
Regulatory T Cell as a Biomarker of Treatment-Free Remission in Patients with Chronic Myeloid Leukemia

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
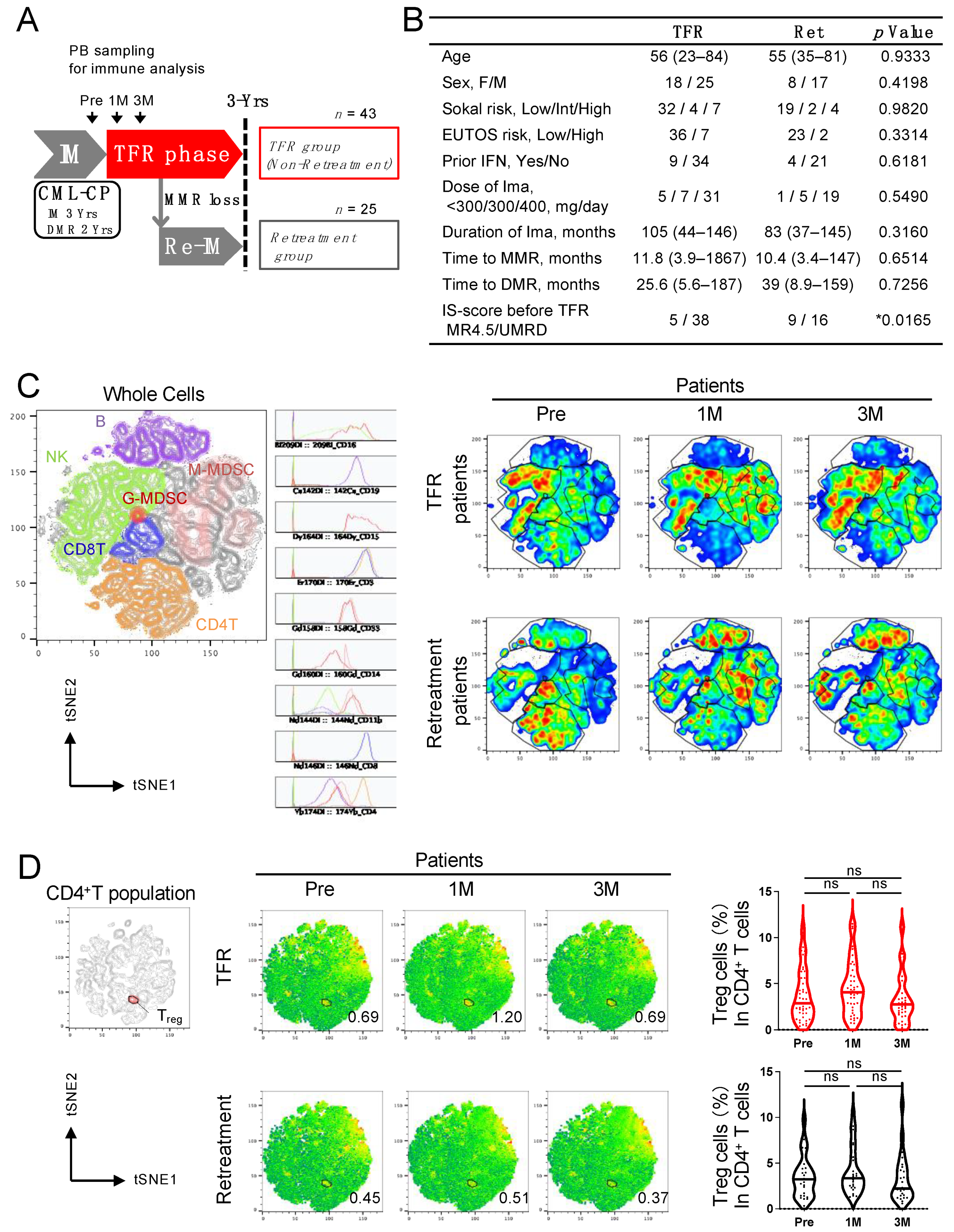
2.1. Patients and Samples
2.2. Mass Cytometry
2.3. Flow Cytometry (FCM)
2.4. ELISA
2.5. Imatinib Concentration
2.6. Cell Culture
2.7. Statistical Analysis
3. Results
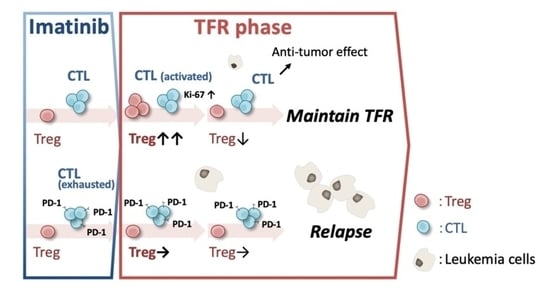
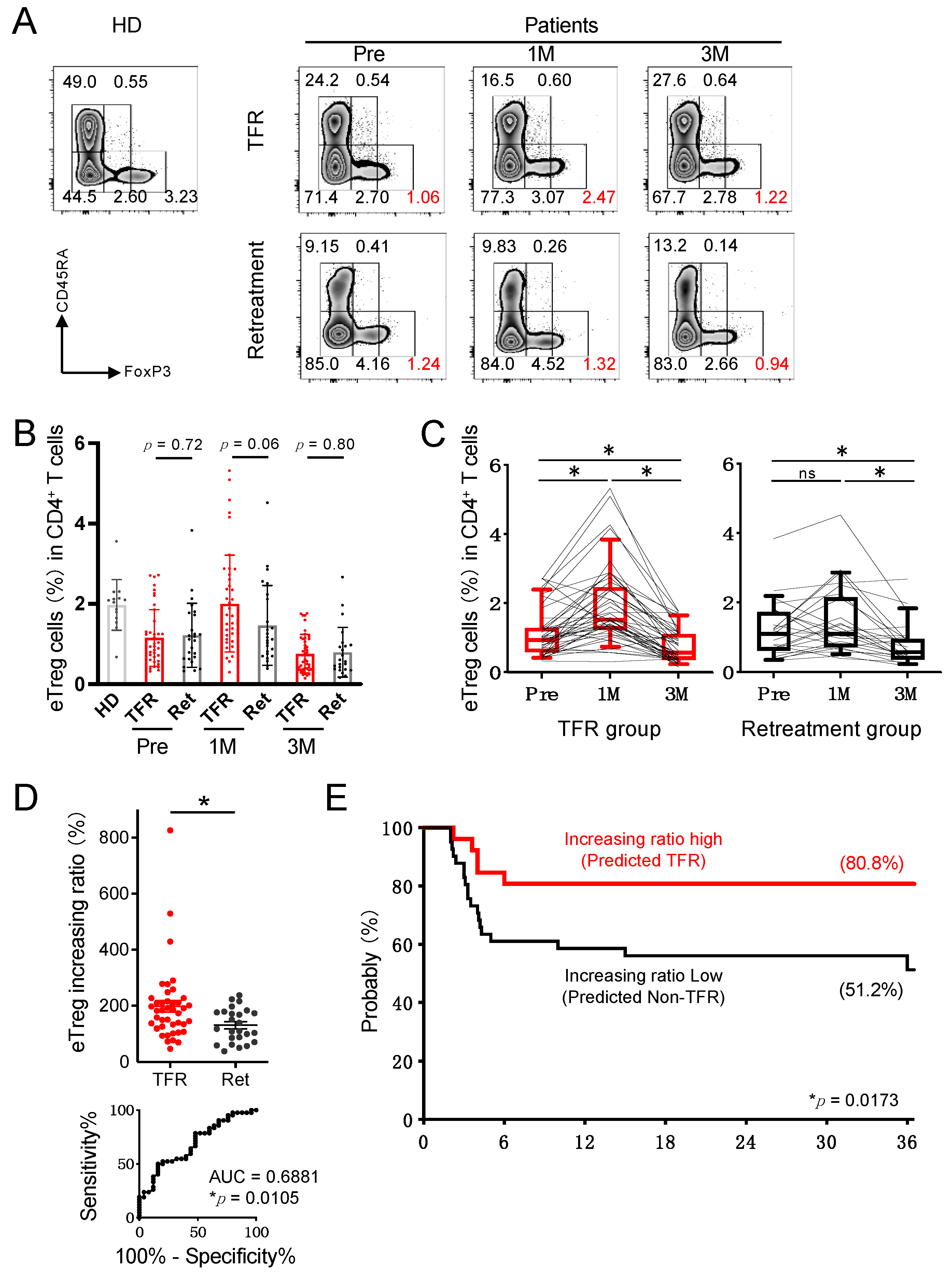
3.1. Analysis of Peripheral Blood in CML Patients Revealed That Treg Cells in the TFR Group Increased 1 Month after Discontinuation of Imatinib
3.2. Treg Variability after Discontinuation of Imatinib May Serve as a Biomarker for TFR Maintenance
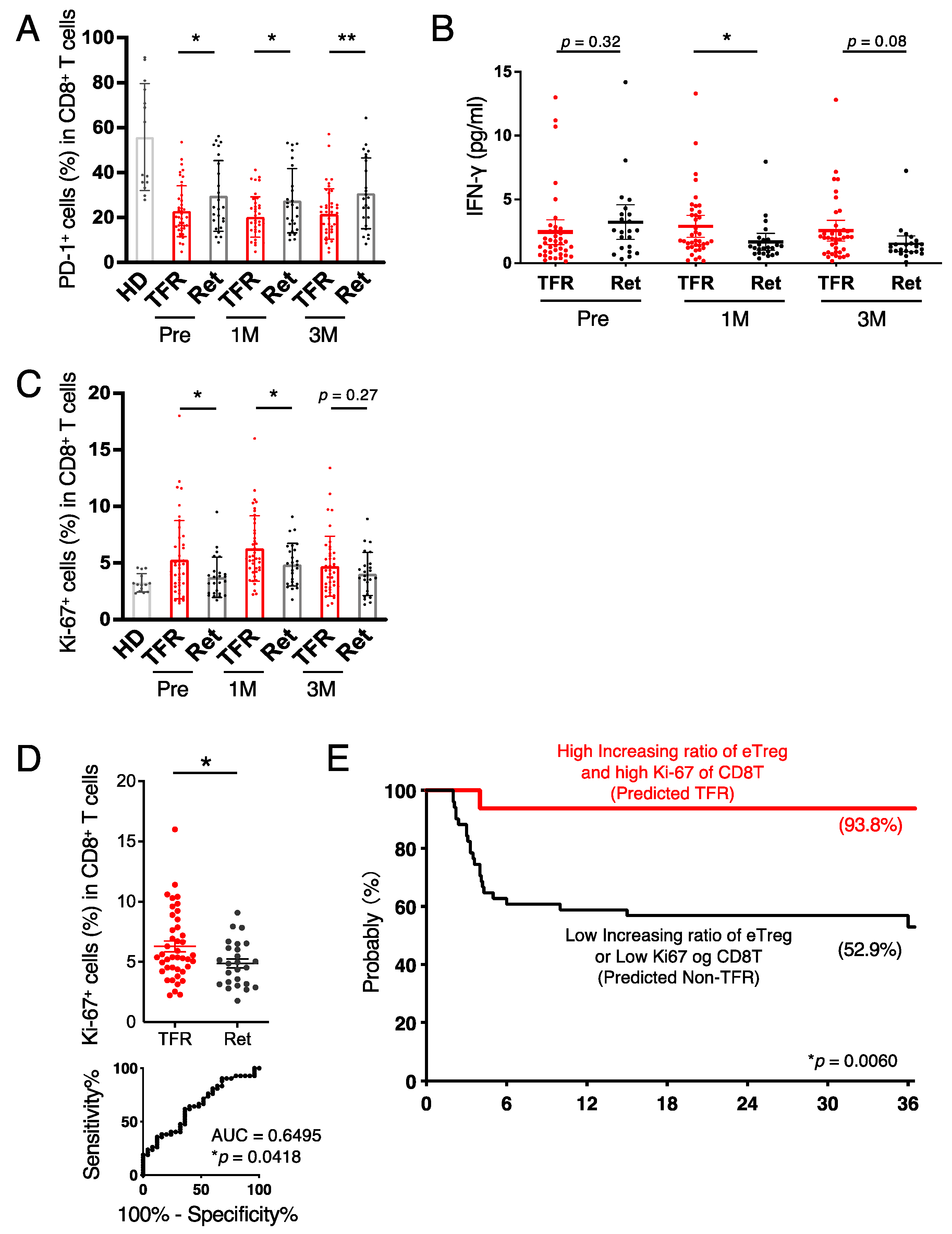
3.3. CD8+ T Cells in the TFR Group Were Activated after Discontinutaion of Imatinib
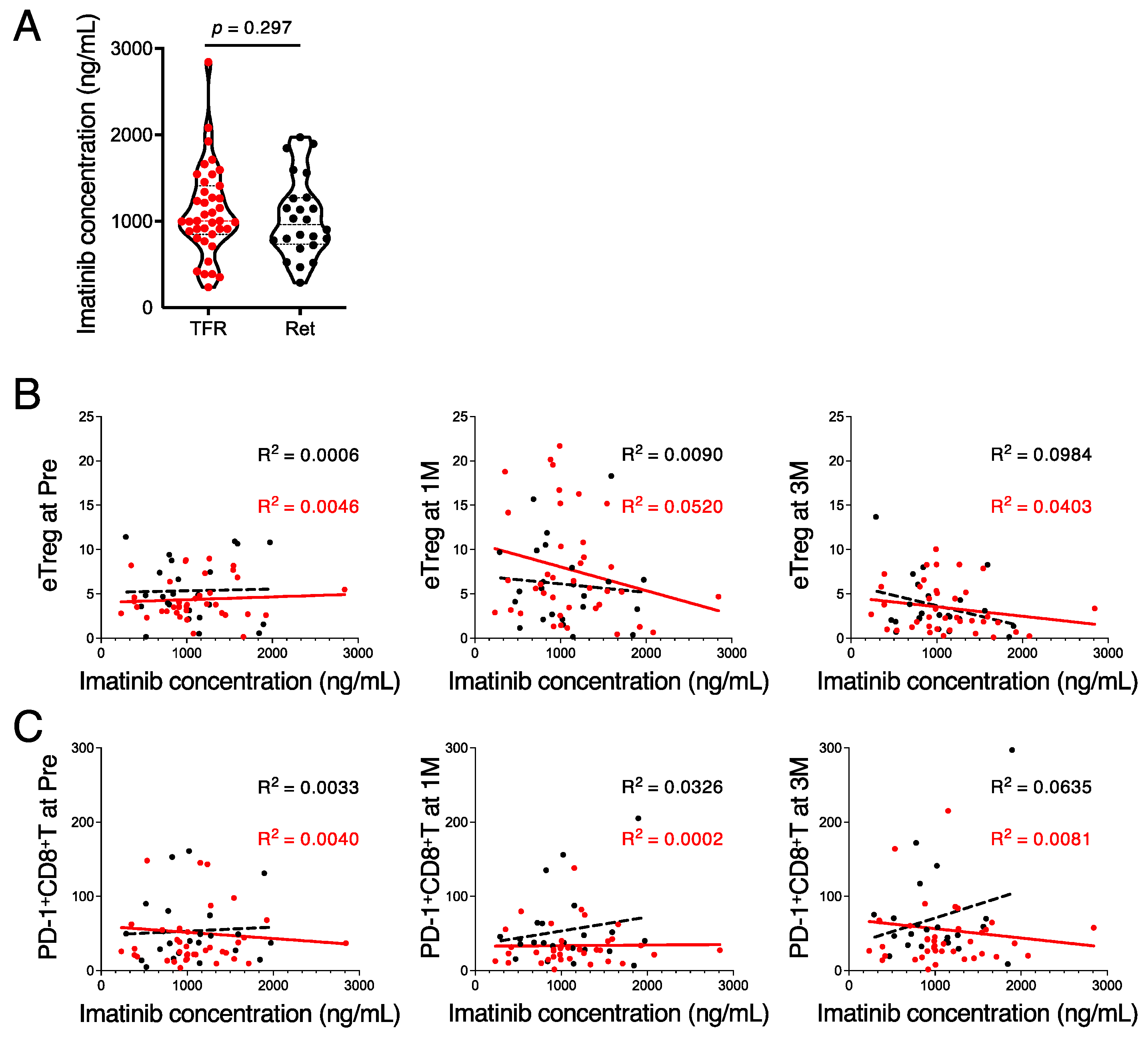
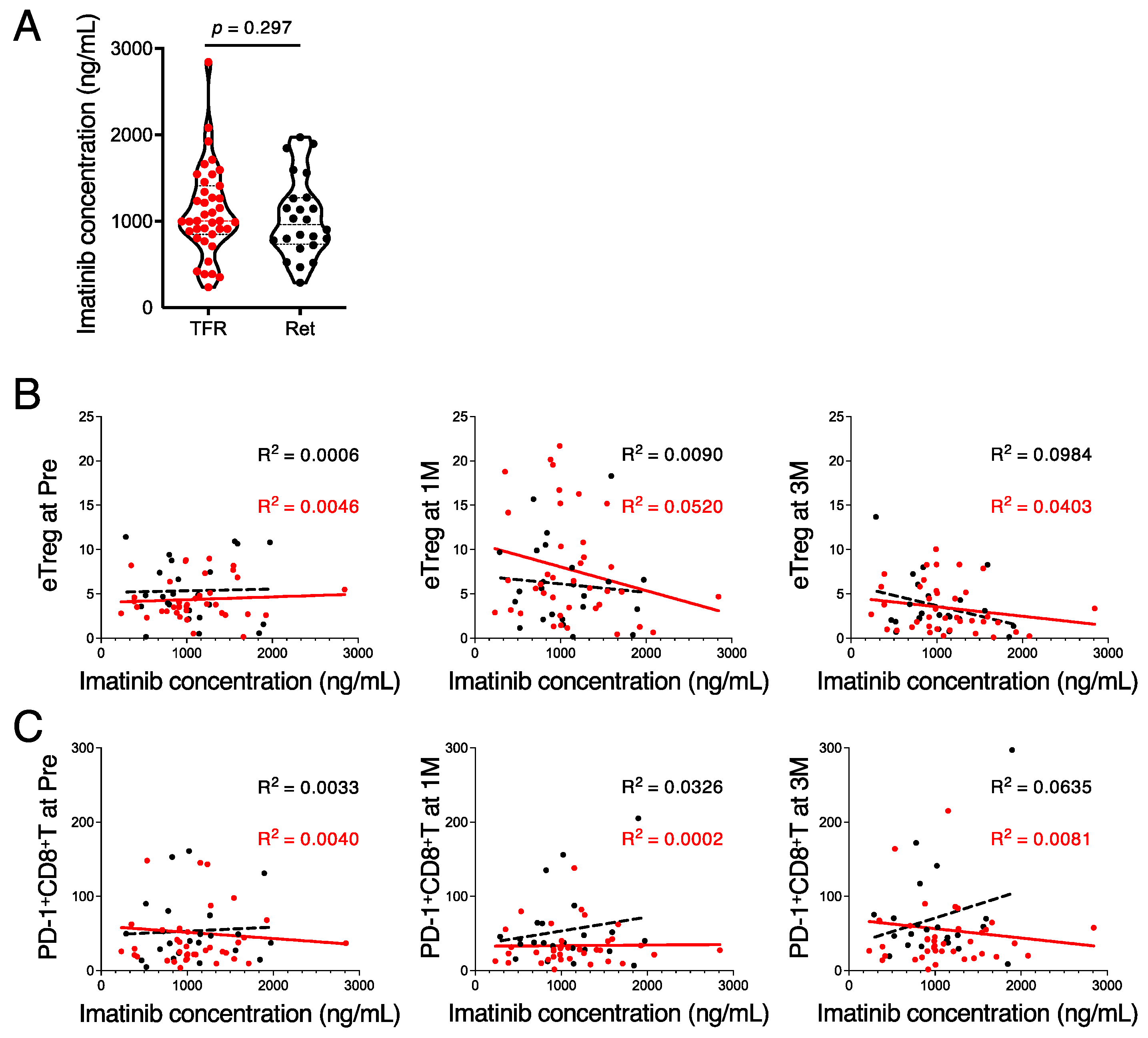
3.4. High Imatinib Concentrations Suppress the Immune Response during the TFR Phase
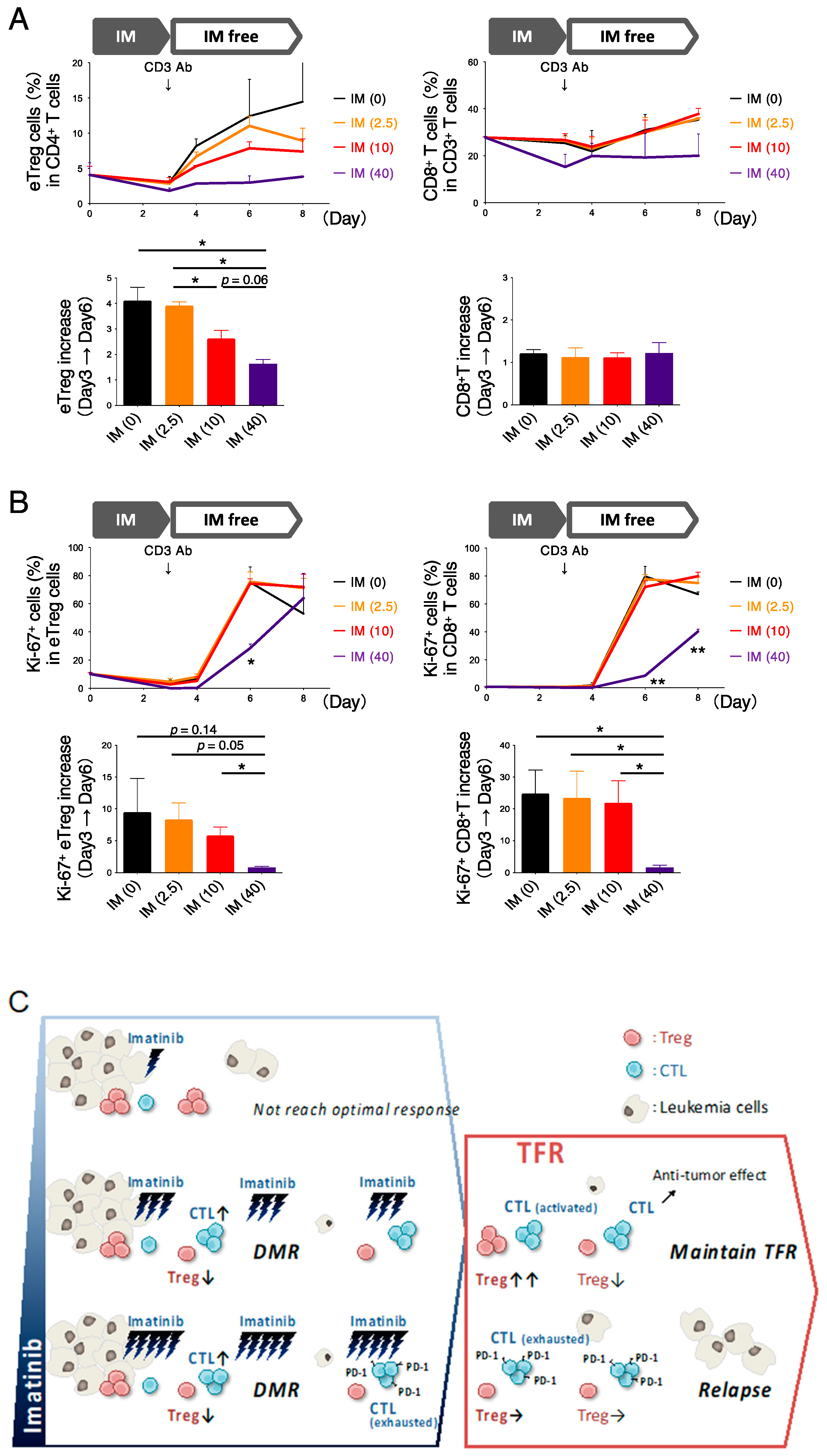
3.5. In Vitro Exposure to High Imatinib Concentrations Suppresses Treg and CD8+ T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Kizaki, M.; Okamoto, S.; Tauchi, T.; Tanaka, H.; Tanimoto, M.; Inokuchi, K.; Murayama, T.; Saburi, Y.; Hino, M.; Tsudo, M.; et al. Current and future perspectives on the TARGET system: The registration system for Glivec established by the JSH. Int. J. Hematol. 2008, 88, 409–417. [Google Scholar] [CrossRef]
