1. Introduction
Glioblastoma (GBM) is the most malignant form of primary brain tumor, with an average survival time of 14.6 months after initial diagnosis [
1]. Following surgery and radiation therapy, temozolomide (TMZ) is the current mainstay chemotherapy for GBM [
2]. However, patients develop drug resistance over time, which results in cancer recurrence and progression [
3,
4,
5]. In addition, aggressive drug regimens cause considerable toxicities and side effects without advancing therapeutic efficacy [
6]. New therapeutic agents that sensitize GBM cells to current chemotherapy drugs, such as TMZ, at subtoxic doses could improve overall therapeutic outcomes [
7,
8,
9].
Nuclear factor-erythroid factor 2-related factor 2 (Nrf2) is an important transcription factor that plays a major role in GBM therapy by negatively impacting chemotherapy through activation of endogenous phase II detoxification mechanisms. In normal cells, Nrf2 is expressed at physiological levels to maintain the cellular homeostasis and to prevent diseases including cancer [
10]. Nrf2 protein is maintained in the cytoplasm as an inactive complex by binding to a repressor molecule known as Keap1 (Klech-like ECH-associated protein 1) [
11]. During redox stress, the Nrf2-Keap1 pathway is activated. Nrf2 is phosphorylated by protein kinase C and translocases to the nucleus, where it binds to DNA binding domains of the phase-II enzyme to regulate their expression through antioxidant response elements (AREs) [
12]. This process prevents reactive oxygen species (ROS)-mediated DNA damage, and maintains cellular homeostasis. However, previous studies have shown that somatic mutations to Nrf2 in cancer cells disrupt its interaction with Keap1 and prevent ubiquitination that leads to higher accumulation of Nrf2 in cells [
13]. In cancer therapy, higher levels of Nrf2 with the upregulated antioxidant pathway lead to cancer cells acquiring protection from the cytotoxic effects of the chemotherapeutic drugs and the development of chemoresistance.
Tumor suppressor p53 is another important transcription factor necessary for the response of cancer cells to various cytotoxic therapies. p53 regulates cellular apoptosis by controlling the expression of genes involved in cell cycle arrest, DNA repair, apoptosis, and senescence [
9].
TP53 gene is mutated in 85% of GBMs and contributes to the loss of apoptotic response in cancer cells [
14]. Many chemotherapeutic drugs, such as doxorubicin (DOX), activate p53-mediated biological mechanisms to induce cytotoxic cell death in cancer therapy [
14]. In addition, mechanisms of inhibition of apoptosis by p53 owing to the upregulation of Nrf2 in cancer cells have been proposed. Thus, understanding the underlying molecular mechanisms of the crosstalk between these two proteins in response to chemotherapy in mutant-p53 GBM cells can shed light on the possibility of using Nrf2 inhibitors/activators to improve GBM treatment [
15,
16].
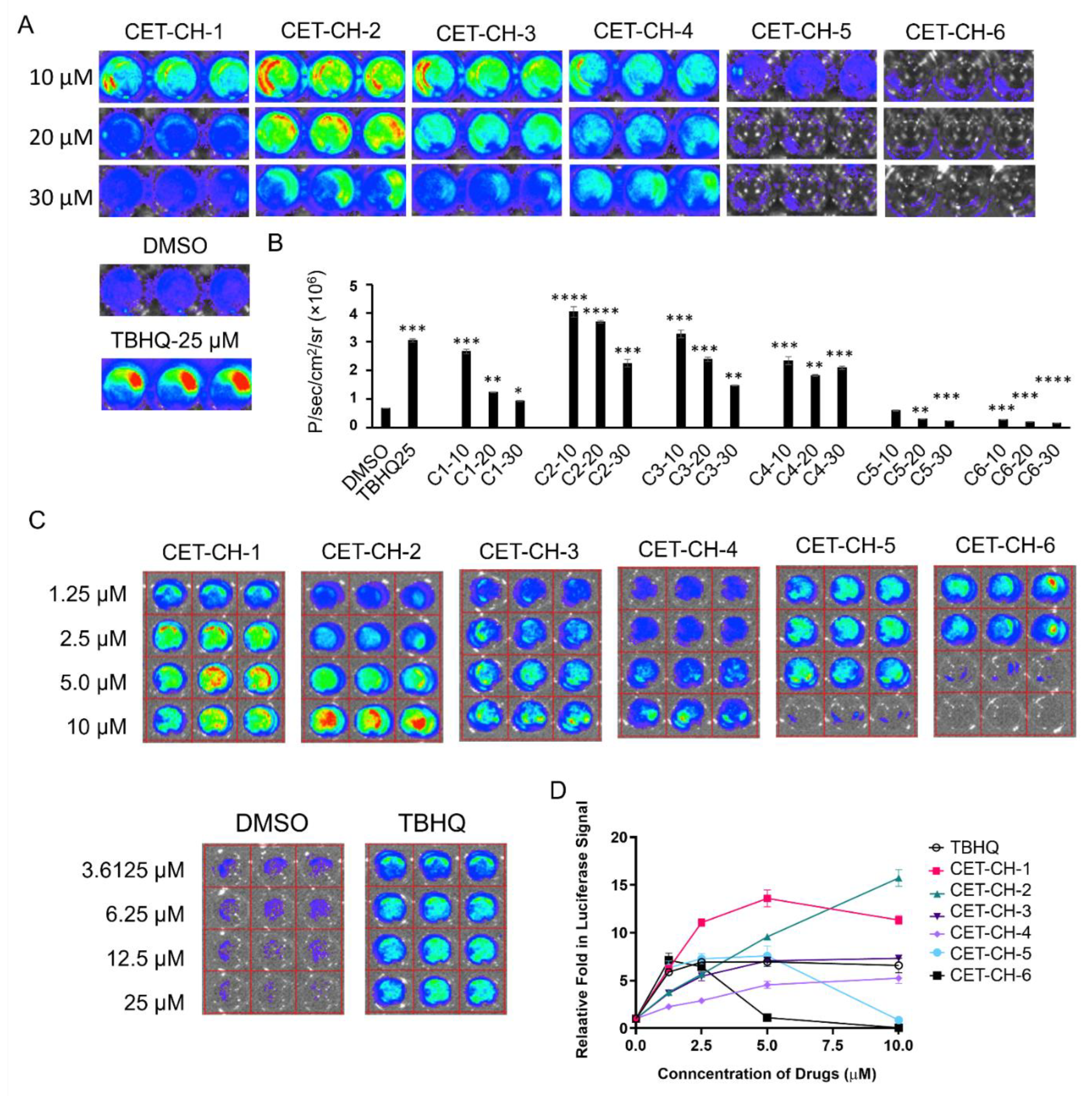
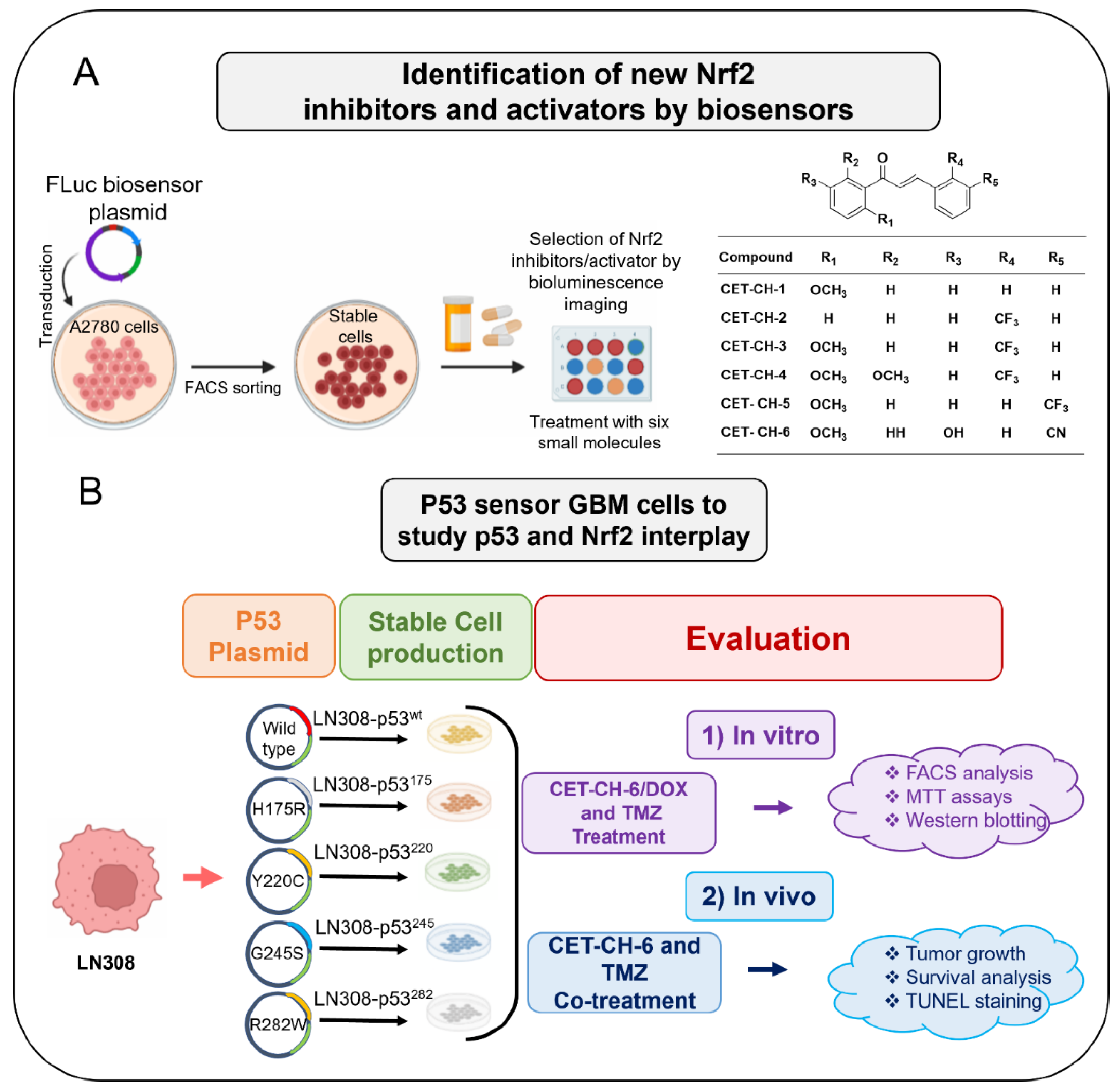
Here, we investigate the interplay between p53 and Nrf2 pathways after treatment using compounds that effectively alter the Nrf2 pathway. We use A2780 cells stably expressing the Firefly luciferase reporter gene under an antioxidant response element (ARE) for screening Nrf2 inhibitors. We use this cell line to study induction of Nrf2 protein upon treatment using small molecule drugs that have been recently studied by Eldhose et al. [
