

Revisiting the Warburg Effect with Focus on Lactate


Abstract
Simple Summary
Abstract
1. Introduction
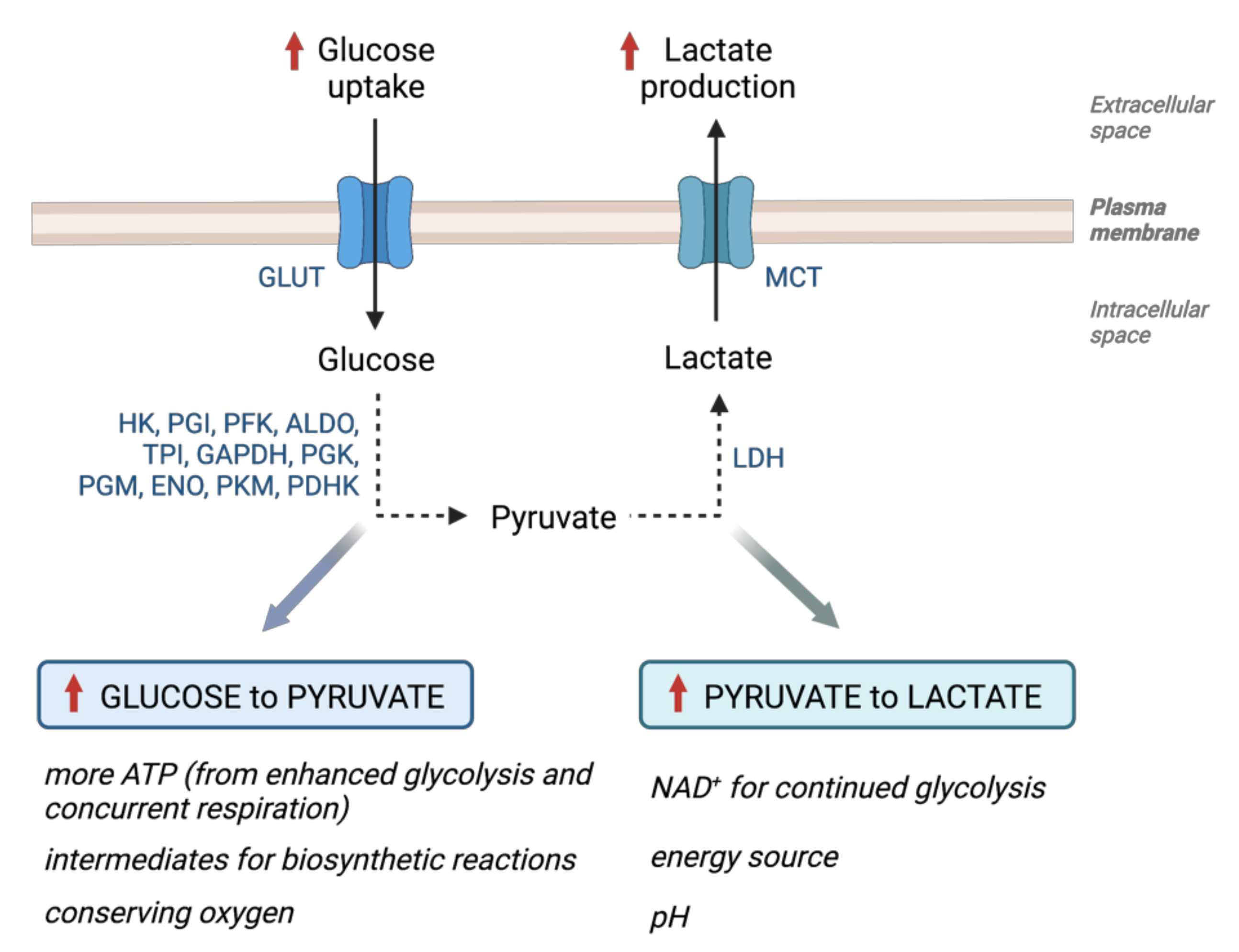
2. Glucose Metabolism in Cancer—Warburg Effect
3. Lactate Dehydrogenase
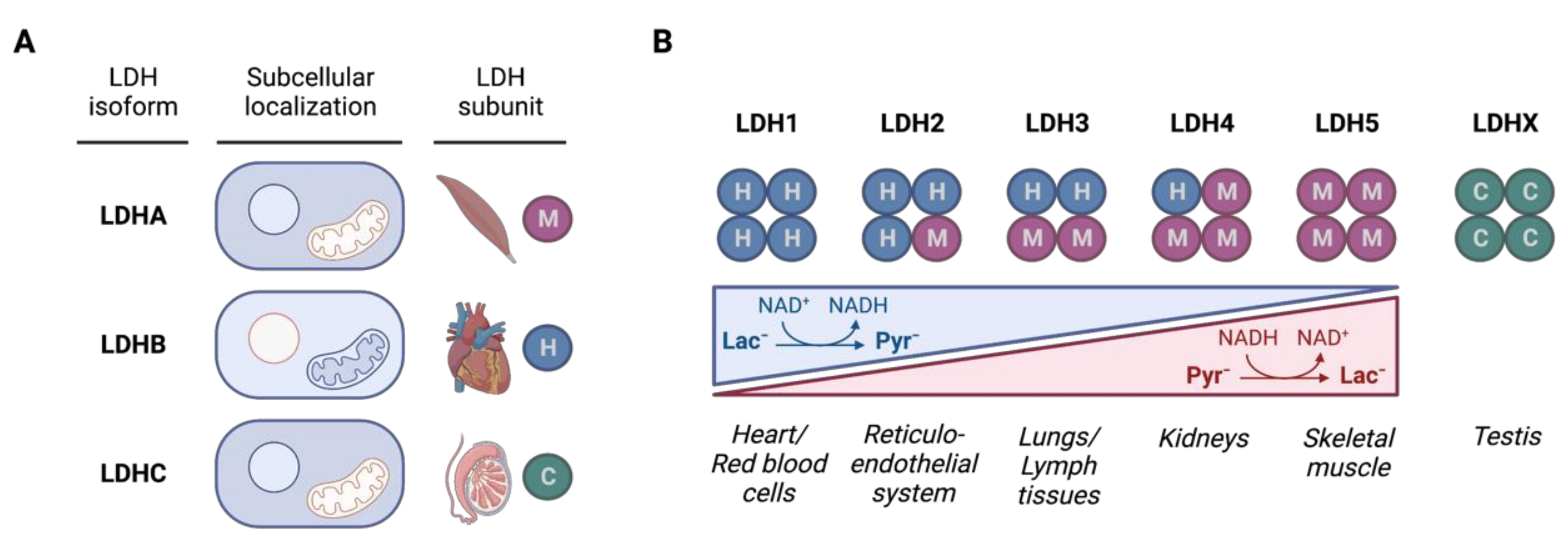
3.1. LDH Isozymes
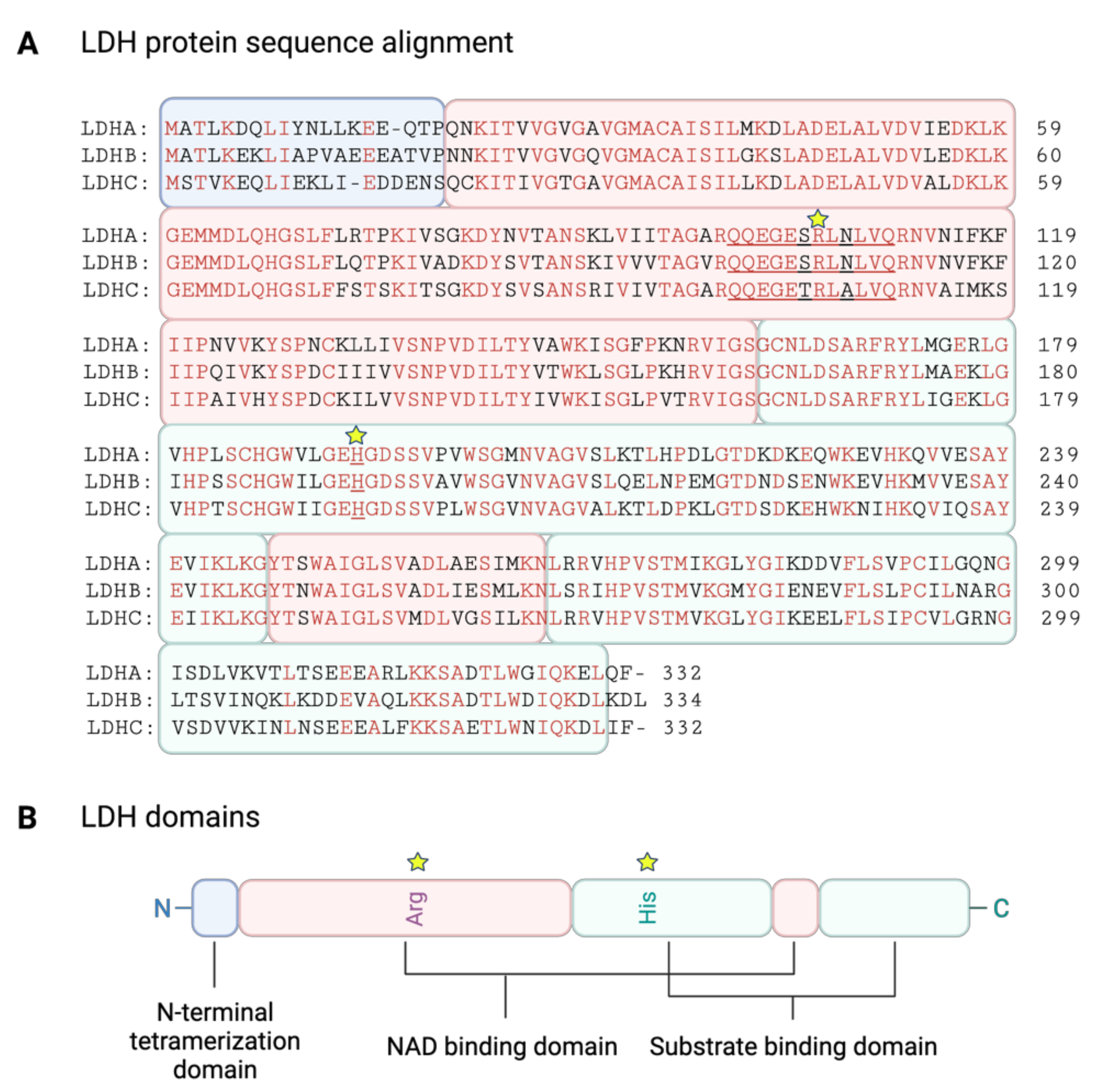
3.2. LDH Structure
3.3. Regulation of LDH Isoforms
3.3.1. LDHA Regulation
3.3.2. LDHB Regulation
3.3.3. LDHC Regulation
3.4. Role of LDH Isoforms in Carcinogenesis
3.4.1. LDHA in Cancer
3.4.2. LDHB in Cancer
3.4.3. LDHC in Germ Cells and Cancer
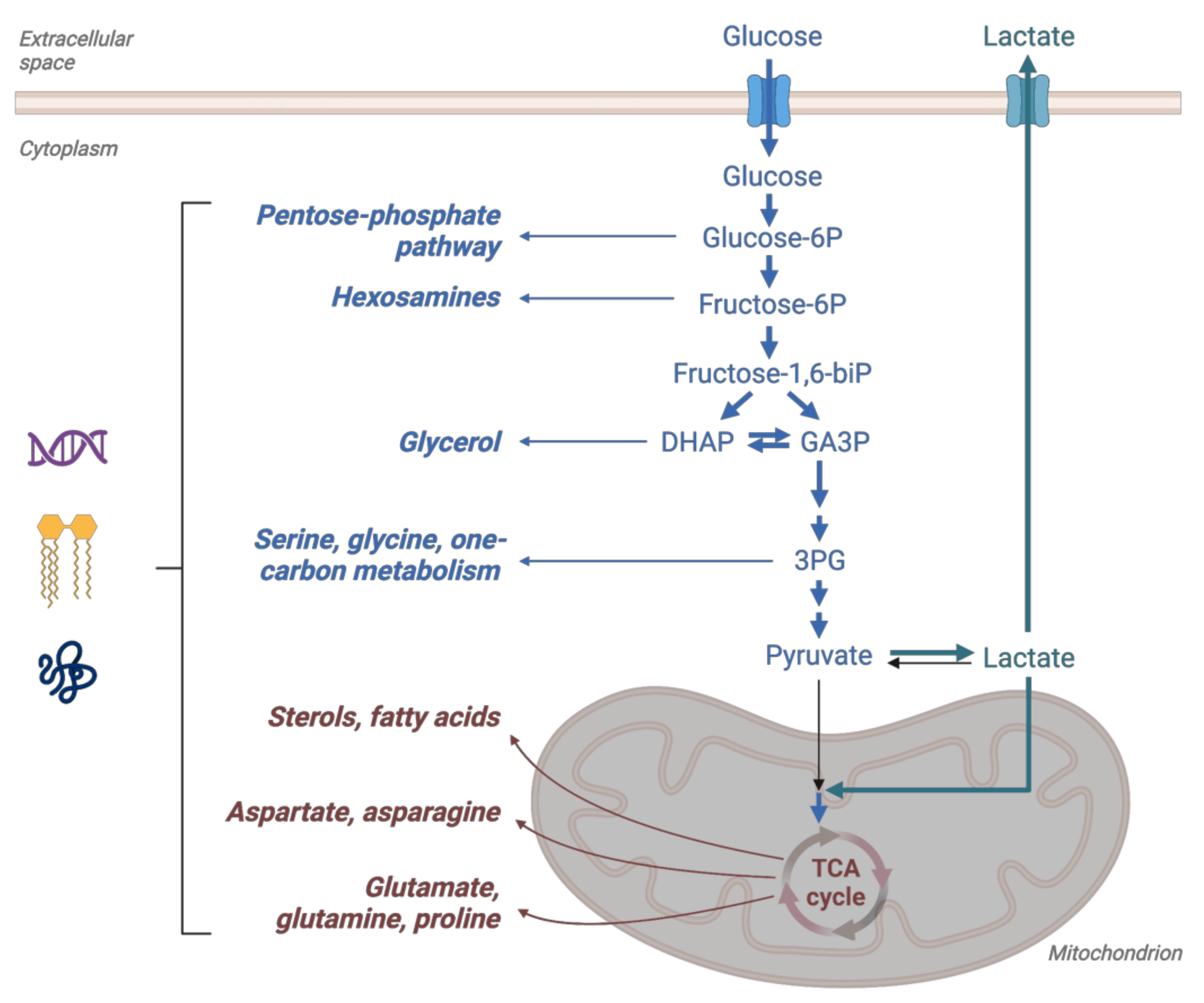
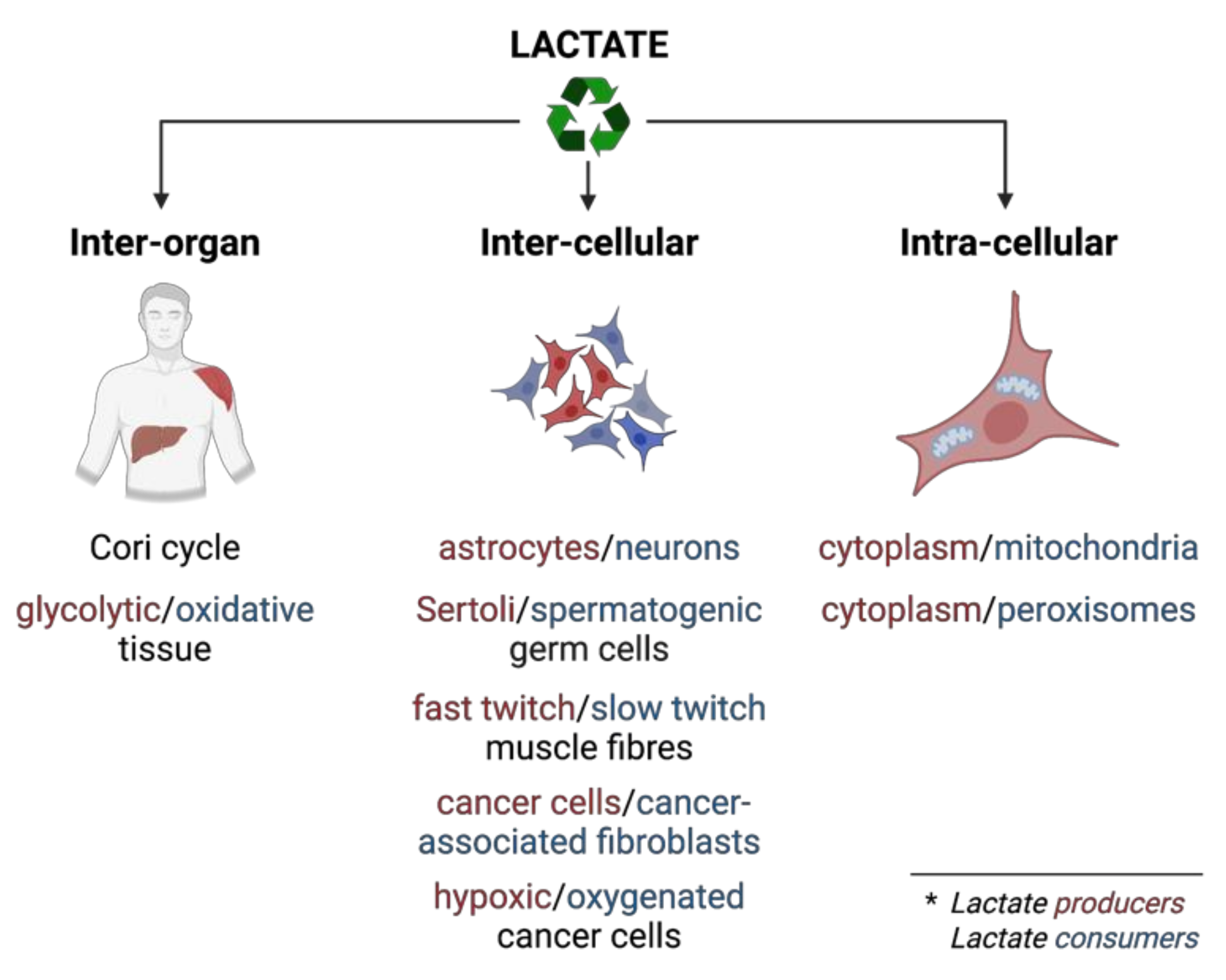
4. Lactate
4.1. Lactate as an Integrating Metabolite and the Role of Monocarboxylate Transporters
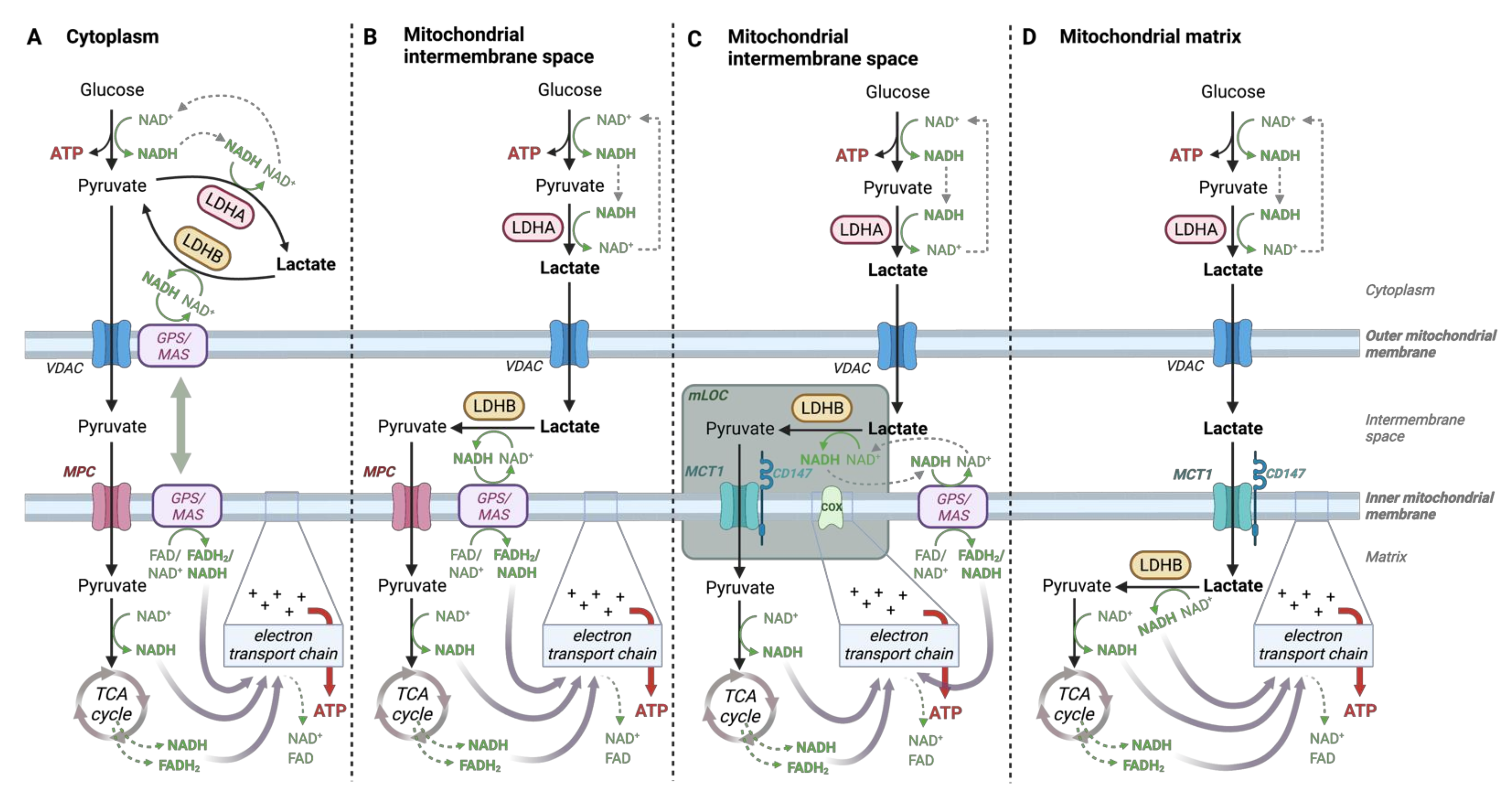
4.2. Lactate-Pyruvate Shuttle and Redox Homeostasis
4.3. Tumor Microenvironment Acidity Coupled to Lactate Production and Transport
4.4. Lactate-Induced Posttranslational Protein Modifications
4.5. Lactate Receptor
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The tortuous path of lactate shuttle discovery: From cinders and boards to the lab and ICU. J. Sport Health Sci. 2020, 9, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Baba, N.; Sharma, H.M. Histochemistry of lactic dehydrogenase in heart and pectoralis muscles of rat. J. Cell Biol. 1971, 51, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef]
- Dubouchaud, H.; Butterfield, G.E.; Wolfel, E.E.; Bergman, B.C.; Brooks, G.A. Endurance training, expression, and physiology of LDH, MCT1, and MCT4 in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E571–E579. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L. The Warburg effect and metabolic efficiency: Re-crunching the numbers. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta 2010, 1797, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Zampieri, L.X.; Capeloa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454. [Google Scholar]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Firth, J.D.; Ratcliffe, P.J. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J. Biol. Chem. 1995, 270, 29083–29089. [Google Scholar] [CrossRef]
- Maltepe, E.; Schmidt, J.V.; Baunoch, D.; Bradfield, C.A.; Simon, M.C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature 1997, 386, 403–407. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.F.; Pugh, C.W.; Bartlett, S.M.; Ratcliffe, P.J. Identification of hypoxically inducible mRNAs in HeLa cells using differential-display PCR. Role of hypoxia-inducible factor-1. Eur. J. Biochem. 1996, 241, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: Similarities with the erythropoietin 3′ enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 6496–6500. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, S.C.; Chen, K.F.; Lai, Y.Y.; Tsai, S.J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Farhana, A.; Lappin, S.L. Biochemistry, Lactate Dehydrogenase. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Holmes, R.S.; Goldberg, E. Computational analyses of mammalian lactate dehydrogenases: Human, mouse, opossum and platypus LDHs. Comput. Biol. Chem. 2009, 33, 379–385. [Google Scholar] [CrossRef]
