
Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain

Abstract
Simple Summary
Abstract
1. Introduction
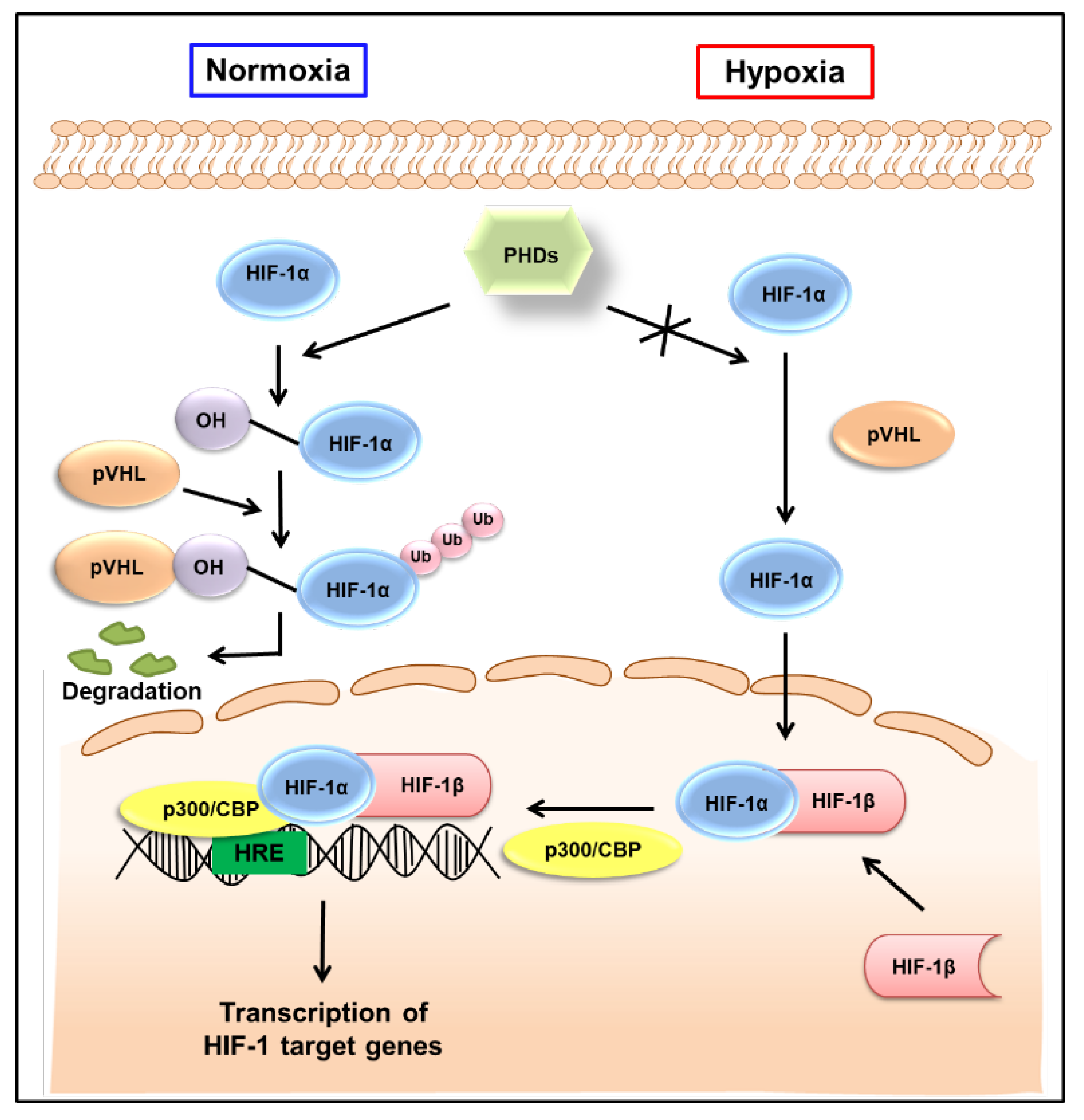
2. Regulation of HIF-1 Activity in Normoxic and Hypoxic Conditions
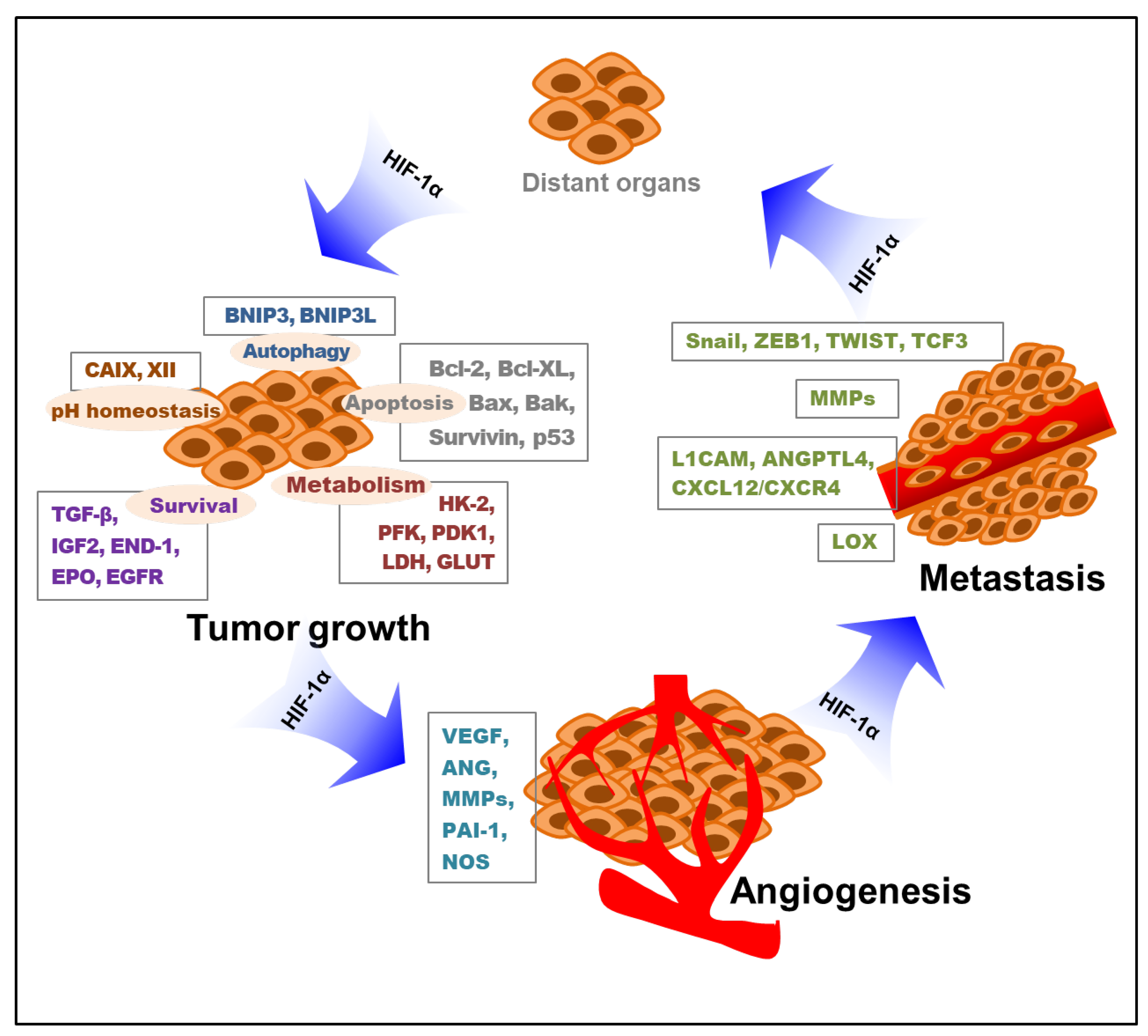
3. Roles of HIF-1 in Tumor Progression
3.1. Role of HIF-1 in Tumor Cell Proliferation and Survival
3.2. Role of HIF-1 in Angiogenesis
3.3. Role of HIF-1 in Tumor Cell Metastasis
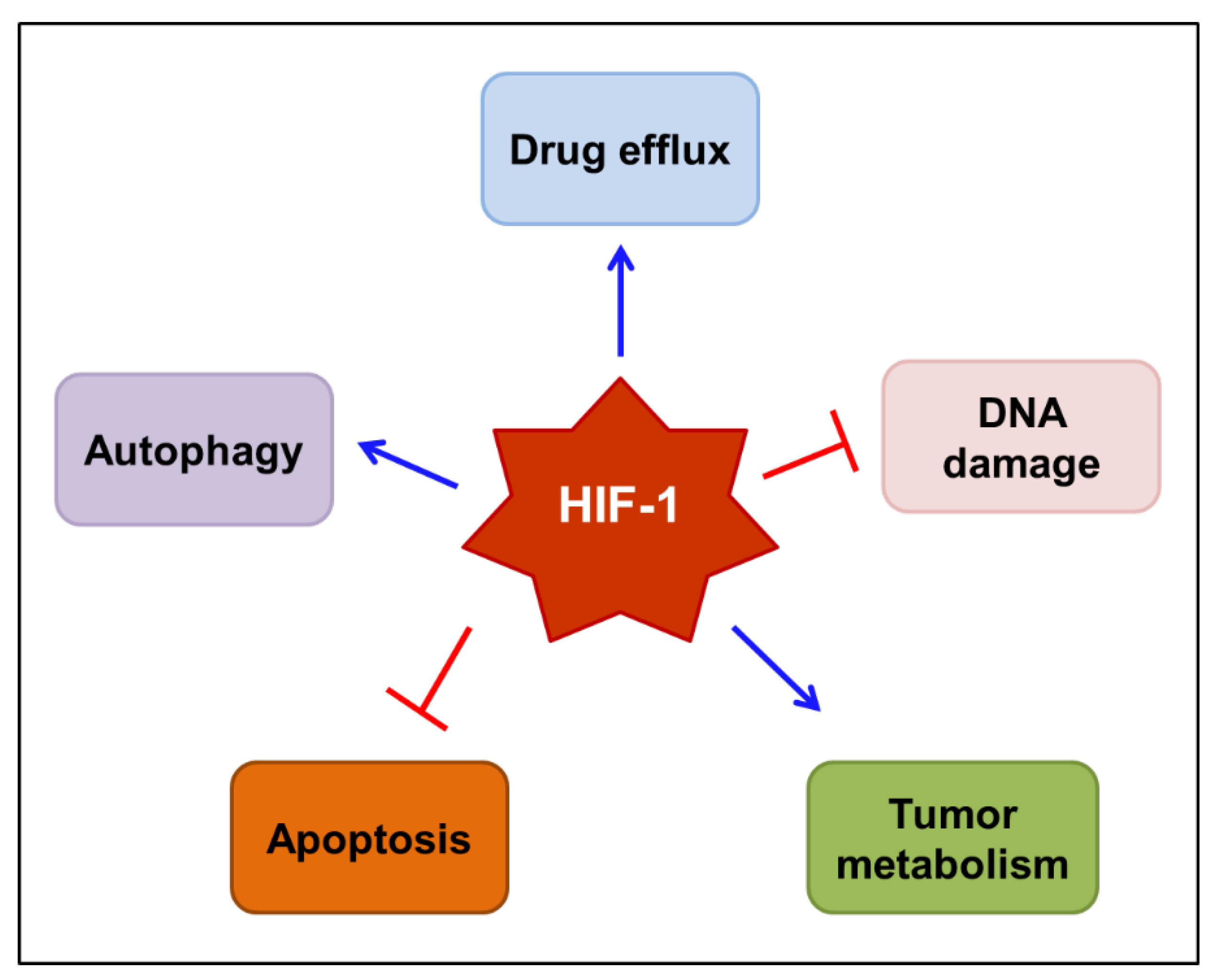
4. Roles of HIF-1 in Anticancer Drug Resistance and Cancer-Related Pain
4.1. Role of HIF-1 in Anticancer Drug Resistance
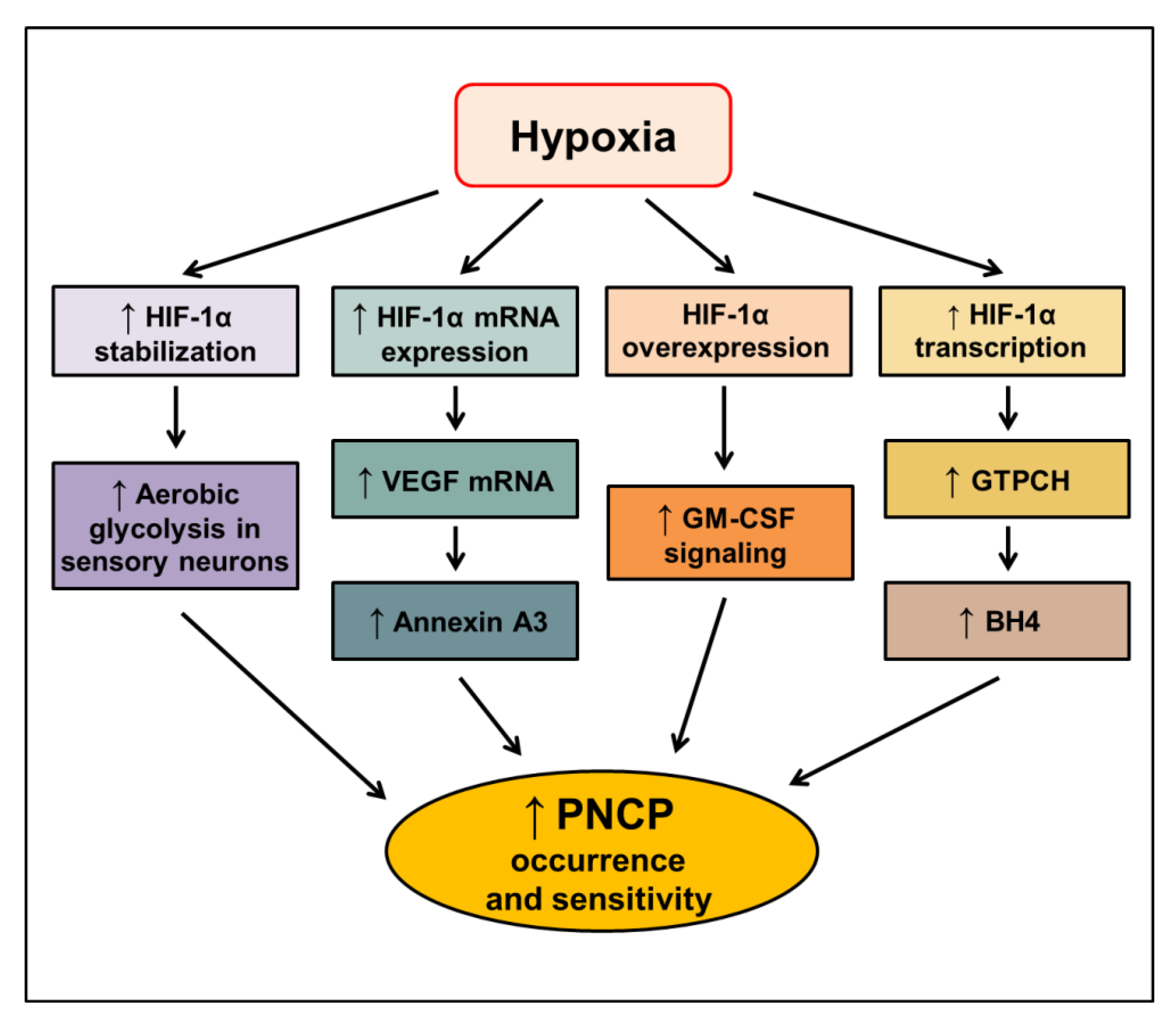
4.2. Role of HIF-1 in Cancer-Related Pain
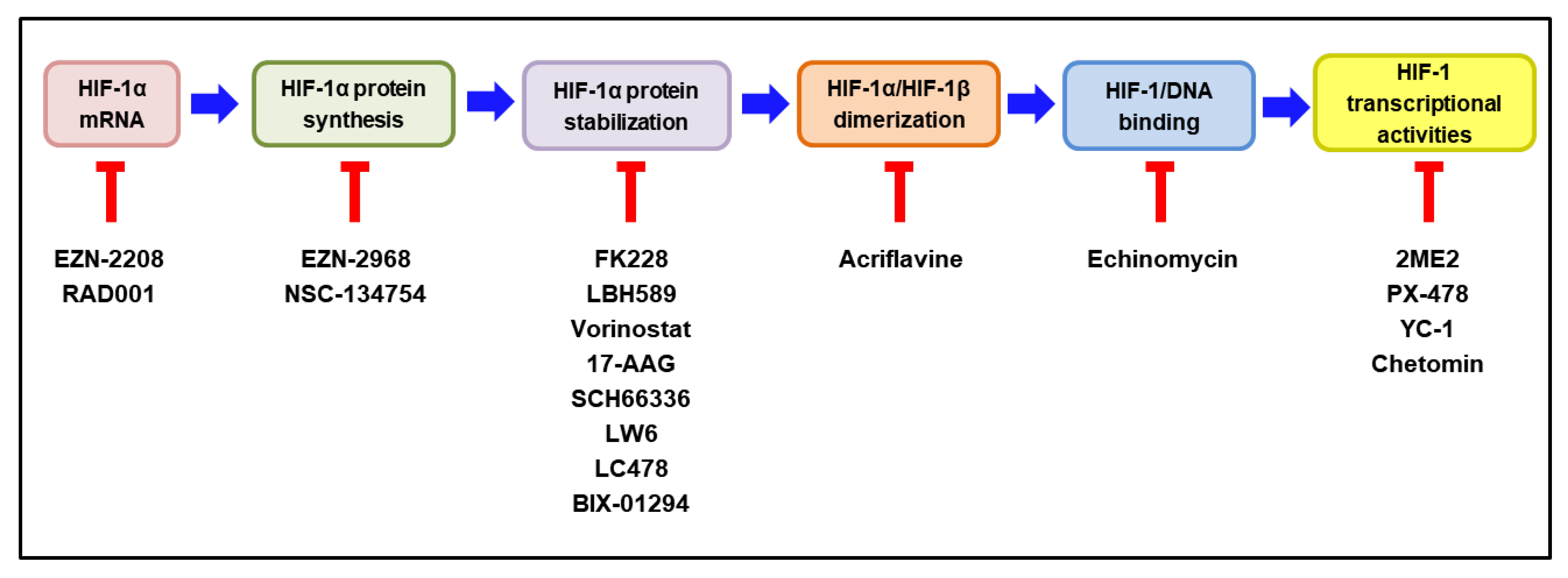