17] (CET-CH-1 to CET-CH-5), along with a novel high potential anticancer agent that we synthesized (CET-CH-6) based on this compound library (
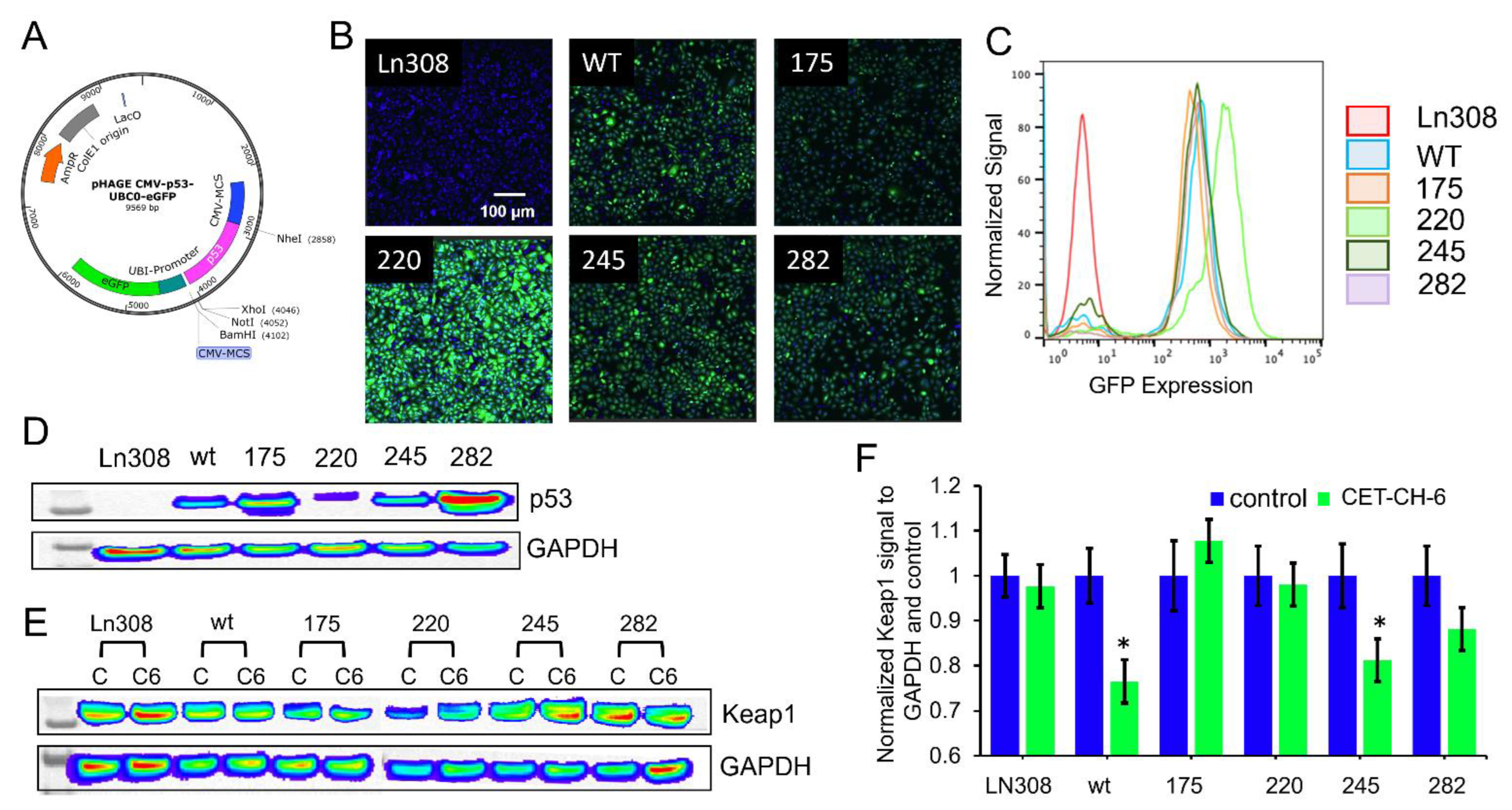
Figure 1A). To study the relationship between mutant p53 and Nrf2, we engineer p53-null LN308 GBM cells to stably express clinically important p53 mutants (
Figure 1B). Through in vitro experiments, we select an Nrf2 inhibitor that enhances the therapeutic effect of TMZ and DOX on GBM cell lines of different p53 status. Tumors of LN308 cells carrying a p53 mutation at amino acid 175, a common missense structural mutation with no effective treatment available, show significant inhibition in growth in vitro and in vivo to the selected Nrf2 inhibitor when combined with TMZ.
2. Materials and Methods
2.1. Chemicals, Enzymes, and Reagents
We purchased A2780 human ovarian cancer cells, U87-MG (#HTB-14), HEK293T (#CRL-3216), and MDA-MB-231 (#CRM-HTB-26) from ATCC (Manassas, VA, USA). LN308 cells (p53 null) were a kind gift from Markus Weiler (Heidelberg University, Germany). We purchased propidium iodide (#P4864), TBHQ (#112941), and doxorubicin (D1515) from Sigma-Aldrich (St. Louis, MO, USA), and DMSO (D159-4) from Fisher Scientific (Santa Clara, CA, USA). We used FBS (#26140087), Penicillin-Streptomycin-Glutamine (#10378016), and sodium bicarbonate (#25080094) from GIBCO BRL (Frederick, MD, USA). We used plasmid extraction kits and DNA gel elution kits from Qiagen (Valencia, CA, USA) and Epoch Life Sciences (Missouri City, TX, USA). We purchased D-Luciferin from Biosynth (Staad, Switzerland). Antibiotics for bacterial and cell culture experiments were from Sigma (St. Louis, MO, USA), and bacterial culture media was from Difco (Franklin Lakes, NJ, USA).
2.2. Synthesis of the Compounds
Synthesis and characterization of chalcone derivatives (CET-CH-1 to CET-CH-5) were performed as described previously [
17]. Synthesis of CET-CH-6 is described in the
supporting information.
2.3. Cell Culture
We cultured the cells in high-glucose Dulbecco’s Modified Eagle’s Medium (#11965092) (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin solution. We maintained cells at 37 °C in 5% CO2, and plated them for experiments to an optimal confluency during their exponential growth phase.
2.4. Construction of Plasmid and Lentiviral Vectors Expressing Nrf2-ARE-FLuc Sensor
We constructed an Nrf2-ARE-FLuc biosensor using a standard PCR based cloning strategy. In brief, we first constructed ARE from Nrf2 target gene (NQO1) by replacing the CMV promoter from pcDNA 3.1+ vector backbone. Then, we introduced the full-length luciferase reporter gene downstream of ARE in NheI and XhoI sites to achieve the pcDNA-NQO1-FLuc construct. The NQO1-FLuc fragment released from the pcDNA-NQO1-FLuc construct using SpeI/XhoI was subcloned into pHAGE lentiviral backbone for producing lentiviruses to create A2780 stable cells for screening Nrf2 activators/inhibitors. We used the sequenced vector constructs (confirmed by Sequetech, Mountain View, CA, USA) in cell culture transient transfection experiments to produce lentivirus. We used the three-vector based transfection system described later for lentivirus production and further transduction in creating stable cells.