- Tauchi, T.; Kizaki, M.; Okamoto, S.; Tanaka, H.; Tanimoto, M.; Inokuchi, K.; Murayama, T.; Saburi, Y.; Hino, M.; Tsudo, M.; et al. Seven-year follow-up of patients receiving imatinib for the treatment of newly diagnosed chronic myelogenous leukemia by the TARGET system. Leuk. Res. 2011, 35, 585–590. [Google Scholar] [CrossRef]
- Mahon, F.-X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Rousselot, P.; Charbonnier, A.; Cony-Makhoul, P.; Agape, P.; Nicolini, F.E.; Varet, B.; Gardembas, M.; Etienne, G.; Rea, D.; Roy, L.; et al. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J. Clin. Oncol. 2014, 32, 424–430. [Google Scholar] [CrossRef]
- Rea, D.; Nicolini, F.E.; Tulliez, M.; Guilhot, F.; Guilhot, J.; Guerci-Bresler, A.; Gardembas, M.; Coiteux, V.; Guillerm, G.; Legros, L.; et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: Interim analysis of the STOP 2G-TKI study. Blood 2017, 129, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Saussele, S.; Richter, J.; Guilhot, J.; Gruber, F.X.; Hjorth-Hansen, H.; Almeida, A.; Janssen, J.J.W.M.; Mayer, J.; Koskenvesa, P.; Panayiotidis, P.; et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): A prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018, 19, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Imagawa, J.; Tanaka, H.; Okada, M.; Nakamae, H.; Hino, M.; Murai, K.; Ishida, Y.; Kumagai, T.; Sato, S.; Ohashi, K.; et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): A multicentre phase 2 trial. Lancet Haematol. 2015, 2, e528–e535. [Google Scholar] [CrossRef]
- Takahashi, N.; Tauchi, T.; Kitamura, K.; Miyamura, K.; Saburi, Y.; Hatta, Y.; Miyata, Y.; Kobayashi, S.; Usuki, K.; Matsumura, I.; et al. Deeper molecular response is a predictive factor for treatment-free remission after imatinib discontinuation in patients with chronic phase chronic myeloid leukemia: The JALSG-STIM213 study. Int. J. Hematol. 2018, 107, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Kirschner, K.; Copland, M. Improving outcomes in chronic myeloid leukemia through harnessing the immunological landscape. Leukemia 2021, 35, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nishikawa, H.; Noguchi, S.; Sugiyama, D.; Morikawa, H.; Takeuchi, Y.; Ha, D.; Shigeta, N.; Kitawaki, T.; Maeda, Y.; et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory T cells. J. Exp. Med. 2020, 217, e20191009. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.; Clarson, J.; Tang, C.; Vidovic, L.; White, D.L.; Hughes, T.P.; Yong, A.S. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood 2017, 129, 1166–1176. [Google Scholar] [CrossRef]
- Kok, C.H.; Yeung, D.T.; Lu, L.; Watkins, D.B.; Leclercq, T.M.; Dang, P.; Saunders, V.A.; Reynolds, J.; White, D.L.; Hughes, T.P. Gene expression signature that predicts early molecular response failure in chronic-phase CML patients on frontline imatinib. Blood Adv. 2019, 3, 1610–1621. [Google Scholar] [CrossRef] [Green Version]
- Ilander, M.; Olsson-Stromberg, U.; Schlums, H.; Guilhot, J.; Bruck, O.; Lahteenmaki, H.; Kasanen, T.; Koskenvesa, P.; Soderlund, S.; Hoglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2017, 31, 1108–1116. [Google Scholar] [CrossRef]
- Rea, D.; Henry, G.; Khaznadar, Z.; Etienne, G.; Guilhot, F.; Nicolini, F.; Guilhot, J.; Rousselot, P.; Huguet, F.; Legros, L.; et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: The IMMUNOSTIM study. Haematologica 2017, 102, 1368–1377. [Google Scholar] [CrossRef]
- Okada, M.; Imagawa, J.; Tanaka, H.; Nakamae, H.; Hino, M.; Murai, K.; Ishida, Y.; Kumagai, T.; Sato, S.; Ohashi, K.; et al. Final 3-year Results of the Dasatinib Discontinuation Trial in Patients With