5. Current Clinical Status of HIF-1 Inhibitors as Potential Anticancer Therapy
5.1. HIF-1 Inhibitor Decreasing HIF-1α mRNA Expression
EZN-2208
5.2. HIF-1 Inhibitor Decreasing HIF-1α Protein Synthesis
EZN-2968
5.3. HIF-1 Inhibitors Decreasing HIF-1α Stabilization
5.3.1. FK228 (Romidepsin)
5.3.2. LBH589 (Panobinostat)
5.3.3. Vorinostat
5.3.4. 17-AAG (Tanespimycin)
5.3.5. SCH66336 (Lonafarnib)
5.4. HIF-1 Inhibitor Decreasing HIF-1α/HIF-1β Dimerization
Acriflavine
5.5. HIF-1 Inhibitor Decreasing HIF-1α/DNA Binding
Echinomycin
5.6. HIF-1 Inhibitor Decreasing HIF-1α Transcriptional Activity
2-Methoxyestradiol (2ME2, Panzem)
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Licona, A.; Pérez-Añorve, I.X.; Flores-Fortis, M.; Moral-Hernández, O.D.; González-de la Rosa, C.H.; Suárez-Sánchez, R.; Chávez-Saldaña, M.; Aréchaga-Ocampo, E. Deciphering the epigenetic network in cancer radioresistance. Radiother. Oncol. 2021, 159, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Cho, J.; Lee, K. Tumour Regression via Integrative Regulation of Neurological, Inflammatory, and Hypoxic Tumour Microenvironment. Biomol. Ther. 2020, 28, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Padma, V.V. An overview of targeted cancer therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef]
- Akanji, M.A.; Rotimi, D.; Adeyemi, O.S. Hypoxia-Inducible Factors as an Alternative Source of Treatment Strategy for Cancer. Oxid. Med. Cell. Longev. 