2.5. Construction of Plasmid and Lentiviral Vectors Expressing p53 Variants
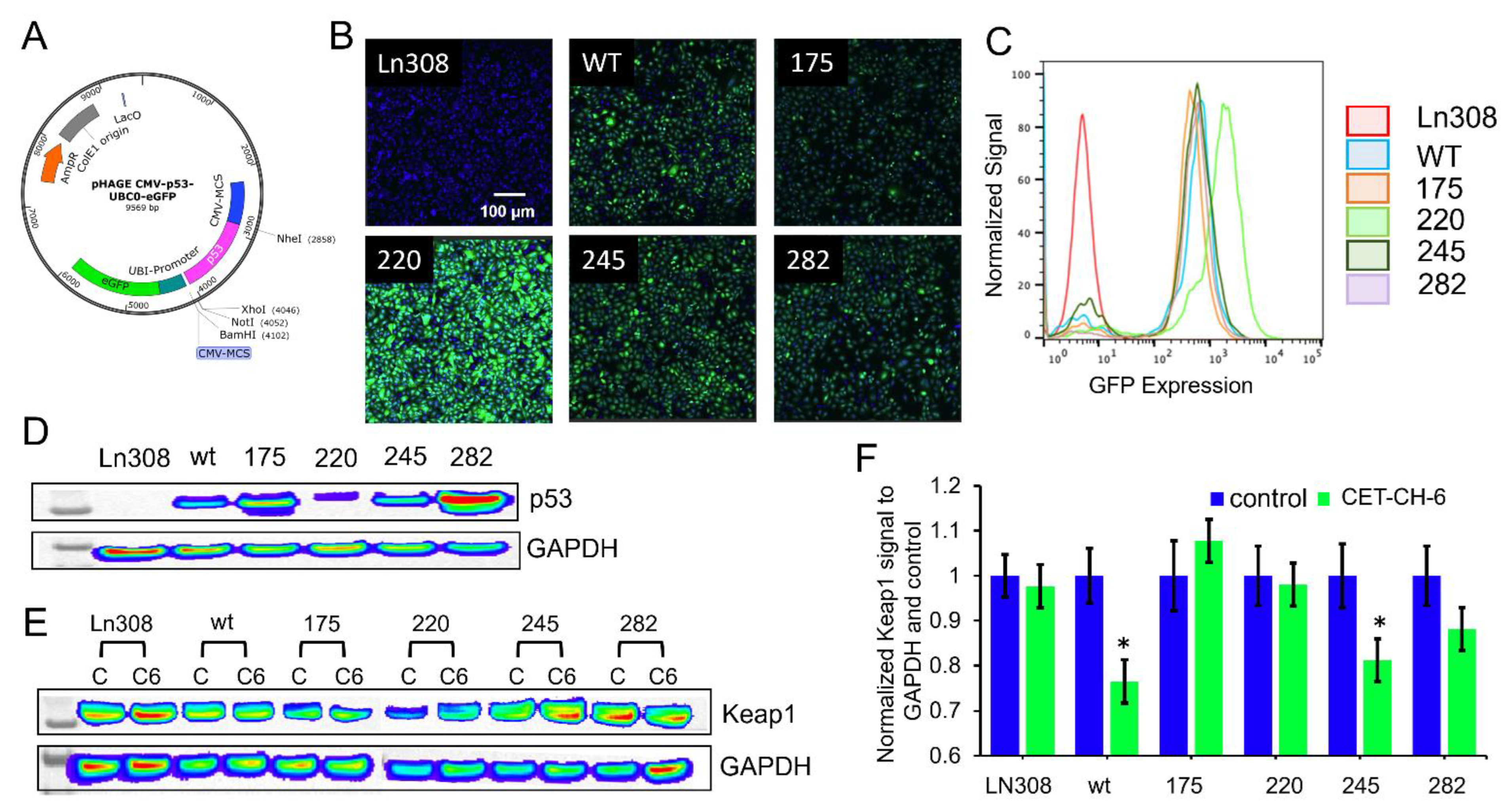
We constructed plasmid vector expressing p53 proteins of wildtype (wt) and mutants under a ubiquitin promoter in a pcDNA 3.1 (+) backbone with a puromycin antibiotic selection marker. We first constructed a vector with p53-wt protein using a standard PCR based cloning strategy. We used site-directed mutagenesis to create a single amino acid change at positions 175 (p53R175H), 220 (p53Y220C), 245 (p53G245S), and 282 (p53R282W) using a site-directed mutagenesis kit from Stratagene (Agilent Technologies, Santa Clara, CA, USA). We used the sequence confirmed vector constructs for transient transfection experiments and for sub-cloning into a lentiviral backbone. These five constructs were further released along with the constitutive Ubiquitin promoter (Ubiquitin-p53) and sub-cloned into a pHAGE-lentiviral backbone at the SpeI/XhoI restriction enzyme sites. We then chose the sequence confirmed vectors for lentiviral production using the three-vector transfection system as described later.
2.6. Lentiviral Production and Stable Cell Generation by Lentiviral Transduction
We maintained A2780 and LN308 cells in DMEM-high glucose medium with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. For lentiviral production, we used HEK293-FT cells grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. We used a three-vector transfection system (pHAGE + VPR + VSVG) to produce lentivirus for NQO1-ARE-FLuc and p53 variants with a GFP biosensor expressing p53wt, p53175, p53220, p53245, and p53282 in a 3:2:1 ratio using a standard calcium phosphate transfection method. Twenty-four hours later, we washed the cells once with PBS and replaced with 8 mL of complete medium containing 10 mM Hepes buffer, pH 7.5. We enriched the virus from the medium after 48 h by filtration followed by ultracentrifugation. After titration, we used viruses for creating the A2780-Nrf2-ARE-FLuc stable clone and LN308 cells stably expressing p53 variants. In brief, we transduced the cells plated to 80% confluence in 10-cm plates 24 h before transduction. We washed the cells once with PBS, and then added 2 mL of 1 × 108 PFU virus mixed with 3 mL of serum-free Opti-MEM and 5 μL of 1 mg/mL polybrene. Each plate was incubated for 4 h at 37 °C and 5% CO2 with intermittent mixing. Four hours later, we washed the plates once with PBS and supplemented with 10 mL of complete medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. We sub-cultured the cells twice before we selected stable cells using FACS sorting. The clonal populations of cells expressing equal levels of p53 constructs were used in further experiments.
2.7. Evaluation of Nrf2 Activation Using Are Signaling Characterized by FLuc Biosensor Bioluminescence Imaging
We treated A2780 cells stably expressing NQO1-FLuc biosensor using different Nrf2 modulators (CET-CH-1 to CET-CH6) along with TBHQ as a positive control, and DMSO as a solvent negative control. We used CET-CH-1 to CET-CH-6 in various concentrations (0, 1.25, 2.5, 5,10, 20, and 30 mM) for the screening studies. We plated 5000 cells/well in 96-well black walled plates 24 h before treating the cells using these Nrf2 modulators. After 48 h, we washed the plates once with PBS and added 30 μg of D-Luc (30 mg/mL) in 50 μL of PBS to each well using a multichannel pipette. We imaged the plates by continuous acquisition of 1 min integration times for a total of 15 min. We quantified luciferase signal from each well using Living Image software by drawing regions of interest (ROIs) over each well. We plotted the relative signal in response to treatment as a graph. The FLuc signal was proportional to Nrf2 activation.
2.8. Immunoblot Analysis for the Evaluation of Downstream Target Gene Signaling
For the analyses of each corresponding pathway, we collected cells after appropriate treatment by centrifuging at 5000 rpm for 5 min and lysed the cell pellets in RIPA buffer containing protease inhibitor cocktail and EDTA by sonication three times at 40% amplitude for 15 s each in ice. We centrifuged the cell lysates at 15,000 rpm for 15 min at 4 °C to remove any insoluble proteins. We estimated the protein concentration using Nanodrop 2000 (Thermo Scientific, Waltham, MA, USA) and further confirmed this using a BCA protein assay system from BioRad. We prepared 200 μg of total protein in 1× Lamelli loading buffer with 5% β-mercaptoethanol (Life Technologies, Carlsbad, CA, USA), and denatured this at 95 °C for 5 min. We resolved the samples on 4-12% SDS-polyacrylamide pre-cast gels (Life Technologies, Carlsbad, CA, USA) and transferred to a polyvinylidene difluoride nylon membrane (Bio-Rad, Hercules, CA, USA) using wet electroblotting. Membranes were blocked in 5% non-fat dry milk in Tris-buffered saline containing 0.05% Tween-20 (TBST), (#P9416) from Sigma-Aldrich followed by incubation with a primary antibody. As appropriate, we used antibodies suitable for the detection of genes suggested by the manufacturers. Following three TBST washes, we incubated membranes with the appropriate horseradish peroxidase-conjugated secondary antibodies (Sigma Aldrich, St Louis, MO, USA). After three additional TBST washes, we incubated immunoblots with the Pierce ECL Western Blotting Substrate (Thermo Scientific, Waltham, MA, USA) for 1 min, and then collected the chemiluminescence signal using an IVIS Lumina imaging system (PerkinElmer, Bridgeville, PA, USA). We used a GAPDH (#5174) and Lamin B1 (#12586) primary antibodies (CST, Danvers, MA, USA) diluted in PBST at 1:2000 as a loading control house-keeping antibody for cytoplasmic and nuclear, respectively. For the expression of genes in response to CET-CH-6 (5 μM) and DOX (0.5 μM) in different treatment conditions, we used Nrf2 (#sc-365949) and Keap1 (#sc-365626) from Santa Cruz Biotechnology (Santa Cruz, CA, USA) as primary antibodies by diluting in PBST at 1:1000. For the expression of p53 in LN308 cells stably expressing p53 variants, we used p53 primary antibody from Cell Signaling (#9282, CST, 1:2000). Similarly, for measuring the change in response to treatment from LN308 cells stably expressing p53 variants after treating with CET-CH-6 and DOX, we used p53 (#9282), Bcl2 (#4223), Bax (#2774), Noxa (#14766), Puma (#4976S), and p21 (#2947) from Cell Signaling (CST, 1:2000), and SOD2 primary antibody from Santa Cruz Biotechnology (#sc-137254). In MDA-MB-231 cells, where we investigated the distribution of the proteins in the nucleus and cytoplasm, we used a different extraction kit by following the manufacturer’s protocol. We quantified nucleocytoplasmic distribution of Nrf2 and Keap1 proteins upon different treatment conditions using a Thermo Scientific NE-PERTM Nuclear and Cytoplasmic Extraction Reagents kit (# 78833, Thermo ScientificTM). Briefly, cells from different treatment groups were collected and pelleted by centrifugation. The cells were suspended in ice-cold CER II. We vortexed the tubes and centrifuged the samples and transferred the supernatant that contained a cytoplasmic extract. The pellet which contained the nuclei was suspended in NER. We vortexed the tubes (4 times with 10 min interval) and centrifuged at maximum speed and immediately transferred the supernatant (nuclear extract). The protein fractions were used for immunoblotting analysis.
