google Scholar] [CrossRef]
- Huret, J.L. Complex translocations, simple variant translocations and Ph-negative cases in chronic myelogenous leukaemia. Qual. Life Res. 1990, 85, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Gorusu, M.; Benn, P.; Li, Z.; Fang, M. On the genesis and prognosis of variant translocations in chronic myeloid leukemia. Cancer Genet. Cytogenet. 2007, 173, 97–106. [Google Scholar] [CrossRef]
- Marzocchi, G.; Castagnetti, F.; Luatti, S.; Baldazzi, C.; Stacchini, M.; Gugliotta, G.; Amabile, M.; Specchia, G.; Sessarego, M.; Giussani, U.; et al. Variant Philadelphia translocations: Molecular-cytogenetic characterization and prognostic influence on frontline imatinib therapy, a GIMEMA Working Party on CML analysis. Blood 2011, 117, 6793–6800. [Google Scholar] [CrossRef]
- Fitzgerald, P.H.; Morris, C.M. Complex chromosomal translocations in the Philadelphia chromosome leukemias: Serial translocations or a concerted genomic rearrangement? Cancer Genet. Cytogenet. 1991, 57, 143–151. [Google Scholar] [CrossRef]
- Naumann, S.; Decker, H.-J. Genesis of variant philadelphia chromosome translocations in chronic myelocytic leukemia. Cancer Genet. Cytogenet. 2003, 147, 18–22. [Google Scholar] [CrossRef]
- Calabrese, G.; Stuppia, L.; Franchi, P.G.; Peila, R.; Morizio, E.; Liberati, A.M.; Spadano, A.; Di Lorenzo, R.; Donti, E.; Antonucci, A.; et al. Complex translocations of the Ph chromosome and Ph negative CML arise from similar mechanisms, as evidenced by FISH analysis. Cancer Genet. Cytogenet. 1994, 78, 153–159. [Google Scholar] [CrossRef]
- Heim, S.; Billström, R.; Kristoffersson, U.; Mandahl, N.; Strömbeck, B.; Mitelman, F. Variant Ph translocations in chronic myeloid leukemia. Cancer Genet. Cytogenet. 1985, 18, 215–227. [Google Scholar] [CrossRef]
- Sinclair, P.B.; Nacheva, E.P.; Leversha, M.; Telford, N.; Chang, J.; Reid, A.; Bench, A.; Champion, K.; Huntly, B.; Green, A.R. Large deletions at the t(9;22) breakpoint are common and may identify a poor-prognosis subgroup of patients with chronic myeloid leukemia. Blood 2000, 95, 738–743. [Google Scholar] [CrossRef] [PubMed]
- El-Zimaity, M.M.T.; Kantarjian, H.; Talpaz, M.; O’Brien, S.; Giles, F.; Garcia-Manero, G.; Verstovsek, S.; Thomas, D.; Ferrajoli, A.; Hayes, K.; et al. Results of imatinib mesylate therapy in chronic myelogenous leukaemia with variant Philadelphia chromosome. Br. J. Haematol. 2004, 125, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Huntly, B.; Reid, A.G.; Bench, A.J.; Campbell, L.J.; Telford, N.; Shepherd, P.; Szer, J.; Prince, H.M.; Turner, P.; Grace, C.; et al. Deletions of the derivative chromosome 9 occur at the time of the Philadelphia translocation and provide a powerful and independent prognostic indicator in chronic myeloid leukemia. Blood 2001, 98, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Bennour, A.; Sennana, H.; Laatiri, M.A.; Elloumi, M.; Khelif, A.; Saad, A. Molecular cytogenetic characterization of variant Philadelphia translocations in chronic myeloid leukemia: Genesis and deletion of derivative chromosome 9. Cancer Genet. Cytogenet. 2009, 194, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.G.; Huntly, B.; Grace, C.; Green, A.; Nacheva, E.P. Survival implications of molecular heterogeneity in variant Philadelphia-positive chronic myeloid leukaemia. Br. J. Haematol. 2003, 121, 419–427. [Google Scholar] [CrossRef]
- Potter, A.M.; Watmore, A.E.; Cooke, P.; Lilleyman, J.S.; Sokol, R.J. Significance of non-standard Philadelphia chromosomes in chronic granulocytic leukaemia. Br. J. Cancer 1981, 44, 51–54. [Google Scholar] [CrossRef]