Chronic Myeloid Leukemia Who Received Dasatinib as a Second-line Treatment. Clin. Lymphoma Myeloma Leuk. 2018, 18, 353–360.e351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irani, Y.D.; Hughes, A.; Clarson, J.; Kok, C.H.; Shanmuganathan, N.; White, D.L.; Yeung, D.T.; Ross, D.M.; Hughes, T.P.; Yong, A.S.M. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. Br. J. Haematol. 2020, 191, 433–441. [Google Scholar] [CrossRef]
- Schutz, C.; Inselmann, S.; Saussele, S.; Dietz, C.T.; Mu Ller, M.C.; Eigendorff, E.; Brendel, C.A.; Metzelder, S.K.; Bru Mmendorf, T.H.; Waller, C.; et al. Expression of the CTLA-4 ligand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of disease recurrence after treatment discontinuation in CML. Leukemia 2017, 31, 829–836. [Google Scholar] [CrossRef]
- Cayssials, E.; Jacomet, F.; Piccirilli, N.; Lefevre, L.; Roy, L.; Guilhot, F.; Chomel, J.C.; Leleu, X.; Gombert, J.M.; Herbelin, A.; et al. Sustained treatment-free remission in chronic myeloid leukaemia is associated with an increased frequency of innate CD8(+) T-cells. Br. J. Haematol. 2019, 186, 54–59. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Tanemura, A.; Tada, Y.; Katayama, I.; Kumanogoh, A.; Nishikawa, H. Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int. Immunol. 2018, 30, 13–22. [Google Scholar] [CrossRef]