2.9. Confocal Microscopy
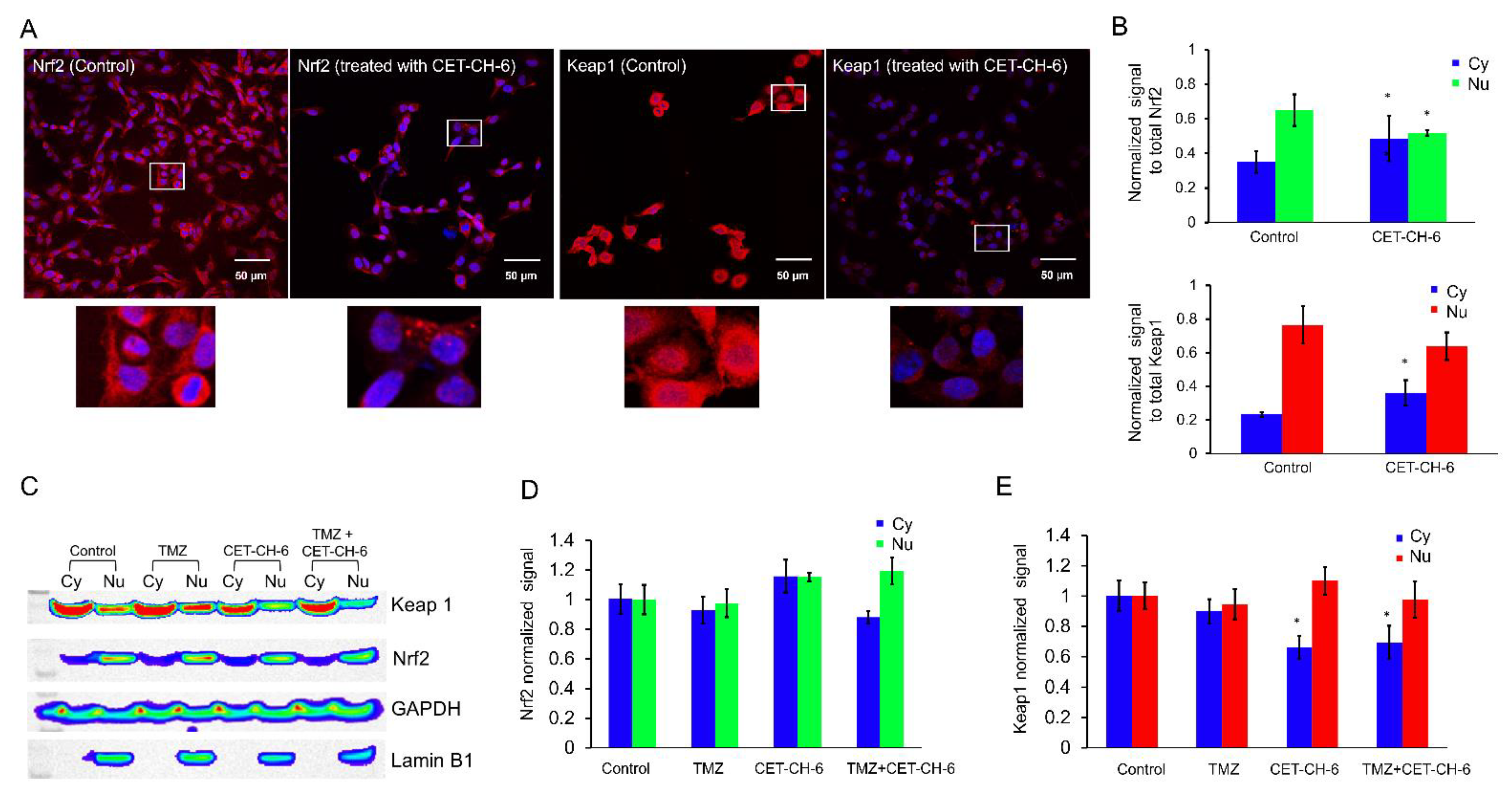
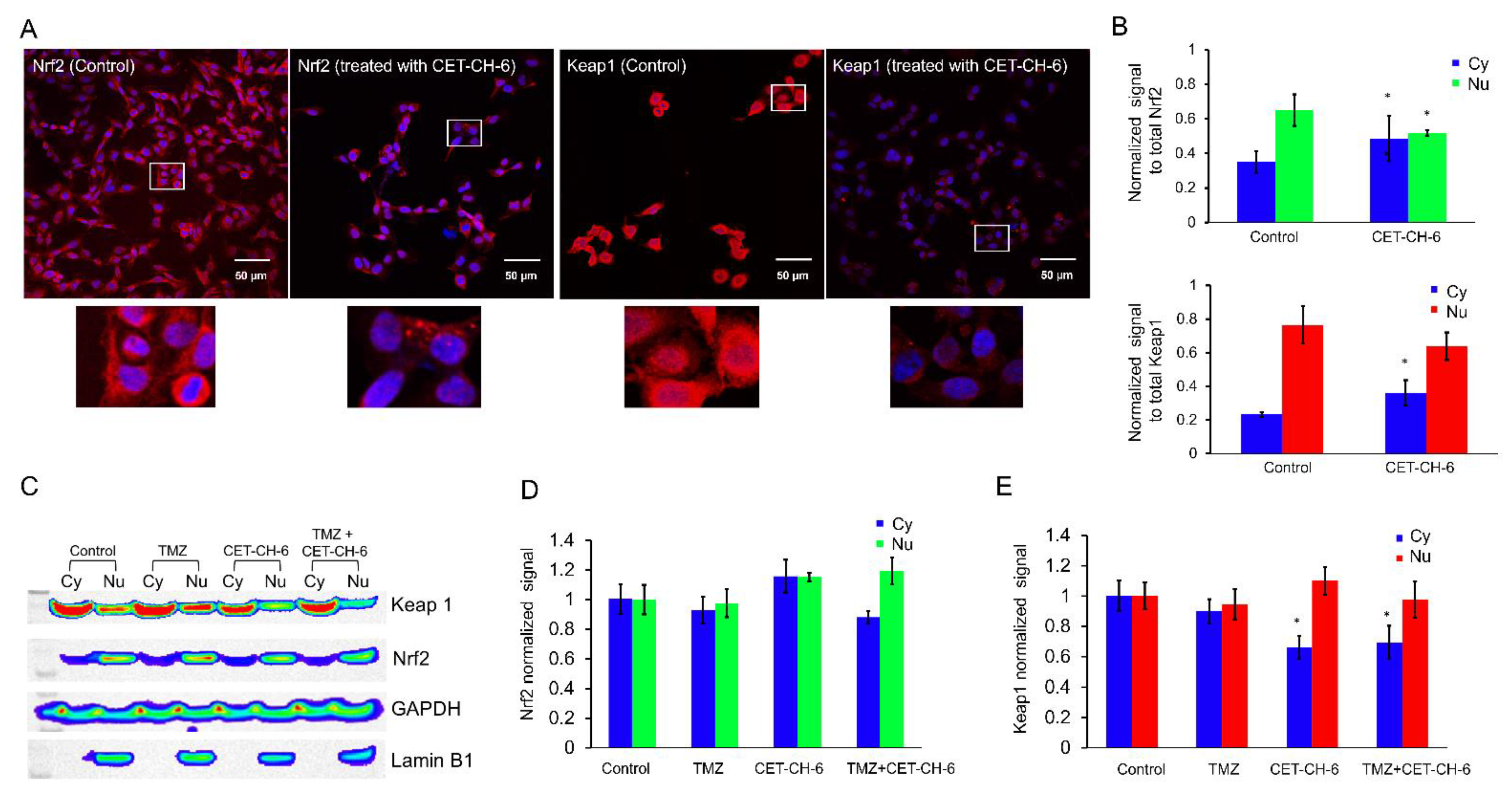
We used confocal microscopy (TCS SP8, Leica, Wetzlar, Germany) to image MDA-MB-231 cells to study the mechanism of activation of Nrf2 in response to CET-CH-6. We seeded 100,000 cells on cover slips (Fisher Scientific, Santa Clara, CA, USA) placed in 6-well plates 24 h before treatment. We treated the cells with CET-CH-6 at 2.5 μM concentration for 24 h. On the day of imaging, we washed the cells with PBS, and fixed them in 1% ice cold PFA by keeping them for 20 min on ice. We then washed them twice with ice cold PBS. We permeabilized the cells by adding 1 mL of permeabilization solution for 1 h. We stained the cells using Nrf2 and Keap1 antibodies (1:200) diluted in PBST with 1% bovine serum albumin and incubated them overnight in a humidified chamber. The next day, we aspirated the solution and washed it with PBST three times, and then stained it with anti-mouse FITC secondary antibody (1:200) for 2 h at room temperature. We then washed the cells with PBST three times, and added Hoechst 33342 to stain the nucleus, mounted cells, and acquired optical images using appropriate imaging channels. We also confirmed the expression of GFP in LN308 clones using confocal microscopy by culturing them on coverslips and imaging them in a GFP channel.
