

Lipid Metabolism Heterogeneity and Crosstalk with Mitochondria Functions Drive Breast Cancer Progression and Drug Resistance


Abstract
Simple Summary
Abstract
1. Introduction
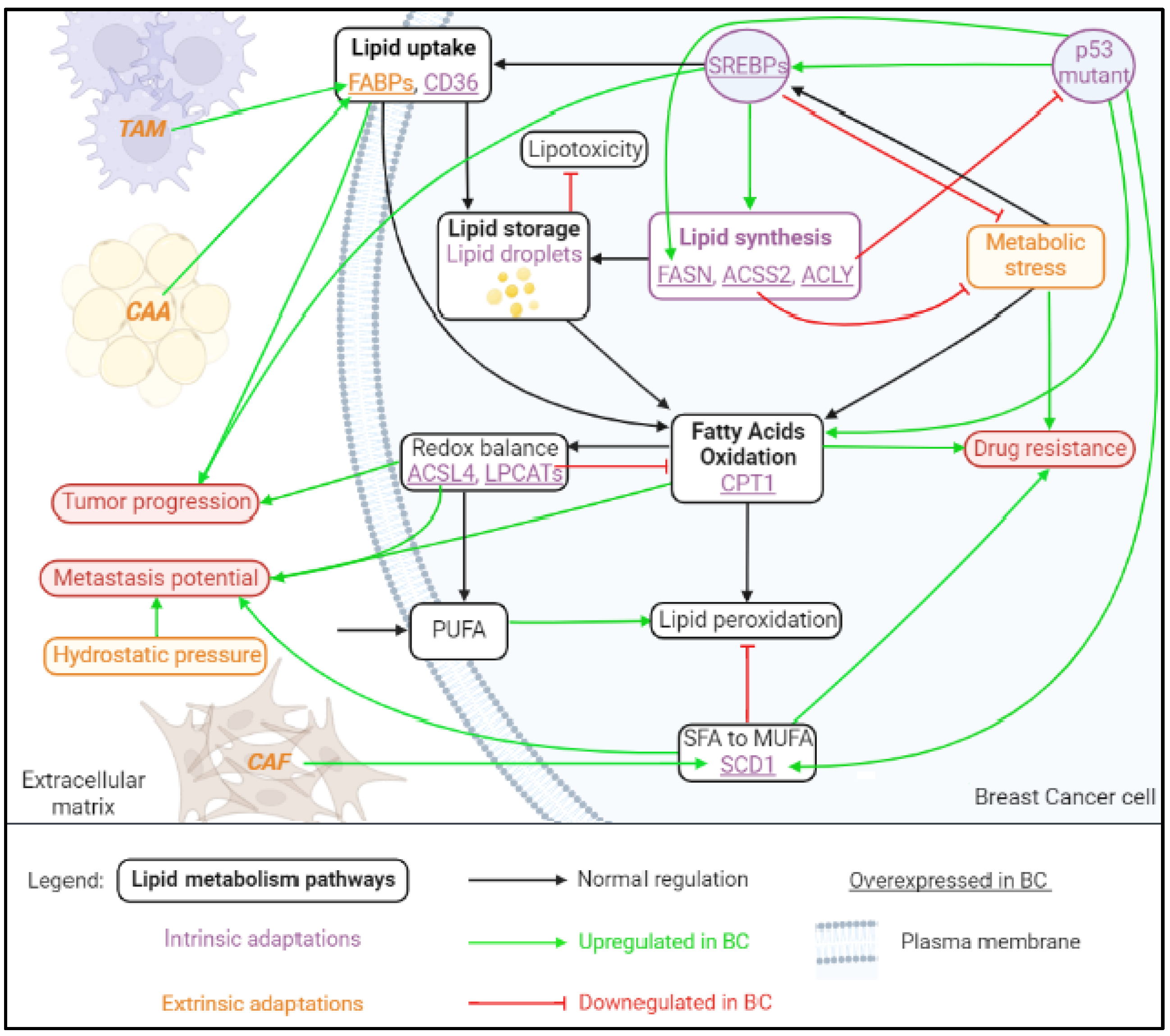
2. Plasticity of Lipid Metabolism
2.1. Fatty Acid Uptake, Storage and De Novo Lipogenesis in Cancer
2.1.1. Lipid Uptake
2.1.2. Lipid Storage
2.1.3. Lipogenesis
2.1.4. Lipid Catabolism
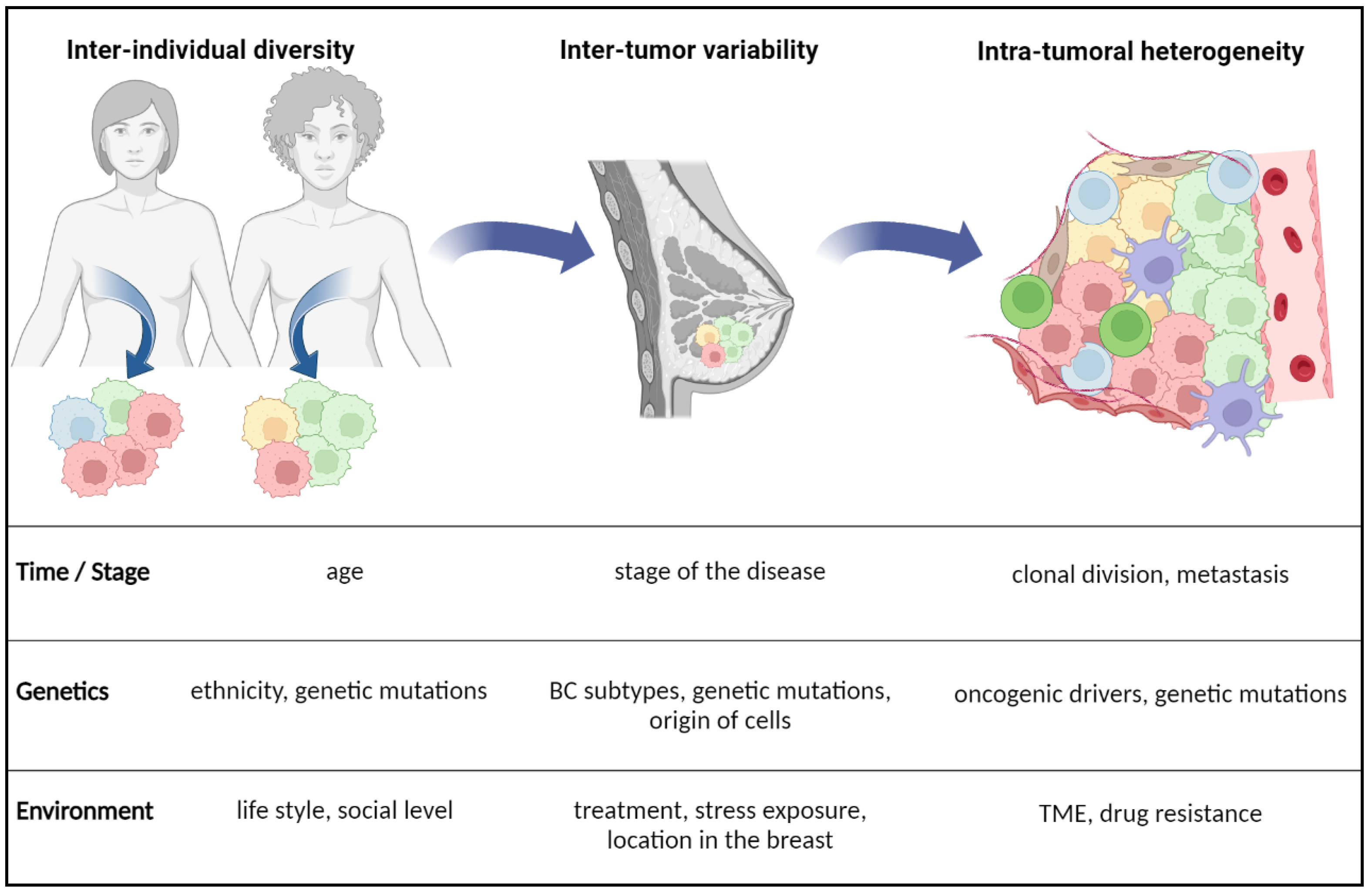
2.2. Factors of BC Plasticity and Lipid Metabolism Reprogramming
2.2.1. Intrinsic Factors of Heterogeneity
Tumor Protein 53
RAS
2.2.2. Extrinsic Factors Regulating Lipid Metabolism in BC
Cancer-Associated Adipocytes (CAA)
Cancer-Associated Fibroblasts (CAF)
Tumor-Associated Macrophages (TAM)
Tumor-Infiltrating Lymphocytes (TIL)
3. Health and Physiologic Consequences
3.1. Hypoxia
3.2. Lipid Peroxidation
3.3. Drug Resistance and Clinical Outcomes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Abdalla, F.; Singh, B.; Bhat, H.K. MicroRNAs and Gene Regulation in Breast Cancer. J. Biochem. Mol. Toxicol. 2020, 34. [Google Scholar] [CrossRef] [PubMed]
- Pranav, P.; Palaniyandi, T.; Baskar, G.; Ravi, M.; Rajendran, B.K.; Sivaji, A.; Ranganathan, M. Gene Expressions and Their Significance in Organoid Cultures Obtained from Breast Cancer Patient-Derived Biopsies. Acta Histochem. 2022, 124, 151910. [Google Scholar] [CrossRef] [PubMed]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in Breast Cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [PubMed]
- Tapia, C.; Savic, S.; Wagner, U.; Schönegg, R.; Novotny, H.; Grilli, B.; Herzog, M.; Barascud, A.D.V.; Zlobec, I.; Cathomas, G.; et al. HER2 Gene Status in Primary Breast Cancers and Matched Distant Metastases. Breast Cancer Res. 2007, 9, R31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lee, A.V.; Rosen, J.M. The Cellular Origin and Evolution of Breast Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a027128. [Google Scholar] [CrossRef] [PubMed]
- De Silva, F.; Alcorn, J. A Tale of Two Cancers: A Current Concise Overview of Breast and Prostate Cancer. Cancers 2022, 14, 2954. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Xu, H.; Ye, D.; Ren, M.; Zhang, H.; Bi, F. Ferroptosis in the Tumor Microenvironment: Perspectives for Immunotherapy. Trends Mol. Med. 2021, 27, 856–867. [Google Scholar] [CrossRef]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Nakagawa, H.; Koike, K. Lipid Metabolism in Oncology: Why It Matters, How to Research, and How to Treat. Cancers 2021, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Sounni, N.E.; Cimino, J.; Blacher, S.; Primac, I.; Truong, A.; Mazzucchelli, G.; Paye, A.; Calligaris, D.; Debois, D.; De Tullio, P.; et al. Blocking Lipid Synthesis Overcomes Tumor Regrowth and Metastasis after Antiangiogenic Therapy Withdrawal. Cell Metab. 2014, 20, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018, 28, 104–117.e5. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.Y.; Kim, H.M.; Koo, J.S. Expression of Lipid Metabolism-Related Proteins in Metastatic Breast Cancer. PLoS ONE 2015, 10, e0137204. [Google Scholar] [CrossRef]