- Everse, J.; Kaplan, N.O. Lactate dehydrogenases: Structure and function. Adv. Enzymol. Relat. Areas Mol. Biol. 1973, 37, 61–133. [Google Scholar] [CrossRef]
- Dawson, D.M.; Goodfriend, T.L.; Kaplan, N.O. Lactic Dehydrogenases: Functions of the Two Types Rates of Synthesis of the Two Major Forms Can be Correlated with Metabolic Differentiation. Science 1964, 143, 929–933. [Google Scholar] [CrossRef]
- Khan, A.A.; Allemailem, K.S.; Alhumaydhi, F.A.; Gowder, S.J.T.; Rahmani, A.H. The Biochemical and Clinical Perspectives of Lactate Dehydrogenase: An Enzyme of Active Metabolism. Endocr. Metab Immune Disord. Drug Targets 2020, 20, 855–868. [Google Scholar] [CrossRef]
- Woodford, M.R.; Chen, V.Z.; Backe, S.J.; Bratslavsky, G.; Mollapour, M. Structural and functional regulation of lactate dehydrogenase-A in cancer. Future Med. Chem. 2020, 12, 439–455. [Google Scholar] [CrossRef]
- Blanco, A.; Zinkham, W.H. Lactate Dehydrogenases in Human Testes. Science 1963, 139, 601–602. [Google Scholar] [CrossRef]
- Goldberg, E. Lactic and Malic Dehydrogenases in Human Spermatozoa. Science 1963, 139, 602–603. [Google Scholar] [CrossRef] [PubMed]
- Flick, M.J.; Konieczny, S.F. Identification of putative mammalian D-lactate dehydrogenase enzymes. Biochem. Biophys. Res. Commun. 2002, 295, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Thabault, L.; Brisson, L.; Brustenga, C.; Martinez Gache, S.A.; Prevost, J.R.C.; Kozlova, A.; Spillier, Q.; Liberelle, M.; Benyahia, Z.; Messens, J.; et al. Interrogating the Lactate Dehydrogenase Tetramerization Site Using (Stapled) Peptides. J. Med. Chem. 2020, 63, 4628–4643. [Google Scholar] [CrossRef] [PubMed]
- Forkasiewicz, A.; Dorociak, M.; Stach, K.; Szelachowski, P.; Tabola, R.; Augoff, K. The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell. Mol. Biol. Lett. 2020, 25, 35. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Wigley, D.B.; Chia, W.N.; Barstow, D.; Atkinson, T.; Holbrook, J.J. Site-directed mutagenesis reveals role of mobile arginine residue in lactate dehydrogenase catalysis. Nature 1986, 324, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Swiderek, K.; Tunon, I.; Marti, S.; Moliner, V. Protein Conformational Landscapes and Catalysis. Influence of Active Site Conformations in the Reaction Catalyzed by L-Lactate Dehydrogenase. ACS Catal. 2015, 5, 1172–1185. [Google Scholar] [CrossRef]