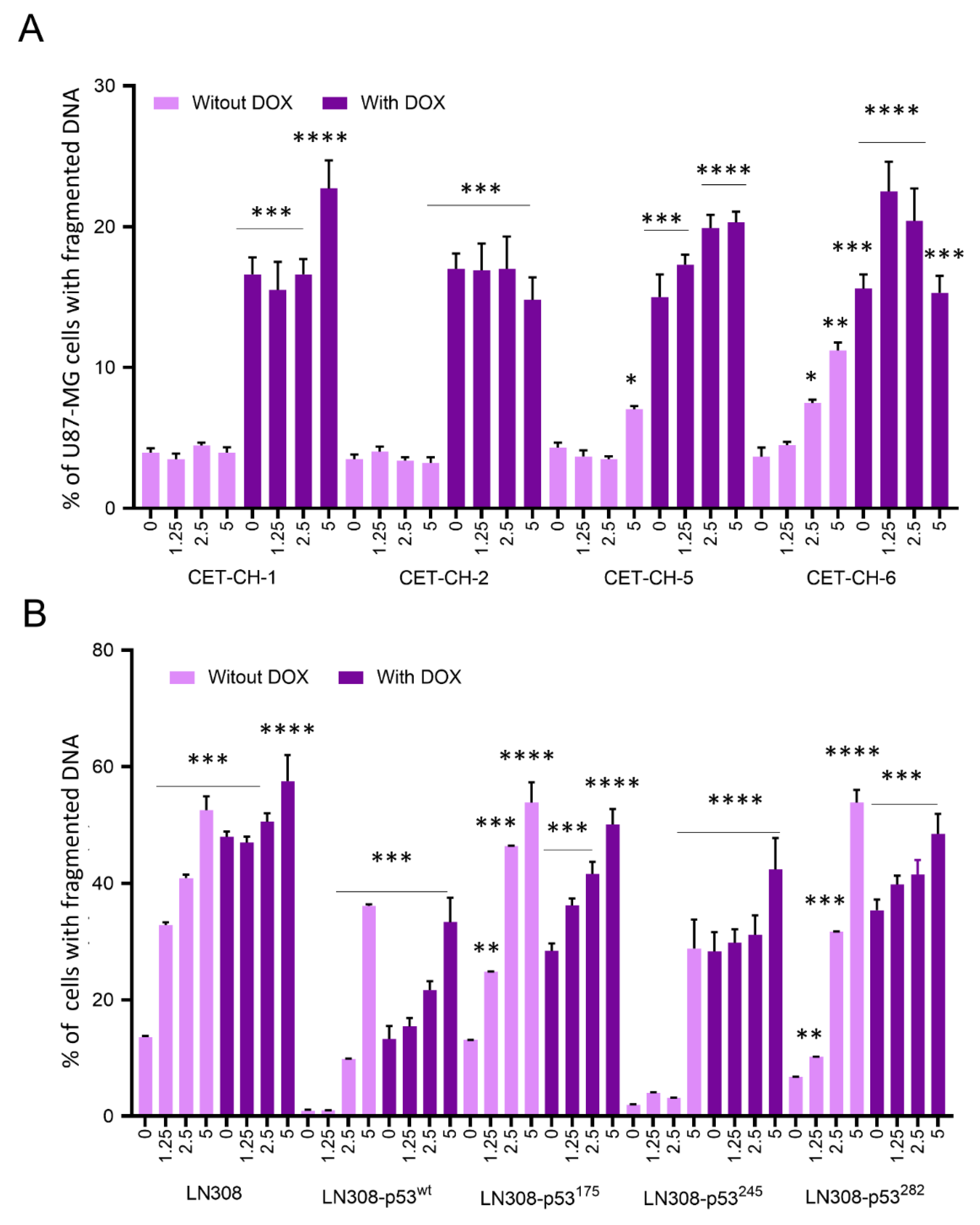
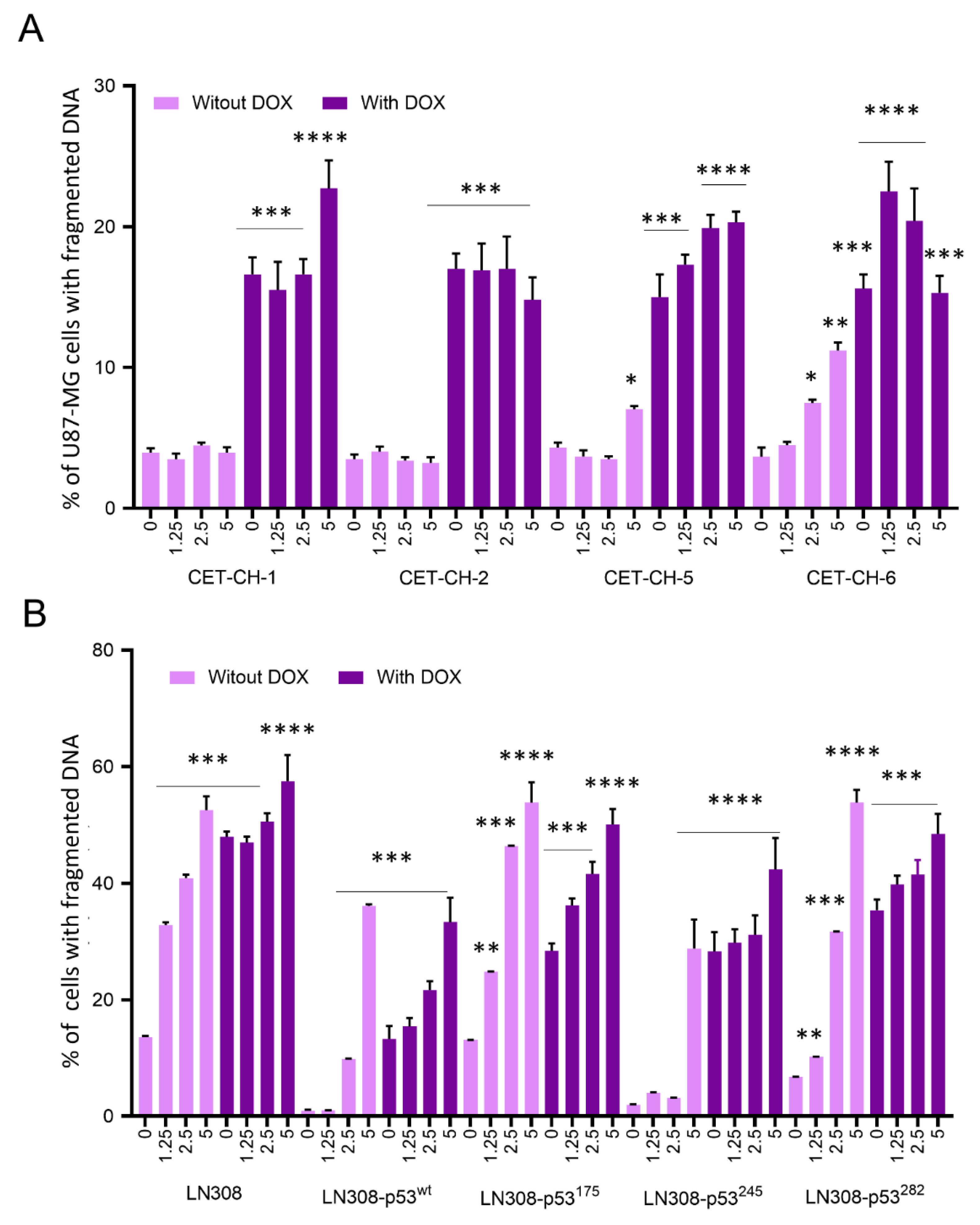
2.10. Evaluation of Apoptosis in U87-MG-p53wt and LN308 Cells Stably Expressing p53 Variants in Response to Treatment with Nrf2 Modulators in the Presence of Chemotherapy Using Propidium Iodide Staining Based Flow Cytometry Analysis
For apoptosis induction in U87-MG and LN308 cells stably expressing different p53 variants, we seeded the cells at 80% confluency in 12-well plates at 1 × 10
5 cells/well in DMEM medium supplemented with 10% FBS for 24 h before co-treatment with CET-CH-1 to CET-CH-6 and DOX. We then treated cells with different concentrations of CET-CH-1 to CET-CH-6 (0, 1.25, 2.5, and 5 μM) in the presence and absence of DOX and incubated them further for 48 h. For the assessment of apoptotic populations, we used two different assays. In the first assay, we collected and fixed the cells for PI-based flow cytometry analysis. In this assay, the apoptotic population is distinguished from the non-apoptotic cells based on the DNA content and its locational and cellular distribution. In non-apoptotic cells, DNA is localized within the nucleus, whereas, in apoptotic cells, the DNA is fragmented. Since we are fixing the cells, the cell membrane is permeable for PI independent of their cellular status. Hence, after PI staining, the apoptotic population shows a weak PI signal from the fragmented DNA while non-apoptotic cells show a strong signal from the intact DNA [
18]. We collected the cells by trypsinization, resuspended them in 0.5 mL ice-cold PBS, and then fixed them by adding 2 mL of 100% ice-cold ethanol while vortexing the sample to achieve a final concentration of ethanol to 70%. We stored the samples at −20 °C until FACS analysis. We have successfully used this assay in many of our previous publications [
19,
20,
21,
22]. Briefly, the cells were collected by centrifugation, washed once in PBS, and re-suspended in 0.5 mL PBS/RNase A (10 µg/mL)/Triton X-100 (0.1%) buffer containing 0.5 µg/mL of PI. After 15 min of incubation at room temperature in the dark, we washed the cells once with PBS and FACS analyzed using a Guava cytometer. In the second assay, we treated the cells in a similar way, but we stained the cells using the Annexin-V/PI kit (Invitrogen, Waltham, MA, USA) right after the collection of cells. We assayed the samples using the Guava cytometer. We analyzed the generated results using FlowJo 8.6.6 software for live and dead cell analysis.