- Kanakasetty, G.B.; Kuntejowdahalli, L.; Thanky, A.H.; Dasappa, L.; Jacob, L.A.; Mallekavu, S.B.; Kumari, P. Predictive and Prognostic Implications of Variant Philadelphia Translocations in CML: Experience from a Tertiary Oncology Center in Southern India. Clin. Lymphoma Myeloma Leuk. 2016, 17, 52–59. [Google Scholar] [CrossRef]
- Koshiyama, D.B.; Capra, M.E.Z.; Paskulin, G.A.; Rosa, R.F.M.; Oliveira, C.A.V.; Vanelli, T.; Fogliatto, L.M.; Zen, P.R.G. Cytogenetic response to imatinib treatment in Southern Brazilian patients with chronic myelogenous leukemia and variant Philadelphia chromosome. Ann. Hematol. 2012, 92, 185–189. [Google Scholar] [CrossRef]
- Fabarius, A.; Leitner, A.; Hochhaus, A.; Müller, M.C.; Hanfstein, B.; Haferlach, C.; Göhring, G.; Schlegelberger, B.; Jotterand, M.; Reiter, A.; et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: Long-term observation of 1151 patients from the randomized CML Study IV. Blood 2011, 118, 6760–6768. [Google Scholar] [CrossRef]
- Eyüpoğlu, D.; Bozkurt, S.; Haznedaroğlu, I.; Büyükaşık, Y.; Güven, D. The Impact of Variant Philadelphia Chromosome Translocations on the Clinical Course of Chronic Myeloid Leukemia. Turk. J. Haematol. 2016, 33, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Richebourg, S.; Eclache, V.; Perot, C.; Portnoi, M.-F.; Akker, J.V.D.; Terre, C.; Maareck, O.; Soenen, V.; Viguié, F.; Lai, J.-L.; et al. Mechanisms of genesis of variant translocation in chronic myeloid leukemia are not correlated with ABL1 or BCR deletion status or response to imatinib therapy. Cancer Genet. Cytogenet. 2008, 182, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Stagno, F.; Vigneri, P.; Del Fabro, V.; Stella, S.; Cupri, A.; Massimino, M.; Consoli, C.; Tambè, L.; Consoli, M.L.; Antolino, A.; et al. Influence of complex variant chromosomal translocations in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Acta Oncol. 2010, 49, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Al-Achkar, W.; Liehr, T.; Wafa, A. Insertion of the 3′ ABL region into the long arm of chromosome 1 in a Philadelphia chromosome-negative chronic myeloid leukemia case. Oncol. Lett. 2010, 1, 951–954. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seong, D.; Kantarjian, H.; Albitar, M.; Arlinghaus, R.; Xu, J.; Talpaz, M.; Rios, M.B.; Guo, J.Q.; O’Brien, S.; Siciliano, M. Analysis of Philadelphia chromosome-negative BCR-ABL-positive chronic myelogenous leukemia by hypermetaphase fluorescence in situ hybridization. Ann. Oncol. 1999, 10, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.H.; Beard, M.E.J.; Morris, C.M.; Heaton, D.C.; Reeve, A.E. Ph-negative chronic myeloid leukaemia. Br. J. Haematol. 1987, 66, 311–314. [Google Scholar] [CrossRef]
- Van Der Plas, D.; Hermans, A.; Soekarman, D.; Smit, E.; De Klein, A.; Smadja, N.; Alimena, G.; Goudsmit, R.; Grosveld, G.; Hagemeijer, A. Cytogenetic and molecular analysis in Philadelphia negative CML. Blood 1989, 73, 1038–1044. [Google Scholar] [CrossRef]
- Virgili, A.; Brazma, D.; Reid, A.G.; Howard-Reeves, J.; Valgañón, M.; Chanalaris, A.; De Melo, V.A.; Marin, D.; Apperley, J.F.; Grace, C.; et al. FISH mapping of Philadelphia negative BCR/ABL1 positive CML. Mol. Cytogenet. 2008, 1, 14. [Google Scholar] [CrossRef]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Huntly, B.J.P.; Bench, A.; Green, A.R. Double jeopardy from a single translocation: Deletions of the derivative chromosome 9 in chronic myeloid leukemia. Blood 2003, 102, 1160–1168. [Google Scholar] [CrossRef][Green Version]