- Rogatzki, M.J.; Ferguson, B.S.; Goodwin, M.L.; Gladden, L.B. Lactate is always the end product of glycolysis. Front. Neurosci. 2015, 9, 22. [Google Scholar] [CrossRef]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Cui, X.G.; Han, Z.T.; He, S.H.; Wu, X.D.; Chen, T.R.; Shao, C.H.; Chen, D.L.; Su, N.; Chen, Y.M.; Wang, T.; et al. HIF1/2alpha mediates hypoxia-induced LDHA expression in human pancreatic cancer cells. Oncotarget 2017, 8, 24840–24852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Wang, X.; Gan, L.; Yu, G.; Chen, Y.; Liu, K.; Li, P.; Pan, J.; Wang, J.; et al. Inhibition of LDH-A by lentivirus-mediated small interfering RNA suppresses intestinal-type gastric cancer tumorigenicity through the downregulation of Oct4. Cancer Lett. 2012, 321, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhou, F.; Li, N.; Li, Q.; Wang, L. FOXM1-LDHA signaling promoted gastric cancer glycolytic phenotype and progression. Int. J. Clin. Exp. Pathol. 2015, 8, 6756–6763. [Google Scholar]
- Shi, M.; Cui, J.; Du, J.; Wei, D.; Jia, Z.; Zhang, J.; Zhu, Z.; Gao, Y.; Xie, K. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin. Cancer Res. 2014, 20, 4370–4380. [Google Scholar] [CrossRef]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro-Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef]
- Kaller, M.; Liffers, S.T.; Oeljeklaus, S.; Kuhlmann, K.; Roh, S.; Hoffmann, R.; Warscheid, B.; Hermeking, H. Genome-wide characterization of miR-34a induced changes in protein and mRNA expression by a combined pulsed SILAC and microarray analysis. Mol. Cell. Proteom. 2011, 10, M111.010462. [Google Scholar] [CrossRef]
- Pullen, T.J.; da Silva Xavier, G.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol. Cell. Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Liu, A.; Fang, C.; Hao, J.; Wang, Z. Lactate dehydrogenase A negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget 2015, 6, 19456–19468. [Google Scholar] [CrossRef]
- Fan, J.; Hitosugi, T.; Chung, T.W.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell. Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef]
- Kwon, O.K.; Bang, I.H.; Choi, S.Y.; Jeon, J.M.; Na, A.Y.; Gao, Y.; Cho, S.S.; Ki, S.H.; Choe, Y.; Lee, J.N.; et al. SIRT5 Is the desuccinylase of LDHA as novel cancer metastatic stimulator in aggressive prostate cancer. Genom. Proteom. Bioinform. 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zou, S.W.; Liu, Y.; Zhou, X.; Mo, Y.; Wang, P.; Xu, Y.H.; Dong, B.; Xiong, Y.; Lei, Q.Y.; et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 2013, 23, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.J.; Li, Y.; Shao, X.X.; Zhou, H.; Zeng, R.; Tang, Z.Y.; Xia, Q.C. Proteome analysis of hepatocellular carcinoma cell strains, MHCC97-H and MHCC97-L, with different metastasis potentials. Proteomics 2004, 4, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Leiblich, A.; Cross, S.S.; Catto, J.W.; Phillips, J.T.; Leung, H.Y.; Hamdy, F.C.; Rehman, I. Lactate dehydrogenase-B is silenced by promoter hypermethylation in human prostate cancer. Oncogene 2006, 25, 2953–2960. [Google Scholar] [CrossRef]
- Brown, N.J.; Higham, S.E.; Perunovic, B.; Arafa, M.; Balasubramanian, S.; Rehman, I. Lactate dehydrogenase-B is silenced by promoter methylation in a high frequency of human breast cancers. PLoS ONE 2013, 8, e57697. [Google Scholar] [CrossRef]
