
Intrinsic Cellular Susceptibility to Barrett’s Esophagus in Adults Born with Esophageal Atresia

 , , ,
, , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
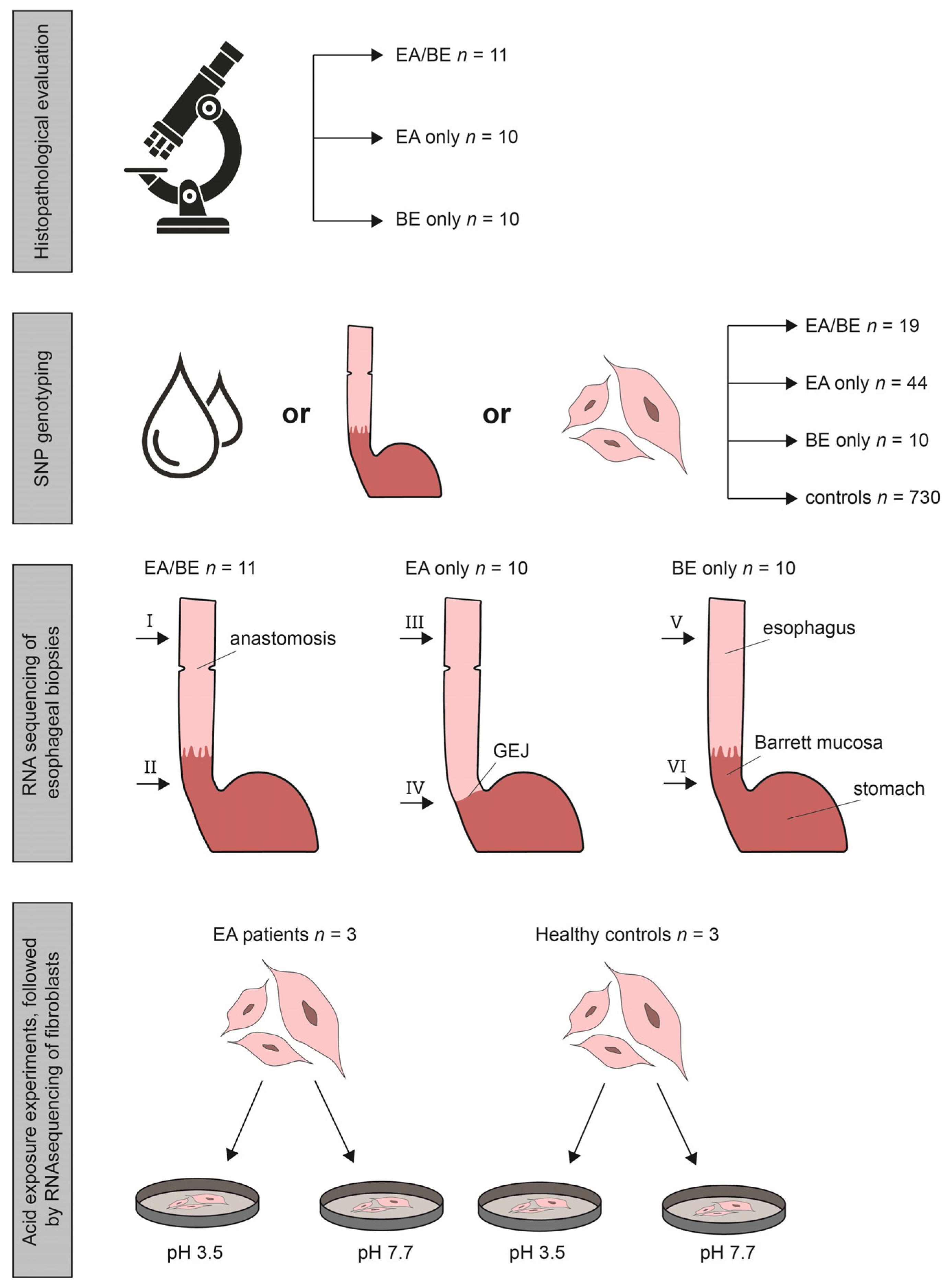
2.1. Study Population
2.2. Histopathological Evaluation
2.3. SNP Genotyping and Calculation of Predisposing SNPs, Associated with BE
2.4. RNA Sequencing, Differential Gene Expression and Pathway Enrichment Analysis
2.5. Acid Exposure Experiments
2.6. Study Approval
3. Results
3.1. Study Population
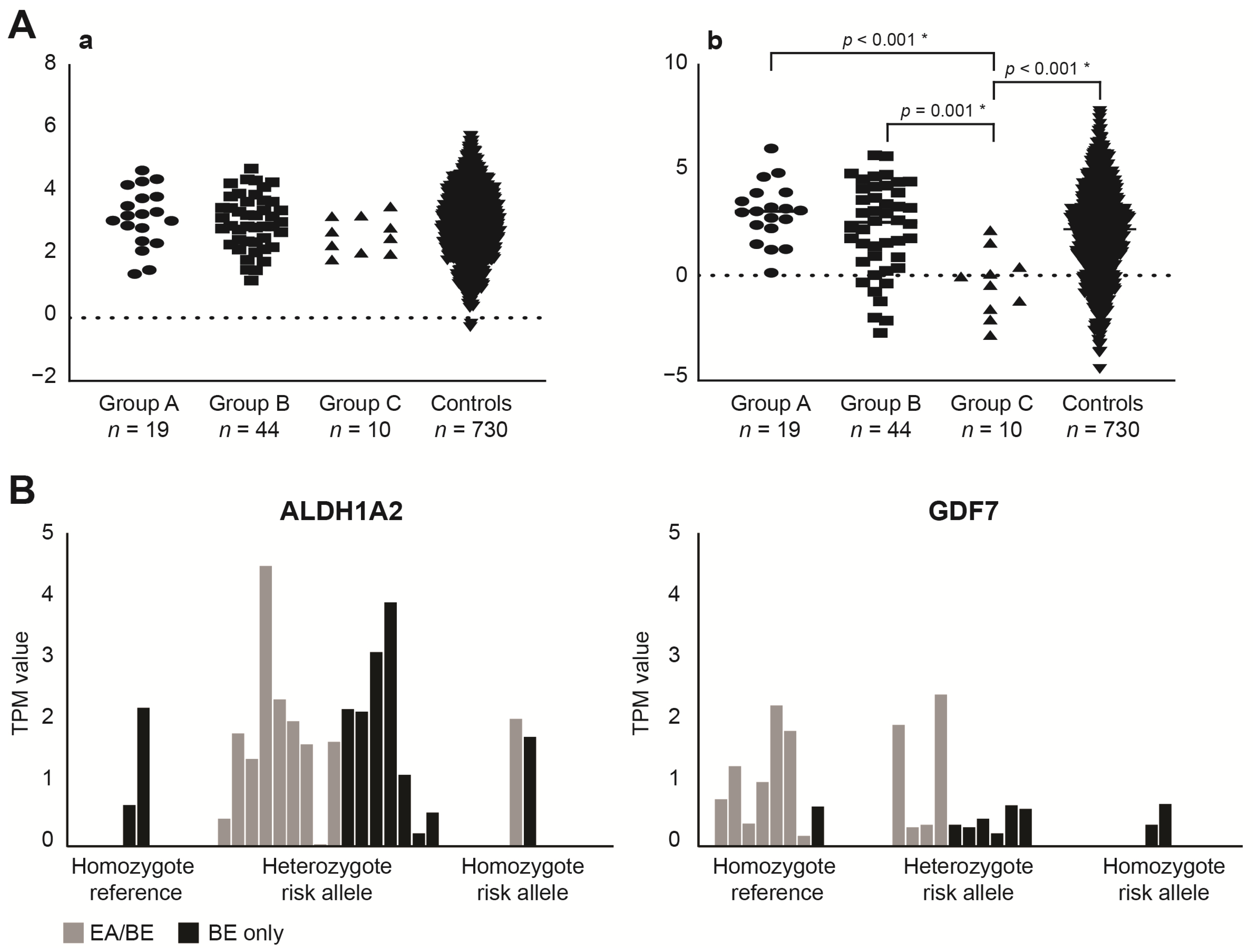
3.2. SNP (Single Nucleotide Polymorphism) Genotyping
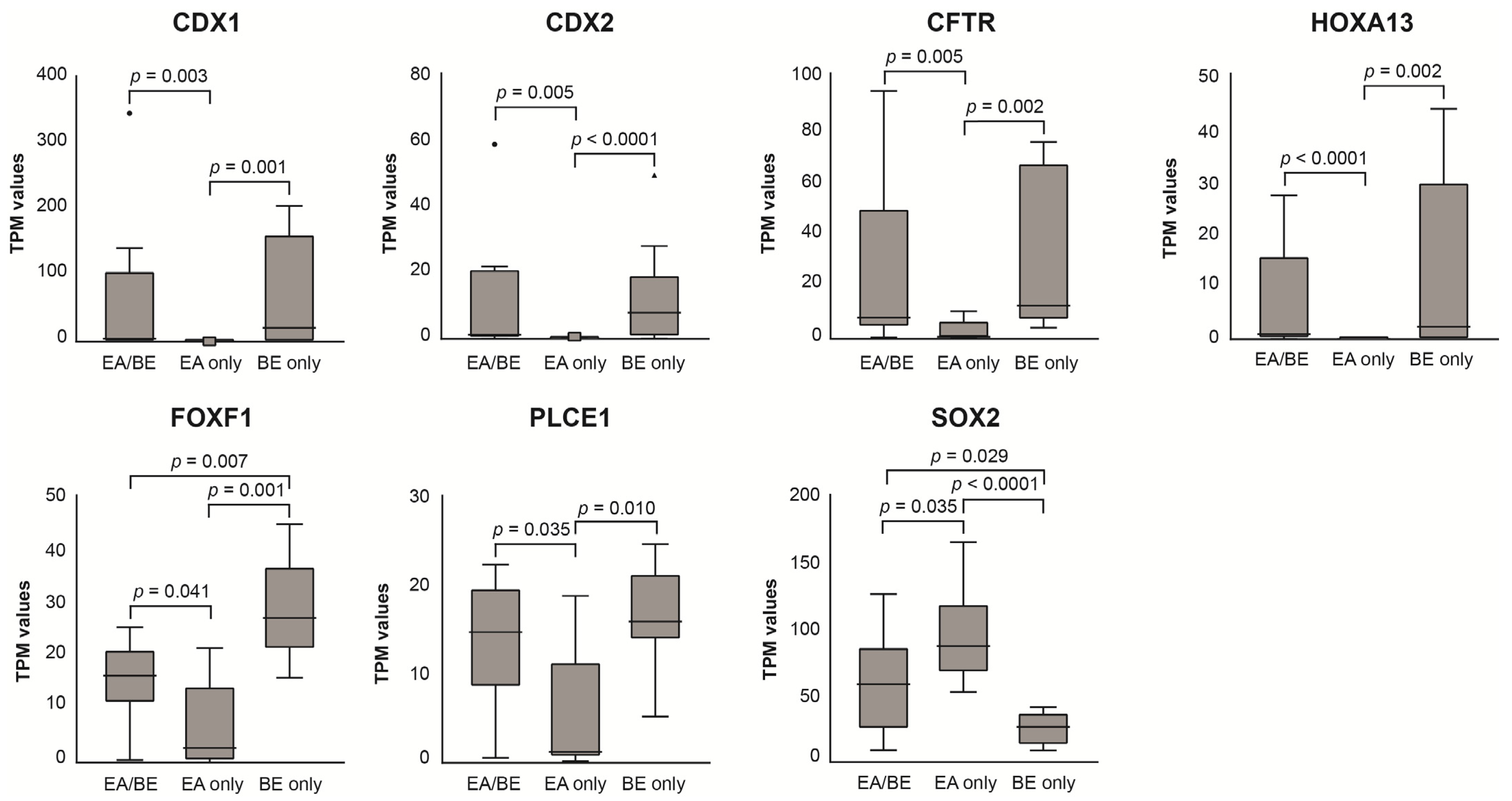
3.3. RNA Sequencing of Esophageal Biopsy Specimens
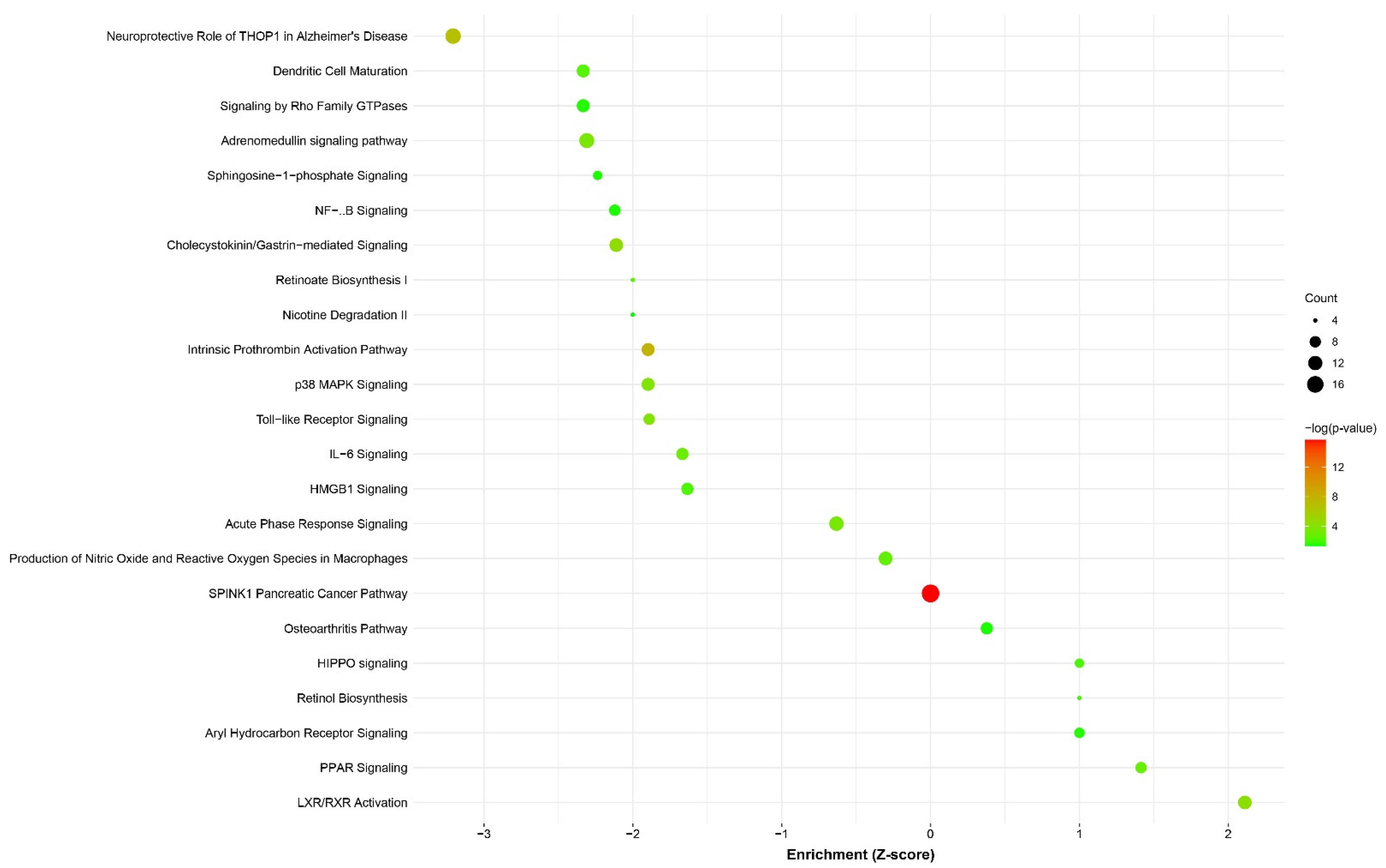
3.4. Differential Expression and Pathway Enrichment Analysis of Esophageal Biopsy Specimens
3.5. Acid Exposure Experiments
4. Discussion
4.1. BE Characteristics of EA/BE Patients and BE-Only Patients
4.2. The Contribution of BE Associated SNPs in EA/BE Patients
4.3. EA/BE Patients Have Comparable Gene Expression of BE Disease Genes as BE-Only Patients
4.4. EA/BE Patients Have an Increased Inflammatory Response
4.5. Dysregulation of RA Metabolism and Signaling
4.6. Downregulation of the Hippo/YAP Pathway
4.7. EA Patients Seem to Be More Sensitive to Acid Reflux Exposure
4.8. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, B.; Tashiro, J.; Allan, B.J.; Sola, J.E.; Parikh, P.P.; Hogan, A.R.; Neville, H.L.; Perez, E.A. A nationwide analysis of clinical outcomes among newborns with esophageal atresia and tracheoesophageal fistulas in the United States. J. Surg. Res. 2014, 190, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Van Lennep, M.; Singendonk, M.M.J.; Dall’Oglio, L.; Gottrand, F.; Krishnan, U.; Terheggen-Lagro, S.W.J.; Omari, T.I.; Benninga, M.A.; van Wijk, M.P. Oesophageal atresia. Nat. Rev. Dis. Primers 2019, 5, 26. [Google Scholar] [CrossRef]
- Vergouwe, F.W.T.; IJsselstijn, H.; Biermann, K.; Erler, N.S.; Wijnen, R.M.H.; Bruno, M.J.; Spaander, M.C.W. High Prevalence of Barrett’s Esophagus and Esophageal Squamous Cell Carcinoma After Repair of Esophageal Atresia. Clin. Gastroenterol. Hepatol. 2018, 16, 513–521.e6. [Google Scholar] [CrossRef]
- Solaymani-Dodaran, M.; Logan, R.F.; West, J.; Card, T.; Coupland, C. Risk of oesophageal cancer in Barrett’s oesophagus and gastro-oesophageal reflux. Gut 2004, 53, 1070–1074. [Google Scholar] [CrossRef]
- Jiang, M.; Li, H.; Zhang, Y.; Yang, Y.; Lu, R.; Liu, K.; Lin, S.; Lan, X.; Wang, H.; Wu, H.; et al. Transitional basal cells at the squamous-columnar junction generate Barrett’s oesophagus. Nature 2017, 550, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Naini, B.V.; Souza, R.F.; Odze, R.D. Barrett’s Esophagus: A Comprehensive and Contemporary Review for Pathologists. Am. J. Surg. Pathol. 2016, 40, e45–e66. [Google Scholar] [CrossRef]