- Luis, G.; Godfroid, A.; Nishiumi, S.; Cimino, J.; Blacher, S.; Maquoi, E.; Wery, C.; Collignon, A.; Longuespée, R.; Montero-Ruiz, L.; et al. Tumor Resistance to Ferroptosis Driven by Stearoyl-CoA Desaturase-1 (SCD1) in Cancer Cells and Fatty Acid Biding Protein-4 (FABP4) in Tumor Microenvironment Promote Tumor Recurrence. Redox Biol. 2021, 43, 102006. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.V.; Anderson, S.M.; Sartorius, C.A. Advances in Analyzing the Breast Cancer Lipidome and Its Relevance to Disease Progression and Treatment. J. Mammary Gland Biol. Neoplasia 2021, 26, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid Metabolism in Cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Wang, W.J. P53 Regulates Lipid Metabolism in Cancer. Int. J. Biol. Macromol. 2021, 192, 45–54. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding Roles for SREBP in Metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a Key Determinant of Metastatic Potential of Ovarian Cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Zakim, D.; Zhang, F.; Noy, N.; Hamilton, J.A. Fatty Acid Flip-Flop in Phospholipid Bilayers Is Extremely Fast. Biochemistry 1995, 34, 11928–11937. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, Y.; Yan, X.; Yan, F.; Sun, Y.; Zeng, J.; Waigel, S.; Yin, Y.; Fraig, M.M.; Egilmez, N.K.; et al. Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab. 2018, 28, 689–705.e5. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid Metabolism in Sickness and in Health: Emerging Regulators of Lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [Google Scholar] [CrossRef]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour Fatty Acid Metabolism in the Context of Therapy Resistance and Obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-Fueled β-Oxidation of Fatty Acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Jr, R.V.F. Lipid Droplet Biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A Highly Regulated Multi-Enzyme Complex Mediates the Catabolism of Cellular Fat Stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 Improves Hepatic Lipotoxicity by Inhibiting Lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy Regulates Lipid Metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Bastien, E.; Santiago de Jesus, J.P.; Dierge, E.; Martherus, R.; Vander Linden, C.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFβ2-Induced Formation of Lipid Droplets Supports Acidosis-Driven EMT and the Metastatic Spreading of Cancer Cells. Nat. Commun. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by MTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xu, X.; Ye, Q. Metabolism and Immunity in Breast Cancer. Front. Med. 2021, 15, 178–207. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive Glutamine Metabolism by IDH1 Mediates Lipogenesis under Hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef]