- Hamadneh, L.; Al-Lakkis, L.; Alhusban, A.A.; Tarawneh, S.; Abu-Irmaileh, B.; Albustanji, S.; Al-Bawab, A.Q. Changes in Lactate Production, Lactate Dehydrogenase Genes Expression and DNA Methylation in Response to Tamoxifen Resistance Development in MCF-7 Cell Line. Genes 2021, 12, 777. [Google Scholar] [CrossRef]
- Zha, X.; Wang, F.; Wang, Y.; He, S.; Jing, Y.; Wu, X.; Zhang, H. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. 2011, 71, 13–18. [Google Scholar] [CrossRef]
- Wu, G.; Yuan, S.; Chen, Z.; Chen, G.; Fan, Q.; Dong, H.; Ye, F.; Li, J.; Zhu, X. The KLF14 Transcription Factor Regulates Glycolysis by Downregulating LDHB in Colorectal Cancer. Int. J. Biol. Sci. 2019, 15, 628–635. [Google Scholar] [CrossRef]
- Liang, X.; Liu, L.; Fu, T.; Zhou, Q.; Zhou, D.; Xiao, L.; Liu, J.; Kong, Y.; Xie, H.; Yi, F.; et al. Exercise Inducible Lactate Dehydrogenase B Regulates Mitochondrial Function in Skeletal Muscle. J. Biol. Chem. 2016, 291, 25306–25318. [Google Scholar] [CrossRef]
- Brisson, L.; Banski, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hee, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Zhang, P.; Wang, B.; Yang, D.; Duan, X.; Jiang, Y.; Xu, T.; Jiang, Y.; Shi, J.; Ding, C.; et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat. Commun. 2019, 10, 5566. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.; Eddy, E.M.; Duan, C.; Odet, F. LDHC: The ultimate testis-specific gene. J. Androl. 2010, 31, 86–94. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Zhang, F.; Wu, H.; Zhang, B.; Wu, X. Correlation between promoter methylation of the LDH-C4 gene and DNMT expression in breast cancer and their prognostic significance. Oncol. Lett. 2022, 23, 35. [Google Scholar] [CrossRef]
- Jethanandani, P.; Goldberg, E. ldhc expression in non-germ cell nuclei is repressed by NF-I binding. J. Biol. Chem. 2001, 276, 35414–35421. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Kontomanolis, E.; Giatromanolaki, A.; Sivridis, E.; Liberis, V. Serum and tissue LDH levels in patients with breast/gynaecological cancer and benign diseases. Gynecol. Obstet Investig. 2009, 67, 162–168. [Google Scholar] [CrossRef]
- Fiume, L. Inhibition of aerobic glycolysis in Yoshida ascites hepatoma by tartronic acid. Nature 1960, 187, 792–793. [Google Scholar] [CrossRef]
- Jafary, F.; Ganjalikhany, M.R.; Moradi, A.; Hemati, M.; Jafari, S. Novel Peptide Inhibitors for Lactate Dehydrogenase A (LDHA): A Survey to Inhibit LDHA Activity via Disruption of Protein-Protein Interaction. Sci. Rep. 2019, 9, 4686. [Google Scholar] [CrossRef]
- Dennison, J.B.; Molina, J.R.; Mitra, S.; Gonzalez-Angulo, A.M.; Balko, J.M.; Kuba, M.G.; Sanders, M.E.; Pinto, J.A.; Gomez, H.L.; Arteaga, C.L.; et al. Lactate dehydrogenase B: A metabolic marker of response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res. 2013, 19, 3703–3713. [Google Scholar] [CrossRef]