- Belkina, A.C.; Ciccolella, C.O.; Anno, R.; Halpert, R.; Spidlen, J.; Snyder-Cappione, J.E. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat. Commun. 2019, 10, 5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanegashima, T.; Togashi, Y.; Azuma, K.; Kawahara, A.; Ideguchi, K.; Sugiyama, D.; Kinoshita, F.; Akiba, J.; Kashiwagi, E.; Takeuchi, A.; et al. Immune Suppression by PD-L2 against Spontaneous and Treatment-Related Antitumor Immunity. Clin. Cancer Res. 2019, 25, 4808–4819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, M.; Takahashi, N.; Sawada, K. Quantitative determination of imatinib in human plasma with high-performance liquid chromatography and ultraviolet detection. J. Chromatogr. Sci. 2011, 49, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Shitara, K.; Nishikawa, H. Regulatory T cells: A potential target in cancer immunotherapy. Ann. N. Y. Acad. Sci. 2018, 1417, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Faitg, T.H.; Lowther, D.E.; Badros, A.Z.; Chagin, K.; Dengel, K.; Iyengar, M.; Melchiori, L.; Navenot, J.M.; Norry, E.; et al. Long-term safety and activity of NY-ESO-1 SPEAR T cells after autologous stem cell transplant for myeloma. Blood Adv. 2019, 3, 2022–2034. [Google Scholar] [CrossRef]