- Han, L.; Chaturvedi, P.; Kishimoto, K.; Koike, H.; Nasr, T.; Iwasawa, K.; Giesbrecht, K.; Witcher, P.C.; Eicher, A.; Haines, L.; et al. Single cell transcriptomics identifies a signaling network coordinating endoderm and mesoderm diversification during foregut organogenesis. Nat. Commun. 2020, 11, 4158. [Google Scholar] [CrossRef] [PubMed]
- Milano, F.; van Baal, J.W.; Buttar, N.S.; Rygiel, A.M.; de Kort, F.; DeMars, C.J.; Rosmolen, W.D.; Bergman, J.J.; van Marle, J.; Wang, K.K.; et al. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology 2007, 132, 2412–2421. [Google Scholar] [CrossRef]
- Wang, D.H.; Clemons, N.J.; Miyashita, T.; Dupuy, A.J.; Zhang, W.; Szczepny, A.; Corcoran-Schwartz, I.M.; Wilburn, D.L.; Montgomery, E.A.; Wang, J.S.; et al. Aberrant epithelial-mesenchymal Hedgehog signaling characterizes Barrett’s metaplasia. Gastroenterology 2010, 138, 1810–1822. [Google Scholar] [CrossRef]
- Chang, C.L.; Lao-Sirieix, P.; Save, V.; De La Cueva Mendez, G.; Laskey, R.; Fitzgerald, R.C. Retinoic acid-induced glandular differentiation of the oesophagus. Gut 2007, 56, 906–917. [Google Scholar] [CrossRef]
- Gharahkhani, P.; Fitzgerald, R.C.; Vaughan, T.L.; Palles, C.; Gockel, I.; Tomlinson, I.; Buas, M.F.; May, A.; Gerges, C.; Anders, M.; et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: A large-scale meta-analysis. Lancet Oncol. 2016, 17, 1363–1373. [Google Scholar] [CrossRef]
- Krishnan, U.; Mousa, H.; Dall’Oglio, L.; Homaira, N.; Rosen, R.; Faure, C.; Gottrand, F. Espghan-Naspghan Guidelines for the Evaluation and Treatment of Gastrointestinal and Nutritional Complications in Children with Esophageal Atresia-Tracheoesophageal Fistula. J. Pediatric Gastroenterol. Nutr. 2016, 63, 550–570. [Google Scholar] [CrossRef] [PubMed]
- Koivusalo, A.I.; Pakarinen, M.P.; Lindahl, H.G.; Rintala, R.J. Endoscopic Surveillance after Repair of Oesophageal Atresia: Longitudinal Study in 209 Patients. J. Pediatric Gastroenterol. Nutr. 2016, 62, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin. Cancer Res. 2018, 24, 4820–4833. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, G.; Wang, J.; Wang, L.; Huang, X.; Cheng, Y. The role of cancer-associated fibroblasts in esophageal cancer. J. Transl. Med. 2016, 14, 30. [Google Scholar] [CrossRef]
- Okawa, T.; Michaylira, C.Z.; Kalabis, J.; Stairs, D.B.; Nakagawa, H.; Andl, C.D.; Johnstone, C.N.; Klein-Szanto, A.J.; El-Deiry, W.S.; Cukierman, E.; et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev. 2007, 21, 2788–2803. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Enerback, S.; Carlsson, P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 2001, 128, 2397–2406. [Google Scholar] [CrossRef]