- Jakobsson, A.; Westerberg, R.; Jacobsson, A. Fatty Acid Elongases in Mammals: Their Regulation and Roles in Metabolism. Prog. Lipid Res. 2006, 45, 237–249. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Fagone, P.; Jackowski, S. Membrane Phospholipid Synthesis and Endoplasmic Reticulum Function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.M.; Frahm, J.L.; Li, L.O.; Coleman, R.A. Acyl-Coenzyme A Synthetases in Metabolic Control. Curr. Opin. Lipidol. 2010, 21, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of Neoplastic Tissue. IV. A Study of Lipid Synthesis in Neoplastic Tissue Slices in Vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Bartolacci, C.; Andreani, C.; El-Gammal, Y.; Scaglioni, P.P. Lipid Metabolism Regulates Oxidative Stress and Ferroptosis in RAS-Driven Cancers: A Perspective on Cancer Progression and Therapy. Front. Mol. Biosci. 2021, 8, 706650. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, T.; Dong, L.; Li, T.; Xue, H.; Gao, B.; Ding, X.; Wang, H.; Li, H. Fatty Acid Synthase Promotes Breast Cancer Metastasis by Mediating Changes in Fatty Acid Metabolism. Oncol. Lett. 2020, 21, 27. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef]
- Curtarello, M.; Tognon, M.; Venturoli, C.; Silic-Benussi, M.; Grassi, A.; Verza, M.; Minuzzo, S.; Pinazza, M.; Brillo, V.; Tosi, G.; et al. Rewiring of Lipid Metabolism and Storage in Ovarian Cancer Cells after Anti-VEGF Therapy. Cells 2019, 8, 1601. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of Glycerolipides; a Comparison of Lecithin and Triglyceride Synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol Metabolism: New Functions and Therapeutic Approaches in Cancer. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Dos Santos, C.; Domingues, G.; Matias, I.; Matos, J.; Fonseca, I.; De Almeida, J.M.; Dias, S. LDL-Cholesterol Signaling Induces Breast Cancer Proliferation and Invasion. Lipids Health Dis. 2014, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Basu, S.; Soni, S.; Pandey, A.; Roy, B.; Oh, M.S.; Chin, K.T.; Paraskar, A.S.; Sarangi, S.; Connor, Y.; et al. Cholesterol-Tethered Platinum II-Based Supramolecular Nanoparticle Increases Antitumor Efficacy and Reduces Nephrotoxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 11294–11299. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Liu, Z.; Zhao, Z.; Zhang, X.; Tao, S.; Yuan, B.; Zhang, J.; Wang, D.; Liu, Q.; Ding, Y. Emerging Roles of Low-Density Lipoprotein in the Development and Treatment of Breast Cancer. Lipids Health Dis. 2019, 18, 137. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Ruiter, J.P.N.; Ijlst, L.; Waterham, H.R.; Houten, S.M. The Enzymology of Mitochondrial Fatty Acid Beta-Oxidation and Its Application to Follow-up Analysis of Positive Neonatal Screening Results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef]
- Tőkés, A.M.; Vári-Kakas, S.; Kulka, J.; Törőcsik, B. Tumor Glucose and Fatty Acid Metabolism in the Context of Anthracycline and Taxane-Based (Neo)Adjuvant Chemotherapy in Breast Carcinomas. Front. Oncol. 2022, 12, 850401. [Google Scholar] [CrossRef]
- Pucci, S.; Zonetti, M.J.; Fisco, T.; Polidoro, C.; Bocchinfuso, G.; Palleschi, A.; Novelli, G.; Spagnoli, L.G.; Mazzarelli, P. Carnitine Palmitoyl Transferase-1A (CPT1A): A New Tumor Specific Target in Human Breast Cancer. Oncotarget 2016, 7, 19982–19996. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a Biomarker and Contributor of Ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Li, J.Y.; Yao, Y.M.; Tian, Y.P. Ferroptosis: A Trigger of Proinflammatory State Progression to Immunogenicity in Necroinflammatory Disease. Front. Immunol. 2021, 12, 701163. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic Mutation in Cancer and Normal Cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496. [Google Scholar] [CrossRef]
- Raza, S.; Ohm, J.E.; Dhasarathy, A.; Schommer, J.; Roche, C.; Hammer, K.D.P.; Ghribi, O. The Cholesterol Metabolite 27-Hydroxycholesterol Regulates P53 Activity and Increases Cell Proliferation via MDM2 in Breast Cancer Cells. Mol. Cell. Biochem. 2015, 410, 187–195. [Google Scholar] [CrossRef]
- Frezza, C. Metabolism and Cancer: The Future Is Now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef]
- Hu, D.; Li, Z.; Zheng, B.; Lin, X.; Pan, Y.; Gong, P.; Zhuo, W.; Hu, Y.; Chen, C.; Chen, L.; et al. Cancer-Associated Fibroblasts in Breast Cancer: Challenges and Opportunities. Cancer Commun. 2022, 42, 401–434. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Tissue-Specific and Convergent Metabolic Transformation of Cancer Correlates with Metastatic Potential and Patient Survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant P53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-Genome Analysis Informs Breast Cancer Response to Aromatase Inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. P53 Regulates Biosynthesis through Direct Inactivation of Glucose-6-Phosphate Dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. P53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Yu, G.; Jiang, L.; Li, T.; Lin, Q.; Tang, Y.; Gu, W. P53-Dependent Regulation of Metabolic Function through Transcriptional Activation of Pantothenate Kinase-1 Gene. Cell Cycle 2013, 12, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Rozeveld, C.N.; Johnson, K.M.; Zhang, L.; Razidlo, G.L. KRAS Controls Pancreatic Cancer Cell Lipid Metabolism and Invasive Potential through the Lipase HSL. Cancer Res. 2020, 80, 4332–4345. [Google Scholar] [CrossRef]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef]