- Shima, H.; Kada, A.; Tanizawa, A.; Yuza, Y.; Watanabe, A.; Ito, M.; Uryu, H.; Koh, K.; Imai, C.; Yoshida, N.; et al. Discontinuation of Tyrosine Kinase Inhibitor in Children with Chronic Myeloid Leukemia (JPLSG STKI-14 study). Blood 2019, 134 (Suppl. S1), 25. [Google Scholar] [CrossRef]
- De Lavallade, H.; Khoder, A.; Hart, M.; Sarvaria, A.; Sekine, T.; Alsuliman, A.; Mielke, S.; Bazeos, A.; Stringaris, K.; Ali, S.; et al. Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood 2013, 122, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Helgason, G.V.; Karvela, M.; Holyoake, T.L. Kill one bird with two stones: Potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood 2011, 118, 2035–2043. [Google Scholar] [CrossRef]
- Seggewiss, R.; Lore, K.; Greiner, E.; Magnusson, M.K.; Price, D.A.; Douek, D.C.; Dunbar, C.E.; Wiestner, A. Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood 2005, 105, 2473–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Lee, B.N.; Talpaz, M.; Donato, N.J.; Cortes, J.E.; Kantarjian, H.M.; Reuben, J.M. Imatinib mesylate suppresses cytokine synthesis by activated CD4 T cells of patients with chronic myelogenous leukemia. Leukemia 2005, 19, 