- Quintás-Cardama, A.; Kantarjian, H.; Shan, J.; Jabbour, E.; Abruzzo, L.V.; Verstovsek, S.; Garcia-Manero, G.; O’Brien, S.; Cortes, J. Prognostic impact of deletions of derivative chromosome 9 in patients with chronic myelogenous leukemia treated with nilotinib or dasatinib. Cancer 2011, 117, 5085–5093. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, M.; Wang, X.; Ren, Y.; Kim, Y.M.; Wang, X.; Lu, X.; Pang, H.; Liu, G.; Gu, Y.; et al. Genomic Copy Number Variants in CML Patients with the Philadelphia Chromosome (Ph+): An Update. Front. Genet. 2021, 12, 697009. [Google Scholar] [CrossRef] [PubMed]
- Kolomietz, E.; Al-Maghrabi, J.; Brennan, S.; Karaskova, J.; Minkin, S.; Lipton, J.; Squire, J.A. Primary chromosomal rearrangements of leukemia are frequently accompanied by extensive submicroscopic deletions and may lead to altered prognosis. Blood 2001, 97, 3581–3588. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Kantarjian, H.; Talpaz, M.; O’Brien, S.; Garcia-Manero, G.; Verstovsek, S.; Rios, M.B.; Hayes, K.; Glassman, A.; Bekele, B.N.; et al. Imatinib mesylate therapy may overcome the poor prognostic significance of deletions of derivative chromosome 9 in patients with chronic myelogenous leukemia. Blood 2005, 105, 2281–2286. [Google Scholar] [CrossRef]
- Castagnetti, F.; Testoni, N.; Luatti, S.; Marzocchi, G.; Mancini, M.; Kerim, S.; Giugliano, E.; Albano, F.; Cuneo, A.; Abruzzese, E.; et al. Deletions of the Derivative Chromosome 9 Do Not Influence the Response and the Outcome of Chronic Myeloid Leukemia in Early Chronic Phase Treated with Imatinib Mesylate: GIMEMA CML Working Party Analysis. J. Clin. Oncol. 2010, 28, 2748–2754. [Google Scholar] [CrossRef]
- Baccarani, M.; Saglio, G.; Goldman, J.; Hochhaus, A.; Simonsson, B.; Appelbaum, F.; Apperley, J.; Cervantes, F.; Cortes, J.; Deininger, M.; et al. Evolving concepts in the management of chronic myeloid leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2006, 108, 1809–1820. [Google Scholar] [CrossRef]
- Švabek, T.; Josipović, M.; Horvat, I.; Zadro, R.; Davidović-Mrsić, S. The incidence of atypical patterns ofBCR-ABL1rearrangement and molecular-cytogenetic response to tyrosine kinase inhibitor therapy in newly diagnosed cases with chronic myeloid leukemia (CML). Blood Res. 2018, 53, 152–159. [Google Scholar] [CrossRef]
- Bacher, U.; Schnittger, S.; Kern, W.; Hiddemann, W.; Haferlach, T.; Schoch, C. The incidence of submicroscopic deletions in reciprocal translocations is similar in acute myeloid leukemia, BCR-ABL positive acute lymphoblastic leukemia, and chronic myeloid leukemia. Haematologica 2005, 90, 558–559. [Google Scholar]
- Moon, H.W.; Chang, Y.H.; Kim, T.Y.; Oh, B.R.; Min, H.C.; Kim, B.K.; Ahn, H.S.; Cho, H.I.; Lee, D.S. Incidence of submicroscopic deletions vary according to disease entities and chromosomal translocations in hematologic malignancies: Investigation by fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2007, 175, 166–168. [Google Scholar] [CrossRef]
- Kim, J.C.; Zuzarte, P.C.; Murphy, T.; Chan-Seng-Yue, M.; Brown, A.M.K.; Krzyzanowski, P.M.; Smith, A.C.; Notta, F.; Minden, M.D.; McPherson, J.D. Cryptic genomic lesions in adverse-risk acute myeloid leukemia identified by integrated whole genome and transcriptome sequencing. Leukemia 2019, 34, 306–311. [Google Scholar] [CrossRef]