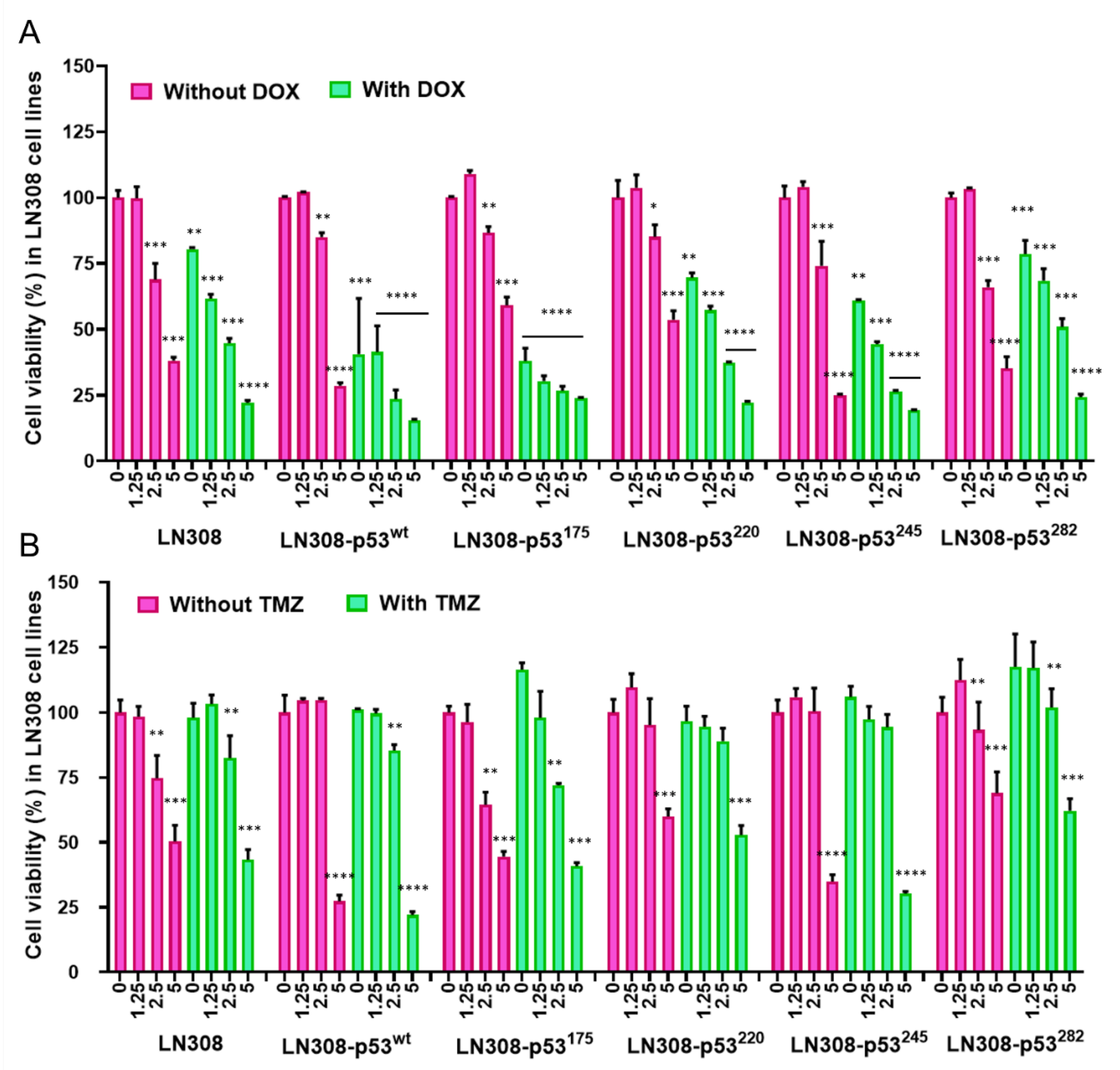
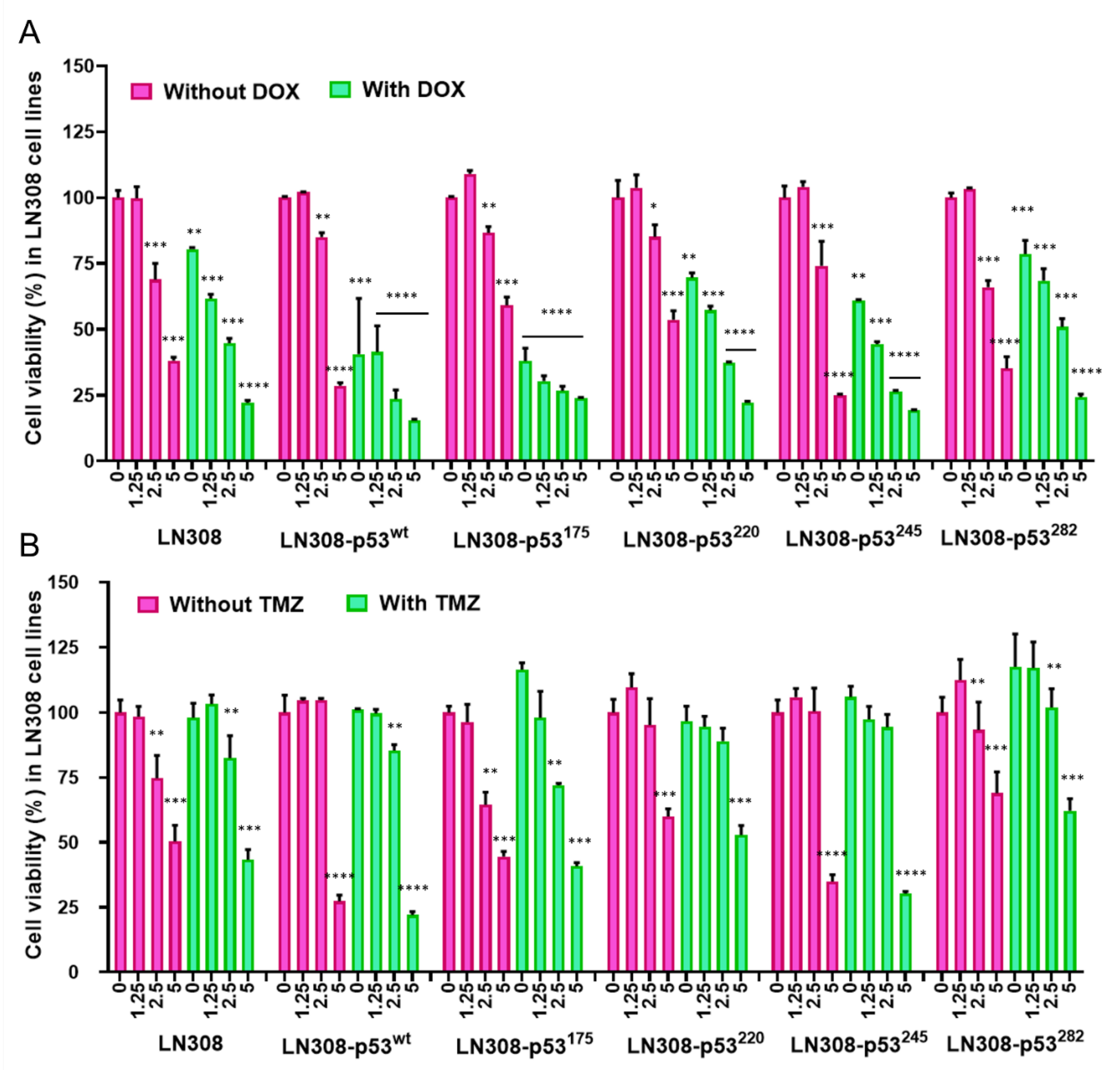
2.11. Evaluation of Cell Viability Using MTT Assay
We used MTT assays to assess the viability of cells after exposure to different treatment conditions. We seeded the cells in 96-well plates at a density of 2500 cells/well 24 h before treatment. We then treated cells with different concentrations of CET-CH-6 (0, 0.125, 0.25, 0.5 μM) in the absence and presence of DOX (0.5 μM), and TMZ (250 μM). At 24, 48, and 72 h after treatment, we aspirated the DMEM, and added 50 μL of MTT solution (diluted in phenol red free RPMI to a final concentration of 1.25 μg/mL) per well of 96-well plates. We then centrifuged plates at 1000 rpm for 3 min before incubating with the MTT solution at 37 °C in the dark for 2 h. After 2 h, we carefully removed the MTT solution without disturbing the formazan crystals and added 100 μL DMSO per well, and allowed for dissolving by keeping it in the incubator for 30 min. After incubation, the spectroscopic signal at 565 nm wavelength was measured on a Tecan machine in accordance with manufacturer instructions. The results were given as percentages of viable cells relative to the control condition.