1905–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujioka, Y.; Sugiyama, D.; Matsumura, I.; Minami, Y.; Miura, M.; Atsuta, Y.; Ohtake, S.; Kiyoi, H.; Miyazaki, Y.; Nishikawa, H.; et al. Regulatory T Cell as a Biomarker of Treatment-Free Remission in Patients with Chronic Myeloid Leukemia. Cancers 2021, 13, 5904. https://doi.org/10.3390/cancers13235904
Fujioka Y, Sugiyama D, Matsumura I, Minami Y, Miura M, Atsuta Y, Ohtake S, Kiyoi H, Miyazaki Y, Nishikawa H, et al. Regulatory T Cell as a Biomarker of Treatment-Free Remission in Patients with Chronic Myeloid Leukemia. Cancers. 2021; 13(23):5904. https://doi.org/10.3390/cancers13235904
Chicago/Turabian StyleFujioka, Yuki, Daisuke Sugiyama, Itaru Matsumura, Yosuke Minami, Masatomo Miura, Yoshiko Atsuta, Shigeki Ohtake, Hitoshi Kiyoi, Yasushi Miyazaki, Hiroyoshi Nishikawa, and et al. 2021. "Regulatory T Cell as a Biomarker of Treatment-Free Remission in Patients with Chronic Myeloid Leukemia" Cancers 13, no. 23: 5904. https://doi.org/10.3390/cancers13235904
APA StyleFujioka, Y., Sugiyama, D., Matsumura, I., Minami, Y., Miura, M., Atsuta, Y., Ohtake, S., Kiyoi, H., Miyazaki, Y., Nishikawa, H., & Takahashi, N. (2021). Regulatory T Cell as a Biomarker of Treatment-Free Remission in Patients with Chronic Myeloid Leukemia. Cancers, 13(23), 5904. https://doi.org/10.3390/cancers13235904