- Ma, E.; Wan, T.S.; Au, C.H.; Ho, D.N.; Ma, S.Y.; Ng, M.H.; Chan, T.L. Next-generation sequencing and molecular cytogenetic characterization of ETV6-LYN fusion due to chromosomes 1, 8 and 12 rearrangement in acute myeloid leukemia. Cancer Genet. 2017, 218-219, 15–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ishii, Y.; Sashida, G.; Takaku, T.-I.; Sumi, M.; Nakajima, A.; Ohyashiki, K. Cryptic chromosomal anomaly in a patient with acute myeloid leukemia leading to AML1/ETO fusion with unfavorable prognostic factors. Cancer Genet. Cytogenet. 2005, 160, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ding, L.; Liang, J.; Tang, Y.; Xue, H.; Gu, L.; Shen, S.; Li, B.; Chen, J. Relatively favorable prognosis for MLL -rearranged childhood acute leukemia with reciprocal translocations. Pediatr. Blood Cancer 2018, 65, e27266. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.D.; de Borja, R.; Young, M.D.; Fuligni, F.; Rosic, A.; Roberts, N.D.; Hajjar, S.; Layeghifard, M.; Novokmet, A.; Kowalski, P.E.; et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science 2018, 361, eaam8419. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M. Chromoplexy: A New Category of Complex Rearrangements in the Cancer Genome. Cancer Cell 2013, 23, 567–569. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; Macdonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Kim, T.; Tyndel, M.S.; Kim, H.J.; Ahn, J.-S.; Choi, S.H.; Park, H.J.; Kim, Y.-K.; Kim, S.Y.; Lipton, J.H.; Zhang, Z.; et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood 2017, 129, 38–47. [Google Scholar] [CrossRef]
- Awad, S.A.; Kankainen, M.; Ojala, T.; Koskenvesa, P.; Eldfors, S.; Ghimire, B.; Kumar, A.; Kytölä, S.; Kamel, M.M.; Heckman, C.A.; et al. Mutation accumulation in cancer genes relates to nonoptimal outcome in chronic myeloid leukemia. Blood Adv. 2020, 4, 546–559. [Google Scholar] [CrossRef]
- Wu, W.; Xu, N.; Zhou, X.; Liu, L.; Tan, Y.; Luo, J.; Huang, J.; Qin, J.; Wang, J.; Li, Z.; et al. Integrative Genomic Analysis Reveals Cancer-Associated Gene Mutations in Chronic Myeloid Leukemia Patients with Resistance or Intolerance to Tyrosine Kinase Inhibitor. OncoTargets Ther. 2020, 13, 8581–8591. [Google Scholar] [CrossRef]
- Ko, T.K.; Javed, A.; Lee, K.L.; Pathiraja, T.N.; Liu, X.; Malik, S.; Soh, S.X.; Heng, X.T.; Takahashi, N.; Tan, J.H.J.; et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood 2020, 135, 2337–2353. [Google Scholar] [CrossRef]
- Magistroni, V.; Mauri, M.; D’Aliberti, D.; Mezzatesta, C.; Crespiatico, I.; Nava, M.; Fontana, D.; Sharma, N.; Parker, W.; Schreiber, A.; et al. De novo UBE2A mutations are recurrently acquired during chronic myeloid leukemia progression and interfere with myeloid differentiation pathways. Haematologica 2019, 104, 1789–1797. [Google Scholar] [CrossRef]
- Mangan, J.K.; Speck, N.A. RUNX1 Mutations in Clonal Myeloid Disorders: From Conventional Cytogenetics to Next Generation Sequencing, A Story 40 Years in the Making. Crit. Rev. Oncog. 2011, 16, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Osato, M.; Asou, N.; Abdalla, E.; Hoshino, K.; Yamasaki, H.; Okubo, T.; Suzushima, H.; Takatsuki, K.; Kanno, T.; Shigesada, K.; et al. Biallelic and Heterozygous Point Mutations in the Runt Domain of the AML1/PEBP2α B Gene Associated with Myeloblastic Leukemias. Blood J. Am. Soc. Hematol. 1999, 93, 1817–1824. [Google Scholar]