- Yang, A.; Herter-Sprie, G.; Zhang, H.; Lin, E.Y.; Biancur, D.; Wang, X.; Deng, J.; Hai, J.; Yang, S.; Wong, K.K.; et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov. 2018, 8, 276–287. [Google Scholar] [CrossRef]
- Giltnane, J.M.; Balko, J.M. Rationale for Targeting the Ras/MAPK Pathway in Triple-Negative Breast Cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar]
- Zhai, D.; Xu, Y.; Abdelghany, L.; Zhang, X.; Liang, J.; Zhang, S.; Guo, C.; Li, T.S. Hydrostatic Pressure Stabilizes HIF-1α Expression in Cancer Cells to Protect against Oxidative Damage during Metastasis. Oncol. Rep. 2021, 46, 211. [Google Scholar] [CrossRef]
- He, M.J.; Pu, W.; Wang, X.; Zhang, W.; Tang, D.; Dai, Y. Comparing DESI-MSI and MALDI-MSI Mediated Spatial Metabolomics and Their Applications in Cancer Studies. Front. Oncol. 2022, 12, 3485. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Sprandio, J.; Avena, P.; Cotzia, P.; Dulau-Florea, A.; Gong, J.; Uppal, G.; Zhan, T.; et al. Hodgkin Lymphoma: A Complex Metabolic Ecosystem with Glycolytic Reprogramming of the Tumor Microenvironment. Semin. Oncol. 2017, 44, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.Z.; Jin, W.L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Malla, R.R.; Padmaraju, V.; Kundrapu, D.B. Tumor-Associated Macrophages: Potential Target of Natural Compounds for Management of Breast Cancer. Life Sci. 2022, 301, 120572. [Google Scholar] [CrossRef] [PubMed]
- Malla, R.R.; Puvalachetty, K.; Vempati, R.K.; Marni, R.; Merchant, N.; Nagaraju, G.P. Cancer Stem Cells and Circulatory Tumor Cells Promote Breast Cancer Metastasis. Clin. Breast Cancer 2022, 22, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef]
- Sacco, A.; Battaglia, A.M.; Botta, C.; Aversa, I.; Mancuso, S.; Costanzo, F.; Biamonte, F. Iron Metabolism in the Tumor Microenvironment-Implications for Anti-Cancer Immune Response. Cells 2021, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Roda, N.; Gambino, V.; Giorgio, M. Metabolic Constrains Rule Metastasis Progression. Cells 2020, 9, 2081. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.e5. [Google Scholar] [CrossRef]
- Ruscitto, F.; Roda, N.; Priami, C.; Migliaccio, E.; Pelicci, P.G. Beyond Genetics: Metastasis as an Adaptive Response in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 6271. [Google Scholar] [CrossRef]