- Woo, J.; Miletich, I.; Kim, B.M.; Sharpe, P.T.; Shivdasani, R.A. Barx1-mediated inhibition of Wnt signaling in the mouse thoracic foregut controls tracheo-esophageal septation and epithelial differentiation. PLoS ONE 2011, 6, e22493. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Lu, M.M.; Zhang, Y.; Tucker, P.W.; Zhou, D.; Morrisey, E.E. Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development 2007, 134, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Banham, A.H.; Beasley, N.; Campo, E.; Fernandez, P.L.; Fidler, C.; Gatter, K.; Jones, M.; Mason, D.Y.; Prime, J.E.; Trougouboff, P.; et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001, 61, 8820–8829. [Google Scholar]
- Shoemaker, M.B.; Husser, D.; Roselli, C.; Al Jazairi, M.; Chrispin, J.; Kuhne, M.; Neumann, B.; Knight, S.; Sun, H.; Mohanty, S.; et al. Genetic Susceptibility for Atrial Fibrillation in Patients Undergoing Atrial Fibrillation Ablation. Circ. Arrhythm. Electrophysiol. 2020, 13, e007676. [Google Scholar] [CrossRef]
- Wang, Z.; Dolle, P.; Cardoso, W.V.; Niederreither, K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev. Biol. 2006, 297, 433–445. [Google Scholar] [CrossRef]
- Chen, F.; Desai, T.J.; Qian, J.; Niederreither, K.; Lu, J.; Cardoso, W.V. Inhibition of Tgf beta signaling by endogenous retinoic acid is essential for primary lung bud induction. Development 2007, 134, 2969–2979. [Google Scholar] [CrossRef]
- Lind, A.; Siersema, P.D.; Kusters, J.G.; Konijn, T.; Mebius, R.E.; Koenderman, L. The Microenvironment in Barrett’s Esophagus Tissue Is Characterized by High FOXP3 and RALDH2 Levels. Front. Immunol. 2018, 9, 1375. [Google Scholar] [CrossRef] [PubMed]
- Domyan, E.T.; Ferretti, E.; Throckmorton, K.; Mishina, Y.; Nicolis, S.K.; Sun, X. Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development 2011, 138, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.; Puig, S.; Iglesias, M.; Seoane, A.; de Bolos, C.; Munitiz, V.; Parrilla, P.; Comerma, L.; Poulsom, R.; Krishnadath, K.K.; et al. Activation of the BMP4 pathway and early expression of CDX2 characterize non-specialized columnar metaplasia in a human model of Barrett’s esophagus. J. Gastrointest. Surg. 2012, 16, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Kaur, B.S.; Triadafilopoulos, G. Acid- and bile-induced PGE(2) release and hyperproliferation in Barrett’s esophagus are COX-2 and PKC-epsilon dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G327–G334. [Google Scholar] [CrossRef]
- Souza, R.F.; Shewmake, K.; Terada, L.S.; Spechler, S.J. Acid exposure activates the mitogen-activated protein kinase pathways in Barrett’s esophagus. Gastroenterology 2002, 122, 299–307. [Google Scholar] [CrossRef]