- Grossmann, V.; Kohlmann, A.; Zenger, M.; Schindela, S.; Eder, C.; Weissmann, S.; Schnittger, S.; Kern, W.; Muller, M.C.; Hochhaus, A.; et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia 2011, 25, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Ochi, Y.; Yoshida, K.; Huang, Y.-J.; Kuo, M.-C.; Nannya, Y.; Sasaki, K.; Mitani, K.; Hosoya, N.; Hiramoto, N.; Ishikawa, T.; et al. Clonal evolution and clinical implications of genetic abnormalities in blastic transformation of chronic myeloid leukaemia. Nat. Commun. 2021, 12, 283. [Google Scholar] [CrossRef] [PubMed]
- Awad, S.A.; Dufva, O.; Ianevski, A.; Ghimire, B.; Koski, J.; Maliniemi, P.; Thomson, D.; Schreiber, A.; Heckman, C.A.; Koskenvesa, P.; et al. RUNX1 mutations in blast-phase chronic myeloid leukemia associate with distinct phenotypes, transcriptional profiles, and drug responses. Leukemia 2020, 35, 1087–1099. [Google Scholar] [CrossRef]
- Roche-Lestienne, C.; Marceau, A.; Labis, E.; Nibourel, O.; Coiteux, V.; Guilhot, J.; Legros, L.; Nicolini, F.; Rousselot, P.; Gardembas, M.; et al. Mutation analysis of TET2, IDH1, IDH2 and ASXL1 in chronic myeloid leukemia. Leukemia 2011, 25, 1661–1664. [Google Scholar] [CrossRef][Green Version]
- Adnan-Awad, S.; Kankainen, M.; Mustjoki, S. Mutational landscape of chronic myeloid leukemia: More than a single oncogene leukemia. Leuk. Lymphoma 2021, 62, 2064–2078. [Google Scholar] [CrossRef]
- Branford, S.; Kim, D.D.H.; Apperley, J.F.; Eide, C.A.; Mustjoki, S.; Ong, S.T.; Nteliopoulos, G.; Ernst, T.; Chuah, C.; Gambacorti-Passerini, C.; et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia 2019, 33, 1835–1850. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Qin, Y.; Gale, R.P.; Huang, X.; Jiang, Q. Correlations between Mutations in Cancer-Related Genes, Therapy Responses and Outcomes of the 3 rd Generation Tyrosine Kinase-Inhibitor (TKI) in Persons with Chronic Myeloid Leukemia Failing Prior TKI-Therapy. Blood 2021, 138, 308. [Google Scholar] [CrossRef]
- Xue, M.; Zeng, Z.; Wang, Q.; Wen, L.; Xu, Y.; Xie, J.; Wang, Q.; Ruan, C.; Wu, D.; Chen, S. Mutational Profiles during the Progression of Chronic Myeloid Leukemia. Blood 2021, 138, 3596. [Google Scholar] [CrossRef]
- Thomson, D.W.; Shahrin, N.H.; Wang, P.P.S.; Wadham, C.; Shanmuganathan, N.; Scott, H.S.; Dinger, M.E.; Hughes, T.P.; Schreiber, A.; Branford, S. Aberrant RAG-mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia 2020, 34, 2051–2063. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Study | BCR::ABL1 Kinase Domain Mutations Detected in Preclinical Studies | BCR::ABL1 Kinase Domain Mutations Detected in Patient Samples (Hughes et al. [46]) |
|---|---|---|
| Lee and Shah [51] | A337V | A337T G463D P465S V468F I502L |
| P465S | ||
| V468F | ||
| Wylie et al. [7] | A337V | |
| P465S | ||
| V468F | ||
| I502L | ||
| Eide et al. [47] | A344P | |
| Y353C | ||
| P465S |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, A.; Shanmuganathan, N.; Branford, S. Genomic Mechanisms Influencing Outcome in Chronic Myeloid Leukemia. Cancers 2022, 14, 620. https://doi.org/10.3390/cancers14030620
Fernandes A, Shanmuganathan N, Branford S. Genomic Mechanisms Influencing Outcome in Chronic Myeloid Leukemia. Cancers. 2022; 14(3):620. https://doi.org/10.3390/cancers14030620
Chicago/Turabian StyleFernandes, Adelina, Naranie Shanmuganathan, and Susan Branford. 2022. "Genomic Mechanisms Influencing Outcome in Chronic Myeloid Leukemia" Cancers 14, no. 3: 620. https://doi.org/10.3390/cancers14030620
APA StyleFernandes, A., Shanmuganathan, N., & Branford, S. (2022). Genomic Mechanisms Influencing Outcome in Chronic Myeloid Leukemia. Cancers, 14(3), 620. https://doi.org/10.3390/cancers14030620