- McCleland, M.L.; Adler, A.S.; Deming, L.; Cosino, E.; Lee, L.; Blackwood, E.M.; Solon, M.; Tao, J.; Li, L.; Shames, D.; et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin. Cancer Res. 2013, 19, 773–784. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Adler, A.S.; Shang, Y.; Hunsaker, T.; Truong, T.; Peterson, D.; Torres, E.; Li, L.; Haley, B.; Stephan, J.P.; et al. An integrated genomic screen identifies LDHB as an essential gene for triple-negative breast cancer. Cancer Res. 2012, 72, 5812–5823. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhang, X.; Ding, X.; Li, H.; Geng, M.; Xie, Z.; Wu, H.; Huang, M. Lactate dehydrogenase B is associated with the response to neoadjuvant chemotherapy in oral squamous cell carcinoma. PLoS ONE 2015, 10, e0125976. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wu, S.; Du, Q.; Wang, A.; Wang, Z. Integrated analysis of gene expression and genomic aberration data in osteosarcoma (OS). Cancer Gene Ther. 2015, 22, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, Y.; Bai, P.; Wang, J.; Liu, Z.; Wang, T.; Cai, Q. LDHB may be a significant predictor of poor prognosis in osteosarcoma. Am. J. Transl. Res. 2016, 8, 4831–4843. [Google Scholar] [PubMed]
- Shibata, S.; Sogabe, S.; Miwa, M.; Fujimoto, T.; Takakura, N.; Naotsuka, A.; Kitamura, S.; Kawamoto, T.; Soga, T. Identification of the first highly selective inhibitor of human lactate dehydrogenase B. Sci. Rep. 2021, 11, 21353. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhou, X.; Yu, Z.; Liu, J.; Huang, G. Low Expression of LDHB Correlates With Unfavorable Survival in Hepatocellular Carcinoma: Strobe-Compliant Article. Medicine 2015, 94, e1583. [Google Scholar] [CrossRef]
- Cui, J.; Quan, M.; Jiang, W.; Hu, H.; Jiao, F.; Li, N.; Jin, Z.; Wang, L.; Wang, Y.; Wang, L. Suppressed expression of LDHB promotes pancreatic cancer progression via inducing glycolytic phenotype. Med. Oncol. 2015, 32, 143. [Google Scholar] [CrossRef]
- Koh, Y.W.; Lee, S.J.; Park, S.Y. Prognostic significance of lactate dehydrogenase B according to histologic type of non-small-cell lung cancer and its association with serum lactate dehydrogenase. Pathol. Res. Pract. 2017, 213, 1134–1138. [Google Scholar] [CrossRef]
- Lee, D.S.; Park, K.R.; Kim, S.J.; Chung, M.J.; Lee, Y.H.; Chang, J.H.; Kang, J.H.; Hong, S.H.; Kim, M.S.; Kim, Y.S. Serum lactate dehydrogenase levels at presentation in stage IV non-small cell lung cancer: Predictive value of metastases and relation to survival outcomes. Tumour Biol. 2016, 37, 619–625. [Google Scholar] [CrossRef]
- Boussouar, F.; Benahmed, M. Lactate and energy metabolism in male germ cells. Trends Endocrinol. Metab. 2004, 15, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Odet, F.; Duan, C.; Willis, W.D.; Goulding, E.H.; Kung, A.; Eddy, E.M.; Goldberg, E. Expression of the gene for mouse lactate dehydrogenase C (Ldhc) is required for male fertility. Biol. Reprod. 2008, 79, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Coonrod, S.; Vitale, A.; Duan, C.; Bristol-Gould, S.; Herr, J.; Goldberg, E. Testis-specific lactate dehydrogenase (LDH-C4; Ldh3) in murine oocytes and preimplantation embryos. J. Androl. 2006, 27, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Koslowski, M.; Tureci, O.; Bell, C.; Krause, P.; Lehr, H.A.; Brunner, J.; Seitz, G.; Nestle, F.O.; Huber, C.; Sahin, U. Multiple splice variants of lactate dehydrogenase C selectively expressed in human cancer. Cancer Res. 2002, 62, 6750–6755. [Google Scholar] [PubMed]