- Hobbs, E.A.; Chen, N.; Kuriakose, A.; Bonefas, E.; Lim, B. Prognostic/Predictive Markers in Systemic Therapy Resistance and Metastasis in Breast Cancer. Ther. Adv. Med. Oncol. 2022, 14, 7588359221112698. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Li, Z.; Zhu, B. Metabolic Regulatory Crosstalk between Tumor Microenvironment and Tumor-Associated Macrophages. Theranostics 2021, 11, 1016–1030. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte Lipolysis Links Obesity to Breast Cancer Growth: Adipocyte-Derived Fatty Acids Drive Breast Cancer Cell Proliferation and Migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef]
- Attané, C.; Muller, C. Drilling for Oil: Tumor-Surrounding Adipocytes Fueling Cancer. Trends Cancer 2020, 6, 593–604. [Google Scholar] [CrossRef]
- Guaita-Esteruelas, S.; Bosquet, A.; Saavedra, P.; Gumà, J.; Girona, J.; Lam, E.W.F.; Amillano, K.; Borràs, J.; Masana, L. Exogenous FABP4 Increases Breast Cancer Cell Proliferation and Activates the Expression of Fatty Acid Transport Proteins. Mol. Carcinog. 2017, 56, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Elenbaas, B.; Weinberg, R.A. Heterotypic Signaling between Epithelial Tumor Cells and Fibroblasts in Carcinoma Formation. Exp. Cell Res. 2001, 264, 169–184. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast Heterogeneity in the Cancer Wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-Associated Fibroblasts: An Emerging Target of Anti-Cancer Immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Angelucci, C.; D’Alessio, A.; Iacopino, F.; Proietti, G.; Di Leone, A.; Masetti, R.; Sica, G. Pivotal Role of Human Stearoyl-CoA Desaturases (SCD1 and 5) in Breast Cancer Progression: Oleic Acid-Based Effect of SCD1 on Cell Migration and a Novel pro-Cell Survival Role for SCD5. Oncotarget 2018, 9, 24364–24380. [Google Scholar] [CrossRef]
- Hao, J.; Yan, F.; Zhang, Y.; Triplett, A.; Zhang, Y.; Schultz, D.A.; Sun, Y.; Zeng, J.; Silverstein, K.A.T.; Zheng, Q.; et al. Expression of Adipocyte/Macrophage Fatty Acid-Binding Protein in Tumor-Associated Macrophages Promotes Breast Cancer Progression. Cancer Res. 2018, 78, 2343–2355. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A Single-Cell and Spatially Resolved Atlas of Human Breast Cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Kan, C.; Sun, M.; Yang, F.; Wong, M.; Wang, S.; Zheng, H. Mapping Breast Cancer Microenvironment Through Single-Cell Omics. Front. Immunol. 2022, 13, 1439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Y.; Rao, E.; Yan, F.; Li, Q.; Zhang, Y.; Silverstein, K.A.T.; Liu, S.; Sauter, E.; Cleary, M.P.; et al. Fatty Acid-Binding Protein E-FABP Restricts Tumor Growth by Promoting IFN-β Responses in Tumor-Associated Macrophages. Cancer Res. 2014, 74, 2986–2998. [Google Scholar] [CrossRef] [PubMed]