We also calculated drug synergy by the coefficient of drug interaction (CDI): CDI = AB/(A × B). Based on the absorbance of each group, AB is the ratio of the combination group to control group, and A or B is the ratio of the single agent group to control group. CDI of less than 1 represents synergistic effects.
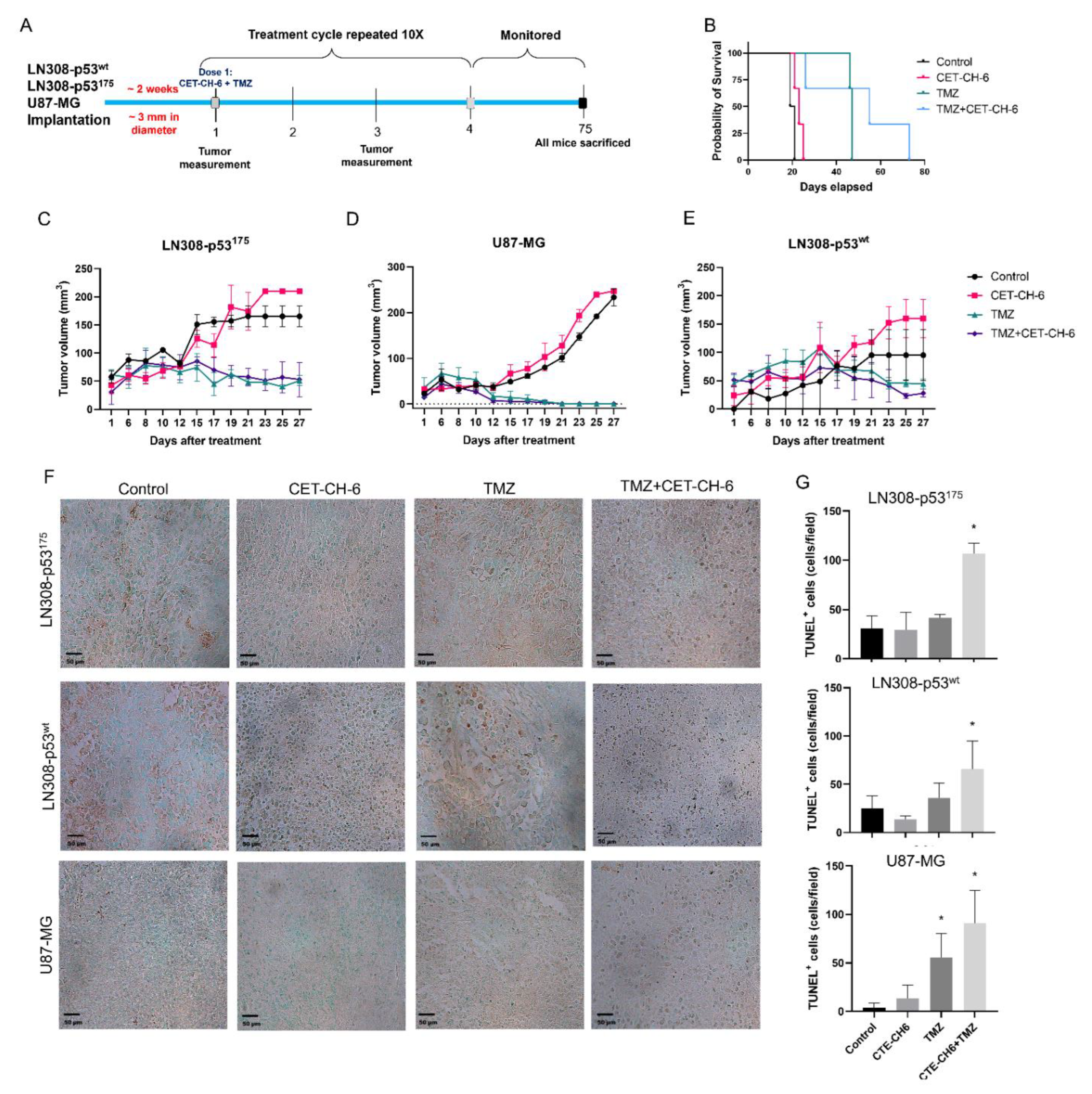
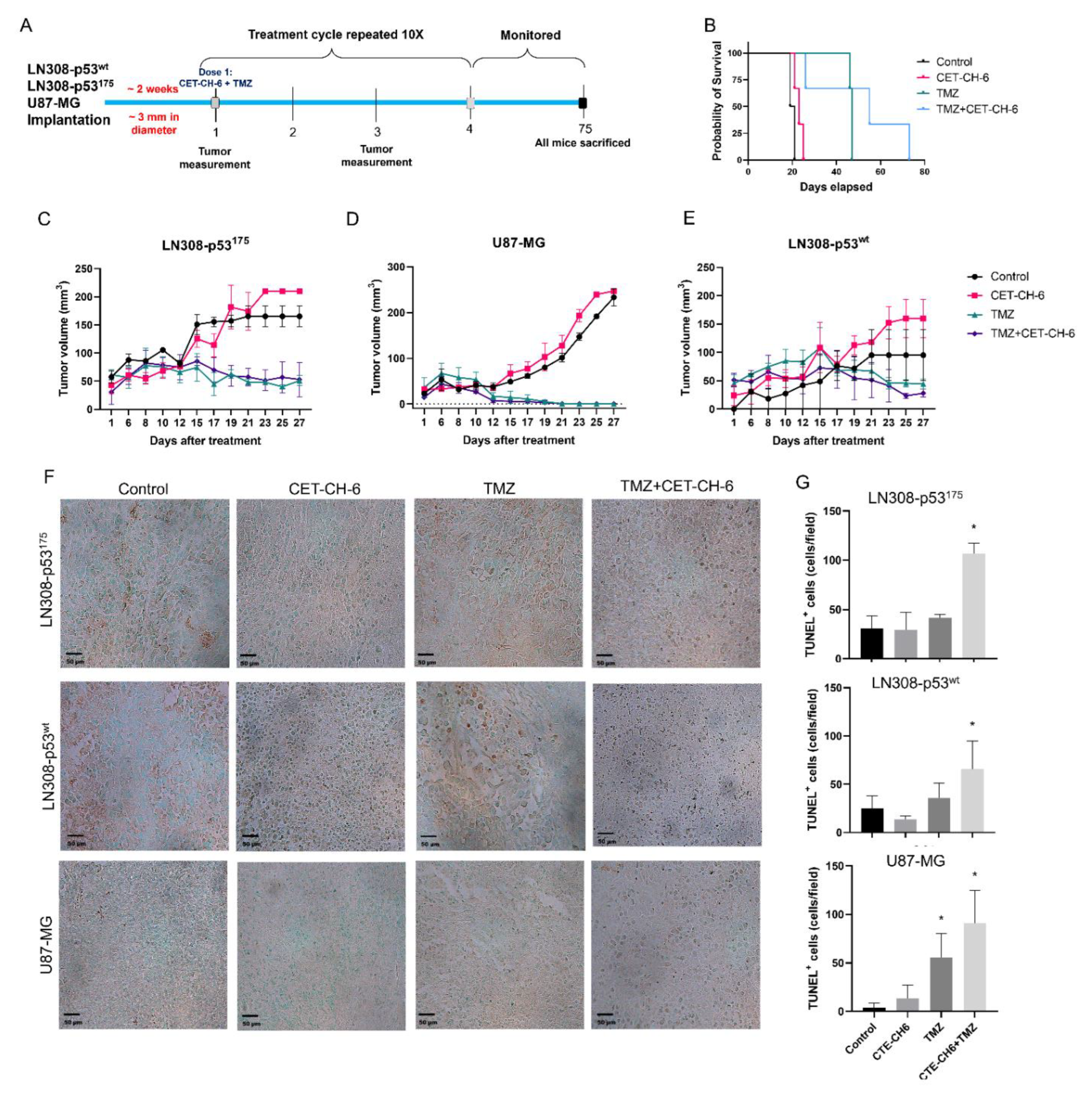
2.12. In Vivo Evaluation of Co-Treatment of CET-CH-6 and TMZ in Subcutaneous GBM Models in Balb/C Nude Mice
All animal experiments were performed in accordance with the Stanford University Administrative Panels on Laboratory Animal Care. We used NSG mice aged 4–6 weeks. We conducted two sets of animal studies. In the first set, we implanted mice with 5 × 106 LN308-p53wt and LN308-p53R175H into the left and right flanks, respectively. We allowed tumors to grow until they reached 3 mm in diameter, and randomized the mice into four groups (number of tumors, n = 4–6 per group). Control mice did not receive any treatment, the TMZ treated group received TMZ (12.5 mg/kg body weight), the CET-CH-6 group received CET-CH-6 (2.5 mg/kg body weight), and the CET-CH-6 + TMZ group received a co-treatment of those drugs at similar concentrations. We treated mice with intraperitoneal injection of drugs diluted in 10% PEG-400 once every two days for five cycles of treatment. We recorded mouse body weight and tumor volume throughout the study. We sacrificed the animals when tumor size exceeded the allowed limit (1.5 cm3), or at day 75 after start of the treatment. We then collected organs and tumors for ex vivo histological, toxicity, and apoptosis analysis. Based on the outcome of the first set, we implanted another set of animals with U87-MG-p53wt and LN308-p53R175H cells in the right and left flanks, respectively, and repeated the same treatment plan. We used TUNEL staining to measure the induced apoptosis.