- Tan, H.; Wang, H.; Ma, J.; Deng, H.; He, Q.; Chen, Q.; Zhang, Q. Identification of human LDHC4 as a potential target for anticancer drug discovery. Acta Pharm. Sin. B 2022, 12, 2348–2357. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; Enerback, S. Lactate: The ugly duckling of energy metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Brooks, G.A.; Arevalo, J.A.; Osmond, A.D.; Leija, R.G.; Curl, C.C.; Tovar, A.P. Lactate in contemporary biology: A phoenix risen. J. Physiol. 2022, 600, 1229–1251. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Perez-Escuredo, J.; Van Hee, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Bergman, B.C.; Wolfel, E.E.; Butterfield, G.E.; Lopaschuk, G.D.; Casazza, G.A.; Horning, M.A.; Brooks, G.A. Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol. 1999, 87, 1684–1696. [Google Scholar] [CrossRef]
- Donovan, C.M.; Brooks, G.A. Endurance training affects lactate clearance, not lactate production. Am. J. Physiol. 1983, 244, E83–E92. [Google Scholar] [CrossRef]
- Mazzeo, R.S.; Brooks, G.A.; Schoeller, D.A.; Budinger, T.F. Disposal of blood [1-13C]lactate in humans during rest and exercise. J. Appl. Physiol. 1986, 60, 232–241. [Google Scholar] [CrossRef]
- Stanley, W.C.; Gertz, E.W.; Wisneski, J.A.; Morris, D.L.; Neese, R.A.; Brooks, G.A. Systemic lactate kinetics during graded exercise in man. Am. J. Physiol. 1985, 249, E595–E602. [Google Scholar] [CrossRef]
- Brooks, G.A.; Dubouchaud, H.; Brown, M.; Sicurello, J.P.; Butz, C.E. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Natl. Acad. Sci. USA 1999, 96, 1129–1134. [Google Scholar] [CrossRef]
- Butz, C.E.; McClelland, G.B.; Brooks, G.A. MCT1 confirmed in rat striated muscle mitochondria. J. Appl. Physiol. 2004, 97, 1059–1066. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hussien, R.; Brooks, G.A. Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: Evidence of a mitochondrial lactate oxidation complex. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1237–E1244. [Google Scholar] [CrossRef]
- Glancy, B.; Kane, D.A.; Kavazis, A.N.; Goodwin, M.L.; Willis, W.T.; Gladden, L.B. Mitochondrial lactate metabolism: History and implications for exercise and disease. J. Physiol. 2021, 599, 863–888. [Google Scholar] [CrossRef] [PubMed]