- Abdel-Latif, M.M.; O’Riordan, J.; Windle, H.J.; Carton, E.; Ravi, N.; Kelleher, D.; Reynolds, J.V. NF-kappaB activation in esophageal adenocarcinoma: Relationship to Barrett’s metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Ann. Surg. 2004, 239, 491–500. [Google Scholar] [CrossRef]
- Morgan, C.; Alazawi, W.; Sirieix, P.; Freeman, T.; Coleman, N.; Fitzgerald, R. In vitro acid exposure has a differential effect on apoptotic and proliferative pathways in a Barrett’s adenocarcinoma cell line. Am. J. Gastroenterol. 2004, 99, 218–224. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Aguirre, T.V.; Davis, S.; Kuebeler, M.; Bhattacharyya, A.; Sampliner, R.E. Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett’s esophagus. Am. J. Gastroenterol. 2004, 99, 1877–1883. [Google Scholar] [CrossRef]
- Kastelein, F.; Spaander, M.C.; Biermann, K.; Steyerberg, E.W.; Kuipers, E.J.; Bruno, M.J. Nonsteroidal anti-inflammatory drugs and statins have chemopreventative effects in patients with Barrett’s esophagus. Gastroenterology 2011, 141, 2000–2008; quiz e2013–e2014. [Google Scholar] [CrossRef]
- Jankowski, J.A.Z.; de Caestecker, J.; Love, S.B.; Reilly, G.; Watson, P.; Sanders, S.; Ang, Y.; Morris, D.; Bhandari, P.; Brooks, C.; et al. Esomeprazole and aspirin in Barrett’s oesophagus (AspECT): A randomised factorial trial. Lancet 2018, 392, 400–408. [Google Scholar] [CrossRef]
- Dvorak, K.; Payne, C.M.; Chavarria, M.; Ramsey, L.; Dvorakova, B.; Bernstein, H.; Holubec, H.; Sampliner, R.E.; Guy, N.; Condon, A.; et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of Barrett’s oesophagus. Gut 2007, 56, 763–771. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Hadengue, A.; Dumonceau, J.M. Retinoids, bile acids and PPARs in Barrett’s oesophagus. Gut 2008, 57, 137. [Google Scholar] [CrossRef]
- Lam-Himlin, D.M.; Daniels, J.A.; Gayyed, M.F.; Dong, J.; Maitra, A.; Pan, D.; Montgomery, E.A.; Anders, R.A. The hippo pathway in human upper gastrointestinal dysplasia and carcinoma: A novel oncogenic pathway. Int. J. Gastrointest. Cancer 2006, 37, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Dietrich, C.; Kaina, B. The aryl hydrocarbon receptor (AhR) in the regulation of cell-cell contact and tumor growth. Carcinogenesis 2010, 31, 1319–1328. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef]
- Koivusalo, A.I.; Rintala, R.J.; Pakarinen, M.P. Outcomes of fundoplication in oesophageal atresia associated gastrooesophageal reflux disease. J. Pediatric Surg. 2018, 53, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, M.; Knuppe, F.; Omari, T.; de Jong, J.; Benninga, M. Evaluation of gastroesophageal function and mechanisms underlying gastroesophageal reflux in infants and adults born with esophageal atresia. J. Pediatric Surg. 2013, 48, 2496–2505. [Google Scholar] [CrossRef] [PubMed]