2.13. H&E Staining of the Organs
We harvested the mice organs and fixed them in 4% paraformaldehyde overnight at 4 °C, and immersed them in 70% ethanol. We sent the samples to the Stanford Animal Histology Services for H&E staining, and imaged them using a Nanozoomer (Hamamatsu, Japan).
2.14. TUNEL Staining of Tumor Tissues
To quantify tumor cell apoptosis upon treatment, we performed a TUNEL assay (TACS 2 TdT-DAB In Situ Apoptosis Detection Kit, Trevigen, Gaithersburg, MD, USA). We sliced paraffin embedded sections and deparaffinized them to perform the assay, labeled according to the manufacturer’s instructions. We scanned the slices under microscope and performed image analysis using ImageJ and the Color Threshold and Particle Analysis functions. We presented the total number of TUNEL positive cells in each sample per field of view to quantitively measure the treatment outcome.
2.15. Statistical Analysis
We performed statistical analyses and prepared graphical presentations using Prism 9 (GraphPad). Results were presented as mean ± SD. Results were representative of at least three independent experiments. Observations with a p-value of less than 0.05 were considered statistically significant. Kaplan–Meier analyses were used for statistical analyses of survival rates.
Note: Additional methods, please refer to
Text S1.
4. Discussion
We identified a novel Nrf2 inhibitor, named CET-CH-6, that synergistically enhances therapeutic effects of TMZ and DOX in GBM cell lines with specific p53 hotspot mutations. To identify this inhibitor, we first developed a biosensor expressing the (NQO1-FLuc) reporter cell line, which indirectly measures the activation of the transcriptional activity of the Nrf2 protein. The testing of this sensor using a known Nrf2 activator, TBHQ, showed a specific activation (using FLuc expression) by 3- to 4-fold. We thus identified four compounds with significant Nrf2 activation while two others showed high and selective inhibition of Nrf2 activity by 4- to 5-fold (
Figure 2). Further validation of these molecules in GBM cells with wild-type and mutant p53 showed differential expression of downstream target proteins of Nrf2.
The presence of structural mutations in the DNA binding domain of p53 gene in GBM makes p53-augmented chemotherapy an attractive strategy to combat GBM. To further facilitate the use of p53 mutant GBM cell lines, we created a library of GBM cell lines that stably express clinically important p53 mutations using p53-null LN308 cells to achieve clonal populations of p53 variant cells in a single genetic background. Chemoresistance in cells with mutant p53 occurs by upregulation of Nrf2 expression in many cancers [
28,
29,
30]. To overcome drug resistance, Nrf2 inhibitors may be used to reduce the activity of Nrf2 while sensitizing the cells to chemotherapies [
31,
32]. For example, treatment of U251-MG and U87-MG cells with FYT720 as an immunosuppressive drug suppressed Nrf2 levels in these cell lines, and significantly sensitized them to TMZ by abolishing the activation of Nrf2 [
33]. Our results support the notion that treatment with CET-CH-6 as an Nrf2 inhibitor promotes cellular apoptosis in GBM cells by 30% more than the baseline levels in LN308, independent of p53 status. However, LN308-p53
wt showed higher sensitivity to co-treatment using CET-CH-6 and DOX compared to p53-null LN308 cells. In addition, the mutant cells carrying the p53
175 variant had the highest inhibition in growth, while p53
220 was the most resistant cell line when co-treated with DOX and CET-CH-6.
Keap1-dependent regulation of Nrf2 in the cytoplasm has been reported as one of the main regulatory mechanisms of Nrf2 [
34], although this is not the only possible explanation for Nrf2 activation [
35]. Keap1 is expressed in the cytoplasm and mediates ubiquitin-mediated degradation of Nrf2 to tightly control its level in cells. Keap1 is also expressed in the nucleus at a lower level [
36]. Upon treatment using CET-CH-6, the expression of Keap1 in the cytoplasm decreased while its expression in the nucleus increased (
Figure 3). We also found that, at the molecular level, Nrf2 showed a similar activity pattern to Keap1. Most studies support the translocation of Nrf2 from cytoplasm to nucleus upon redox stress, and the observation of Keap1 following the same path needs further verification. We observed some variations between confocal and western for the nuclear Nrf2 in control cells. For immunostaining, we rapidly fix the cells, and hence there are no transient stresses during the process for the Nrf2 to rapidly move to the nucleus. In contrast, for western, we collect cells after trypsinization. Hence, there are some additional stresses during this process, which causes the translocation of more Nrf2 into the nucleus. Here, we overall compared the nuclear Nrf2 in control cells vs. treated group. In addition, in the confocal images, we are showing a single slice close to the nucleus, but, in western, the result is from the entire nuclear protein. TMZ alone and in combination with CET-CH-6 significantly reduced the expression of Keap1 and Nrf2 in cells in both the cytoplasm and nucleus. This suggests that TMZ causes ubiquitination and degradation of both molecules.
Cell survival, senescence, and apoptosis signaling regulated by NF-kB, p53, and Nrf2 play important roles in multidrug resistance [
37]. Our Western blotting analysis showed that pathways related to p53 such as p21, Puma, BAX, and Bcl2 become highly activated through co-treatment with DOX and CET-CH-6. We observed upregulation of p21 in LN308 with mutant p53 specifically in LN308-p53
175 and LN308-p53
245, the two cell lines with very effective treatment results. P21 has been shown to stabilize Nrf2 by binding to Keap1 and preventing its degradation [
38]. Puma, which is mainly regulated by p53, showed basal level activation in the LN308 parental cell line, suggesting that there might be pathways involved in the activation of PUMA other than p53-mediated regulation, which also needs further investigation. The level of SOD2 as a downstream target of Nrf2 did not change significantly after treatment with CET-CH-6, except for LN308-p53
wt, which suggests that the Nrf2 pathway was not completely inhibited in cells carrying p53 mutant variants. Here, we mainly focused on activation of the p53 and Nrf2 pathways. Whether Nrf2 inhibition can enhance activation of the downstream genes of NF-kB needs to be investigated in the future studies.
Nrf2 has been known to have a dual nature, which raises concerns when considering it as a promising treatment addition [
39]. Upon toxicity, a cell will activate Nrf2 to transcribe protective genes and regulate cytoprotective mechanisms. Cells will continuously produce Nrf2 until they reach a threshold. Once the threshold is reached, Nrf2 switches from its cytoprotective functions into apoptotic functions. Our access to a set of small molecules enabled us to pay particular attention to monitoring the switches from protective redox signaling into apoptotic signaling. We expected to see higher apoptotic populations when cells were treated with CET-CH-2 as an Nrf2 activator. However, when we treated U87-MG cells with CET-CH-2, we did not observe any inhibition in cell growth (
Figure 5A). This may be because the switching threshold in cells was not achieved by this compound. Unlike U87-MG cells, LN308 cells with mutant p53 showed some response to CET-CH-2 treatment. Co-treatment with CET-CH-2 and DOX showed that CET-CH-2 has some synergistic effects in the inhibition of cell growth, especially on LN308-p53
245. Interestingly, U87-MG-p53
wt and LN308-p53
wt, even though both had wild-type p53, showed differential responses to treatments with CET-CH-2 or CET-CH-6. In addition to genomic differences between these cell lines, differential responses might be owing to the genomic location of p53. Whereas U87-MG cells possess native endogenous p53, LN308-p53
wt possess the same in different genomic locations.
Currently, TMZ is the first line chemotherapy treatment for GBM. It works by alkylation of cellular DNA leading to DNA cross links and cell death. Nrf2 is understood to be one of the key resistance mediators in GBM and melanoma. Indeed, silencing of the Nrf2 pathway greatly enhances cell death in vitro and in vivo via different pathways [
40,
41,
42,
43]. To study the role of our Nrf2 inhibitor on mutant p53 in vivo, we co-treated mice bearing different subcutaneous GBM cells with CET-CH-6 and TMZ. Those findings supported our in vitro results by observing and higher apoptosis in the co-treated mice compared to TMZ alone mice after ten doses of drugs (
Figure 8). We also determined that the CET-CH-6 compound is particularly effective on the p53 mutation at amino acid residue 175 which is a clinically common hotspot mutation in GBM, and the most frequently mutated site at the DNA binding region [
14]. The p53
R175H is a conformational mutant that affects the positioning of L2 and L3, the two amino acid loops that interact with the minor groove of the DNA molecule. p53 in general suppresses the transcriptional activity of Nrf2 and negatively regulates the antioxidant activity to enhance cancer chemotherapy [
44]. In contrast, mutant p53, including R175H, with their gain of functions, results in interacting with the Nrf2 to facilitate its nuclear translocation and activation of antioxidant signaling proteins in cells [
45]. This process negatively impacts the therapeutic outcome of chemotherapy, with the upregulation of antioxidant signaling. TMZ is particularly effective in patients with MGMT promoter methylation; however, more than 50% of GBM patients retain MGMT expression and demonstrate less sensitivity to TMZ [
46]. Further investigations are needed to understand the role of Nrf2 inhibitors in MGMT methylated versus unmethylated tumors.




 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}