- Kane, D.A. Lactate oxidation at the mitochondria: A lactate-malate-aspartate shuttle at work. Front. Neurosci. 2014, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Heslop, K.A.; Milesi, V.; Maldonado, E.N. VDAC Modulation of Cancer Metabolism: Advances and Therapeutic Challenges. Front. Physiol. 2021, 12, 742839. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rai, M.; Buddika, K.; Sterrett, M.C.; Luhur, A.; Mahmoudzadeh, N.H.; Julick, C.R.; Pletcher, R.C.; Chawla, G.; Gosney, C.J.; et al. Lactate dehydrogenase and glycerol-3-phosphate dehydrogenase cooperatively regulate growth and carbohydrate metabolism during Drosophila melanogaster larval development. Development 2019, 146, dev175315. [Google Scholar] [CrossRef]
- Swietach, P. What is pH regulation, and why do cancer cells need it? Cancer Metastasis Rev. 2019, 38, 5–15. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar]
- Svastova, E.; Hulikova, A.; Rafajova, M.; Zat’ovicova, M.; Gibadulinova, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef]
- Ames, S.; Andring, J.T.; McKenna, R.; Becker, H.M. CAIX forms a transport metabolon with monocarboxylate transporters in human breast cancer cells. Oncogene 2020, 39, 1710–1723. [Google Scholar] [CrossRef] [PubMed]
- Forero-Quintero, L.S.; Ames, S.; Schneider, H.P.; Thyssen, A.; Boone, C.D.; Andring, J.T.; McKenna, R.; Casey, J.R.; Deitmer, J.W.; Becker, H.M. Membrane-anchored carbonic anhydrase IV interacts with monocarboxylate transporters via their chaperones CD147 and GP70. J. Biol. Chem. 2019, 294, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Panisova, E.; Kery, M.; Sedlakova, O.; Brisson, L.; Debreova, M.; Sboarina, M.; Sonveaux, P.; Pastorekova, S.; Svastova, E. Lactate stimulates CA IX expression in normoxic cancer cells. Oncotarget 2017, 8, 77819–77835. [Google Scholar] [CrossRef] [PubMed]
- Benej, M.; Svastova, E.; Banova, R.; Kopacek, J.; Gibadulinova, A.; Kery, M.; Arena, S.; Scaloni, A.; Vitale, M.; Zambrano, N.; et al. CA IX Stabilizes Intracellular pH to Maintain Metabolic Reprogramming and Proliferation in Hypoxia. Front. Oncol. 2020, 10, 1462. [Google Scholar] [CrossRef]
- Garcia-Canaveras, J.C.; Chen, L.; Rabinowitz, J.D. The Tumor Metabolic Microenvironment: Lessons from Lactate. Cancer Res. 2019, 79, 3155–3162. [Google Scholar] [CrossRef]
- Hu, X.; Chao, M.; Wu, H. Central role of lactate and proton in cancer cell resistance to glucose deprivation and its clinical translation. Signal Transduct. Target. Ther. 2017, 2, 16047. [Google Scholar] [CrossRef]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Torrini, C.; Nguyen, T.T.T.; Shu, C.; Mela, A.; Humala, N.; Mahajan, A.; Seeley, E.H.; Zhang, G.; Westhoff, M.A.; Karpel-Massler, G.; et al. Lactate is an epigenetic metabolite that drives survival in model systems of glioblastoma. Mol. Cell 2022, 82, 3061–3076.e3066. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Zhang, D.; Wei, W.; Baek, M.; Liu, W.; Gao, J.; Dankova, D.; Nielsen, A.L.; Bolding, J.E.; Yang, L.; et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci. Adv. 2022, 8, eabi6696. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, D.; Jiang, Y.; Hou, J.; Tian, M.; Li, J.; Sun, L.; Zhang, Y.; Zhang, T.; Li, Z.; et al. Lactate Modulates Cellular Metabolism Through Histone Lactylation-Mediated Gene Expression in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 647559. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yin, J.; Shan, L.; Yi, X.; Zhang, W.; Ding, Y. Identification of lysine-lactylated substrates in gastric cancer cells. iScience 2022, 25, 104630. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef]