- Tugay, M.; Yildiz, F.; Utkan, T.; Ulak, G.; Gacar, N.; Erden, F. Impaired esophageal reactivity in adriamycin-induced rat esophageal atresia: An in vitro study. J. Pediatric Surg. 2001, 36, 1569–1573. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kauer, W.K.; Peters, J.H.; DeMeester, T.R.; Feussner, H.; Ireland, A.P.; Stein, H.J.; Siewert, R.J. Composition and concentration of bile acid reflux into the esophagus of patients with gastroesophageal reflux disease. Surgery 1997, 122, 874–881. [Google Scholar] [CrossRef]
- Zaninotto, G.; Di Mario, F.; Costantini, M.; Baffa, R.; Germana, B.; Dal Santo, P.L.; Rugge, M.; Bolzan, M.; Naccarato, R.; Ancona, E. Oesophagitis and pH of refluxate: An experimental and clinical study. Br. J. Surg. 1992, 79, 161–164. [Google Scholar] [CrossRef]
- Leedham, S.J.; Preston, S.L.; McDonald, S.A.; Elia, G.; Bhandari, P.; Poller, D.; Harrison, R.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef]
- Martinez, P.; Mallo, D.; Paulson, T.G.; Li, X.; Sanchez, C.A.; Reid, B.J.; Graham, T.A.; Kuhner, M.K.; Maley, C.C. Evolution of Barrett’s esophagus through space and time at single-crypt and whole-biopsy levels. Nat. Commun. 2018, 9, 794. [Google Scholar] [CrossRef]
- Jego, M.; Volant, A.; Faycal, J.; Doucet, L.; Andlauer, E.; Delalande, A.H.; Cholet, F.; Nousbaum, J.B.; Gouerou, H.; Robaszkiewicz, M. Prevalence and topography of intestinal metaplasia in columnar lined esophagus. Gastroenterol. Clin. Biol. 2007, 31, 601–606. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Wang, X.; Bertrand, D.; Kern, F.; Zhang, T.; Duleba, M.; Srivastava, S.; Khor, C.C.; Hu, Y.; Wilson, L.H.; et al. Mutational spectrum of Barrett’s stem cells suggests paths to initiation of a precancerous lesion. Nat. Commun. 2016, 7, 10380. [Google Scholar] [CrossRef]
- Mourikis, T.P.; Benedetti, L.; Foxall, E.; Temelkovski, D.; Nulsen, J.; Perner, J.; Cereda, M.; Lagergren, J.; Howell, M.; Yau, C.; et al. Patient-specific cancer genes contribute to recurrently perturbed pathways and establish therapeutic vulnerabilities in esophageal adenocarcinoma. Nat. Commun. 2019, 10, 3101. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Ahimaz, P.; Edwards, N.A.; Hagen, J.J.; Faure, C.; Kingma, P.; Middlesworth, W.; Khlevner, J.; Fiky, M.E.; Schindel, D.; et al. Identification and validation of novel candidate risk genes in endocytic vesicular trafficking associated with esophageal atresia and tracheoesophageal fistulas. medRxiv 2021. [Google Scholar] [CrossRef]