- Tolba, M.F.; Omar, H.A. Immunotherapy, an Evolving Approach for the Management of Triple Negative Breast Cancer: Converting Non-Responders to Responders. Crit. Rev. Oncol. Hematol. 2018, 122, 202–207. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, X.; Sha, H.; Xie, L.; Liu, B. Recent Progress on Therapeutic Vaccines for Breast Cancer. Front. Oncol. 2022, 12, 905832. [Google Scholar] [CrossRef]
- Dekker, Y.; Le Dévédec, S.E.; Danen, E.H.J.; Liu, Q. Crosstalk between Hypoxia and Extracellular Matrix in the Tumor Microenvironment in Breast Cancer. Genes 2022, 13, 1585. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, H.; Zhou, Y.; Shang, L.; Zhang, Y.; Yang, F.; Shi, X. The Hypoxia Conditioned Mesenchymal Stem Cells Promote Hepatocellular Carcinoma Progression through YAP Mediated Lipogenesis Reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 228. [Google Scholar] [CrossRef]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P.; et al. Sterol Regulatory Element Binding Protein-Dependent Regulation of Lipid Synthesis Supports Cell Survival and Tumor Growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef]
- Pohl, E.E.; Jovanovic, O. The Role of Phosphatidylethanolamine Adducts in Modification of the Activity of Membrane Proteins under Oxidative Stress. Molecules 2019, 24, 4545. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, W.K.; Bae, K.H.; Lee, S.C.; Lee, E.W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Vellon, L.; Cuyàs, E.; Buxó, M.; Martin-Castillo, B.; Serra, D.; García, J.; Menendez, J.A.; Lupu, R. Clinical and Therapeutic Relevance of the Metabolic Oncogene Fatty Acid Synthase in HER2+ Breast Cancer. Histol. Histopathol. 2017, 32, 687–698. [Google Scholar] [CrossRef]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Orlando, U.D.; Castillo, A.F.; Medrano, M.A.R.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA Synthetase-4 Is Implicated in Drug Resistance in Breast Cancer Cell Lines Involving the Regulation of Energy-Dependent Transporter Expression. Biochem. Pharmacol. 2019, 159, 52–63. [Google Scholar] [CrossRef]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 Is an Independent Prognostic Marker and Promotes Invasion and Migration in Breast Cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef]
- Szutowicz, A.; Kwiatkowski, J.; Angielski, S. Lipogenetic and Glycolytic Enzyme Activities in Carcinoma and Nonmalignant Diseases of the Human Breast. Br. J. Cancer 1979, 39, 681–687. [Google Scholar] [CrossRef]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X.; et al. Author Correction: Fatty Acid Synthesis Is Required for Breast Cancer Brain Metastasis (Nature Cancer, (2021), 2, 4, (414-428), 10.1038/S43018-021-00183-Y). Nat. Cancer 2021, 2, 1243. [Google Scholar] [CrossRef]