- Ahmed, K.; Tunaru, S.; Tang, C.; Muller, M.; Gille, A.; Sassmann, A.; Hanson, J.; Offermanns, S. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 2010, 11, 311–319. [Google Scholar] [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef]
- Ishihara, S.; Hata, K.; Hirose, K.; Okui, T.; Toyosawa, S.; Uzawa, N.; Nishimura, R.; Yoneda, T. The lactate sensor GPR81 regulates glycolysis and tumor growth of breast cancer. Sci. Rep. 2022, 12, 6261. [Google Scholar] [CrossRef]
- San-Millan, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front. Oncol. 2020, 10, 231. [Google Scholar] [CrossRef]
- Certo, M.; Llibre, A.; Lee, W.; Mauro, C. Understanding lactate sensing and signalling. Trends Endocrinol. Metab. 2022, 33, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Peng, W.B.; Zhang, P.; Yang, X.P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. eBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef]
- Grigalavicius, M.; Ezzatpanah, S.; Papakyriakou, A.; Raabe, T.T.H.; Yannakopoulou, K.; Theodossiou, T.A. 5-ALA Is a Potent Lactate Dehydrogenase Inhibitor but Not a Substrate: Implications for Cell Glycolysis and New Avenues in 5-ALA-Mediated Anticancer Action. Cancers 2022, 14, 4003. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulation at the Level of: | LDHA | LDHB | LDHC | ||||
|---|---|---|---|---|---|---|---|
| Induction | Repression | Induction | Repression | Induction | Repression | ||
| RNA | Epigenetically | methylation | methylation | methylation | |||
| Transcription factors | HIF MYC FOXM1 | KLF4 | STAT3 PGC-1α | KLF14 | CREB | NF-I | |
| Protein | Translation | miRNA | |||||
| Degradation | SIRT2 (K5 deacetylation) | K5 acetylation | |||||
| Activity | Post-translational modifications | FGFR1 (Y10, Y83 phosphorylation) K118 succinylation | SIRT5 (K118 desuccinylation) | SIRT5 (K329 deacetylation) Aurora-A (S162 phosphorylation) | K329 acetylation | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocianova, E.; Piatrikova, V.; Golias, T. Revisiting the Warburg Effect with Focus on Lactate. Cancers 2022, 14, 6028. https://doi.org/10.3390/cancers14246028
Kocianova E, Piatrikova V, Golias T. Revisiting the Warburg Effect with Focus on Lactate. Cancers. 2022; 14(24):6028. https://doi.org/10.3390/cancers14246028
Chicago/Turabian StyleKocianova, Eva, Viktoria Piatrikova, and Tereza Golias. 2022. "Revisiting the Warburg Effect with Focus on Lactate" Cancers 14, no. 24: 6028. https://doi.org/10.3390/cancers14246028
APA StyleKocianova, E., Piatrikova, V., & Golias, T. (2022). Revisiting the Warburg Effect with Focus on Lactate. Cancers, 14(24), 6028. https://doi.org/10.3390/cancers14246028