- Brosens, E.; Marsch, F.; de Jong, E.M.; Zaveri, H.P.; Hilger, A.C.; Choinitzki, V.G.; Holscher, A.; Hoffmann, P.; Herms, S.; Boemers, T.M.; et al. Copy number variations in 375 patients with oesophageal atresia and/or tracheoesophageal fistula. Eur. J. Hum. Genet. 2016, 24, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, S.Y.; James, P.D.; Steele, R.J.; Hardcastle, J.D.; Atkinson, M. Length of Barrett’s oesophagus: An important factor in the development of dysplasia and adenocarcinoma. Gut 1992, 33, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Chegwidden, L.; Li, X.; Findlay, J.M.; Farnham, G.; Castro Giner, F.; Peppelenbosch, M.P.; Kovac, M.; Adams, C.L.; Prenen, H.; et al. Polymorphisms near tbx5 and gdf7 are associated with increased risk for barrett’s esophagus. Gastroenterology 2015, 148, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.M.; Ek, W.E.; Zhang, R.; Liu, X.; Onstad, L.; Sather, C.; Lao-Sirieix, P.; Gammon, M.D.; Corley, D.A.; Shaheen, N.J.; et al. A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and barrett’s esophagus. Nat. Genet. 2013, 45, 1487–1493. [Google Scholar] [CrossRef]
- Casson, A.G.; Zheng, Z.; Evans, S.C.; Geldenhuys, L.; van Zanten, S.V.; Veugelers, P.J.; Porter, G.A.; Guernsey, D.L. Cyclin d1 polymorphism (g870a) and risk for esophageal adenocarcinoma. Cancer 2005, 104, 730–739. [Google Scholar] [CrossRef]
- Ren, D.; Zheng, G.; Bream, S.; Tevebaugh, W.; Shaheen, N.J.; Chen, X. Single nucleotide polymorphisms of caudal type homeobox 1 and 2 are associated with barrett’s esophagus. Dig. Dis. Sci. 2014, 59, 57–63. [Google Scholar] [CrossRef]
- Su, Z.; Gay, L.J.; Strange, A.; Palles, C.; Band, G.; Whiteman, D.C.; Lescai, F.; Langford, C.; Nanji, M.; Edkins, S.; et al. Common variants at the mhc locus and at chromosome 16q24.1 predispose to barrett’s esophagus. Nat. Genet. 2012, 44, 1131–1136. [Google Scholar] [CrossRef]
- Becker, J.; May, A.; Gerges, C.; Anders, M.; Veits, L.; Weise, K.; Czamara, D.; Lyros, O.; Manner, H.; Terheggen, G.; et al. Supportive evidence for foxp1, barx1, and foxf1 as genetic risk loci for the development of esophageal adenocarcinoma. Cancer Med. 2015, 4, 1700–1704. [Google Scholar] [CrossRef]
- Kala, Z.; Dolina, J.; Marek, F.; Izakovicova Holla, L. Polymorphisms of glutathione s-transferase m1, t1 and p1 in patients with reflux esophagitis and barrett’s esophagus. J. Hum. Genet. 2007, 52, 527–534. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bull, L.M.; White, D.L.; Bray, M.; Nurgalieva, Z.; El-Serag, H.B. Phase i and ii enzyme polymorphisms as risk factors for barrett’s esophagus and esophageal adenocarcinoma: A systematic review and meta-analysis. Dis. Esophagus 2009, 22, 571–587. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McElholm, A.R.; McKnight, A.J.; Patterson, C.C.; Johnston, B.T.; Hardie, L.J.; Murray, L.J.; Finbar, G. A population-based study of igf axis polymorphisms and the esophageal inflammation, metaplasia, adenocarcinoma sequence. Gastroenterology 2010, 139, 204–212.e3. [Google Scholar] [CrossRef]