- Calligaris, D.; Caragacianu, D.; Liu, X.; Norton, I.; Thompson, C.J.; Richardson, A.L.; Golshan, M.; Easterling, M.L.; Santagata, S.; Dillon, D.A.; et al. Application of Desorption Electrospray Ionization Mass Spectrometry Imaging in Breast Cancer Margin Analysis. Proc. Natl. Acad. Sci. USA 2014, 111, 15184–15189. [Google Scholar] [CrossRef] [PubMed]
- Guenther, S.; Muirhead, L.J.; Speller, A.V.M.; Golf, O.; Strittmatter, N.; Ramakrishnan, R.; Goldin, R.D.; Jones, E.; Veselkov, K.; Nicholson, J.; et al. Spatially Resolved Metabolic Phenotyping of Breast Cancer by Desorption Electrospray Ionization Mass Spectrometry. Cancer Res. 2015, 75, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Laisupasin, P.; Thompat, W.; Sukarayodhin, S.; Sornprom, A.; Sudjaroen, Y. Comparison of Serum Lipid Profiles between Normal Controls and Breast Cancer Patients. J. Lab. Physicians 2013, 5, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous Lipids Promote the Growth of Breast Cancer Cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef]
- Mendes, C.; Lopes-Coelho, F.; Ramos, C.; Martins, F.; Santos, I.; Rodrigues, A.; Silva, F.; André, S.; Serpa, J. Unraveling FATP1, Regulated by ER-β, as a Targeted Breast Cancer Innovative Therapy. Sci. Rep. 2019, 9, 14107. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørile, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Eiriksson, F.F.; Nøhr, M.K.; Costa, M.; Bödvarsdottir, S.K.; Ögmundsdottir, H.M.; Thorsteinsdottir, M. Lipidomic Study of Cell Lines Reveals Differences between Breast Cancer Subtypes. PLoS ONE 2020, 15, e0231289. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Targets | References |
|---|---|
| FASN, Lipin, CD36, CPT1/2, SCD1 | [56] |
| FASN, CD36, CPT1/2, SREBP1 | [25] |
| FASN, ACLY, ACC, SREBP1, SCD1 | [12] |
| FASN, CD36, ACLY, CPT1, ACC, ACS, SREBP1, SCD1 | [24] |
| FASN, ACLY, ACSS, SCD1 | [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azam, A.; Sounni, N.E. Lipid Metabolism Heterogeneity and Crosstalk with Mitochondria Functions Drive Breast Cancer Progression and Drug Resistance. Cancers 2022, 14, 6267. https://doi.org/10.3390/cancers14246267
Azam A, Sounni NE. Lipid Metabolism Heterogeneity and Crosstalk with Mitochondria Functions Drive Breast Cancer Progression and Drug Resistance. Cancers. 2022; 14(24):6267. https://doi.org/10.3390/cancers14246267
Chicago/Turabian StyleAzam, Aurelien, and Nor Eddine Sounni. 2022. "Lipid Metabolism Heterogeneity and Crosstalk with Mitochondria Functions Drive Breast Cancer Progression and Drug Resistance" Cancers 14, no. 24: 6267. https://doi.org/10.3390/cancers14246267
APA StyleAzam, A., & Sounni, N. E. (2022). Lipid Metabolism Heterogeneity and Crosstalk with Mitochondria Functions Drive Breast Cancer Progression and Drug Resistance. Cancers, 14(24), 6267. https://doi.org/10.3390/cancers14246267