- Moons, L.M.; Kusters, J.G.; van Delft, J.H.; Kuipers, E.J.; Gottschalk, R.; Geldof, H.; Bode, W.A.; Stoof, J.; van Vliet, A.H.; Ketelslegers, H.B.; et al. A pro-inflammatory genotype predisposes to barrett’s esophagus. Carcinogenesis 2008, 29, 926–931. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buas, M.F.; He, Q.; Johnson, L.G.; Onstad, L.; Levine, D.M.; Thrift, A.P.; Gharahkhani, P.; Palles, C.; Lagergren, J.; Fitzgerald, R.C.; et al. Germline variation in inflammation-related pathways and risk of barrett’s oesophagus and oesophageal adenocarcinoma. Gut 2017, 66, 1739–1747. [Google Scholar] [CrossRef]
- Yan, C.; Ji, Y.; Huang, T.; Yu, F.; Gao, Y.; Gu, Y.; Qi, Q.; Du, J.; Dai, J.; Ma, H.; et al. An esophageal adenocarcinoma susceptibility locus at 9q22 also confers risk to esophageal squamous cell carcinoma by regulating the function of barx1. Cancer Lett. 2018, 421, 103–111. [Google Scholar] [CrossRef]
- Abnet, C.C.; Freedman, N.D.; Hu, N.; Wang, Z.; Yu, K.; Shu, X.O.; Yuan, J.M.; Zheng, W.; Dawsey, S.M.; Dong, L.M.; et al. A shared susceptibility locus in plce1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nat. Genet. 2010, 42, 764–767. [Google Scholar] [CrossRef]
- Dura, P.; van Veen, E.M.; Salomon, J.; te Morsche, R.H.; Roelofs, H.M.; Kristinsson, J.O.; Wobbes, T.; Witteman, B.J.; Tan, A.C.; Drenth, J.P.; et al. Barrett associated mhc and foxf1 variants also increase esophageal carcinoma risk. Int. J. Cancer 2013, 133, 1751–1755. [Google Scholar] [CrossRef]
- Wideroff, L.; Vaughan, T.L.; Farin, F.M.; Gammon, M.D.; Risch, H.; Stanford, J.L.; Chow, W.H. Gst, nat1, cyp1a1 polymorphisms and risk of esophageal and gastric adenocarcinomas. Cancer Detect. Prev. 2007, 31, 233–236. [Google Scholar] [CrossRef][Green Version]
- Gross, R.E. The Surgery of Infancy and Childhood: Its Principles and Techniques; W.B. Saunders Co.: Philadelphia, PA, USA, 1953. [Google Scholar]
- Lundell, L.R.; Dent, J.; Bennett, J.R.; Blum, A.L.; Armstrong, D.; Galmiche, J.P.; Johnson, F.; Hongo, M.; Richter, J.E.; Spechler, S.J.; et al. Endoscopic assessment of oesophagitis: Clinical and functional correlates and further validation of the los angeles classification. Gut 1999, 45, 172–180. [Google Scholar] [CrossRef]
- Rasanen, K.; Itkonen, O.; Koistinen, H.; Stenman, U.H. Emerging roles of spink1 in cancer. Clin. Chem. 2016, 62, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.E.; Berry, A.A.; Kirkwood-Wilson, R.; Roberts, N.A.; Hearn, T.; Salisbury, R.J.; Blaylock, J.; Piper Hanley, K.; Hanley, N.A. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013, 62, 3514–3522. [Google Scholar] [CrossRef] [PubMed]
- Gene Expression for Spink1 (ensg00000164266.10). Available online: https://gtexportal.org/home/gene/SPINK1 (accessed on 15 October 2021).
- Alexander, D.H.; Lange, K. Enhancements to the admixture algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Goddard, M.E.; Visscher, P.M. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007, 17, 1520–1528. [Google Scholar] [CrossRef]
- Clc Genomics Workbench User Manual. Available online: https://digitalinsights.qiagen.com/technical-support/manuals/ (accessed on 15 October 2021).
- Bus, P.; Siersema, P.D.; Verbeek, R.E.; van Baal, J.W. Upregulation of mirna-143, -145, -192, and -194 in esophageal epithelial cells upon acidic bile salt stimulation. Dis. Esophagus 2014, 27, 591–600. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Esophageal Biopsy Specimens | Fibroblasts from Acid Exposure experiment | |||||||
|---|---|---|---|---|---|---|---|---|
| II vs. VI (n = 353) | EA Patients vs. Controls (Acid-Exposed) (n = 258) | EA Patients vs. Controls (Non-Exposed) (n = 314) | Acid-Exposed vs. Non-Exposed (All Samples) (n = 578) | |||||
| Canonical Pathways | −log(p-Value) | Z-Score | −log(p-Value) | Z-Score | −log(p-Value) | Z-Score | −log(p-Value) | Z-Score |
| Agranulocyte Adhesion and Diapedesis | 1.69 | N/A | - | - | 1.52 | N/A | - | - |
| Altered T Cell and B Cell Signaling in Rheumatoid Arthritis | 3.22 | N/A | 2.57 | N/A | 2.05 | N/A | - | - |
| Atherosclerosis Signaling | 4.93 | N/A | 2.04 | N/A | 2.23 | N/A | - | - |
| Cholecystokinin/Gastrin-mediated Signaling | 4.38 | 2.111 | 2.35 | 0 | 1.39 | N/A | - | - |
| Communication between Innate and Adaptive Immune Cells | 2.39 | N/A | 2.47 | N/A | - | - | - | - |
| Dendritic Cell Maturation | 2.27 | 2.333 | 4.600 | −0.707 | 2.19 | −1.633 | - | - |
| Extrinsic Prothrombin Activation Pathway | 1.36 | N/A | 2.34 | N/A | - | - | - | - |
| Glucocorticoid Receptor Signaling | 4.51 | N/A | 1.53 | N/A | 2.03 | N/A | - | - |
| Graft-versus-Host Disease Signaling | 4.23 | N/A | 3.600 | N/A | - | - | - | - |
| HMGB1 Signaling | 2.09 | 1.633 | 2.37 | N/A | - | - | - | - |
| IL-6 Signaling | 2.89 | 1.667 | 1.33 | N/A | - | - | 5.02 | 2.117 |
| Intrinsic Prothrombin Activation Pathway | 7.92 | 1.897 | 2.61 | N/A | - | - | - | - |
| LXR/RXR Activation | 4.31 | −2.111 | 2.12 | −1 | - | - | - | - |
| MSP-RON Signaling Pathway | 4.58 | N/A | 1.44 | N/A | 1.81 | N/A | - | - |
| Osteoarthritis Pathway | 1.44 | −0.378 | - | - | 6.98 | −1.265 | - | - |
| PPAR Signaling | 2.81 | −1.414 | 2.33 | 0 | - | - | 2.57 | −2.524 |
| Production of Nitric Oxide and Reactive Oxygen Species in Macrophages | 2.68 | 0.302 | 1.48 | N/A | - | - | - | - |
| Retinol Biosynthesis | 1.95 | −1 | 1.49 | N/A | - | - | - | - |
| Role of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis | 2.59 | N/A | 3.710 | N/A | 3.87 | N/A | - | - |
| Role of Osteoblasts, Osteoclasts and Chondrocytes in Rheumatoid Arthritis | 3.10 | N/A | 3.280 | N/A | 4.53 | N/A | - | - |
| Sphingosine-1-phosphate Signaling | 1.44 | −2.236 | 1.40 | N/A | 2.38 | −1 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
ten Kate, C.A.; de Klein, A.; de Graaf, B.M.; Doukas, M.; Koivusalo, A.; Pakarinen, M.P.; van der Helm, R.; Brands, T.; IJsselstijn, H.; van Bever, Y.; et al. Intrinsic Cellular Susceptibility to Barrett’s Esophagus in Adults Born with Esophageal Atresia. Cancers 2022, 14, 513. https://doi.org/10.3390/cancers14030513
ten Kate CA, de Klein A, de Graaf BM, Doukas M, Koivusalo A, Pakarinen MP, van der Helm R, Brands T, IJsselstijn H, van Bever Y, et al. Intrinsic Cellular Susceptibility to Barrett’s Esophagus in Adults Born with Esophageal Atresia. Cancers. 2022; 14(3):513. https://doi.org/10.3390/cancers14030513
Chicago/Turabian Styleten Kate, Chantal A., Annelies de Klein, Bianca M. de Graaf, Michail Doukas, Antti Koivusalo, Mikko P. Pakarinen, Robert van der Helm, Tom Brands, Hanneke IJsselstijn, Yolande van Bever, and et al. 2022. "Intrinsic Cellular Susceptibility to Barrett’s Esophagus in Adults Born with Esophageal Atresia" Cancers 14, no. 3: 513. https://doi.org/10.3390/cancers14030513
APA Styleten Kate, C. A., de Klein, A., de Graaf, B. M., Doukas, M., Koivusalo, A., Pakarinen, M. P., van der Helm, R., Brands, T., IJsselstijn, H., van Bever, Y., Wijnen, R. M. H., Spaander, M. C. W., & Brosens, E. (2022). Intrinsic Cellular Susceptibility to Barrett’s Esophagus in Adults Born with Esophageal Atresia. Cancers, 14(3), 513. https://doi.org/10.3390/cancers14030513