2019, 2019, 8547846. [Google Scholar] [CrossRef]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef]
- Liu, L.; Ning, X.; Sun, L.; Zhang, H.; Shi, Y.; Guo, C.; Han, S.; Liu, J.; Sun, S.; Han, Z.; et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008, 99, 121–128. [Google Scholar] [CrossRef]
- Iijima, M.; Gombodorj, N.; Tachibana, Y.; Tachibana, K.; Yokobori, T.; Honma, K.; Nakano, T.; Asao, T.; Kuwahara, R.; Aoyama, K.; et al. Development of single nanometer-sized ultrafine oxygen bubbles to overcome the hypoxia-induced resistance to radiation therapy via the suppression of hypoxia-inducible factor1α. Int. J. Oncol. 2018, 52, 679–686. [Google Scholar] [CrossRef]
- Ma, Z.; Xiang, X.; Li, S.; Xie, P.; Gong, Q.; Goh, B.C.; Wang, L. Targeting hypoxia-inducible factor-1, for cancer treatment: Recent advances in developing small-molecule inhibitors from natural compounds. Semin. Cancer Biol. 2022, 80, 379–390. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Carver, L.A.; Hogenesch, J.B.; Bradfield, C.A. Tissue specific expression of the rat Ah-receptor and ARNT mRNAs. Nucleic Acids Res. 1994, 22, 3038–3044. [Google Scholar] [CrossRef]
- Graham, A.M.; Presnell, J.S. Hypoxia Inducible Factor (HIF) transcription factor family expansion, diversification, divergence and selection in eukaryotes. PLoS ONE 2017, 12, e0179545. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Schönberger, T.; Fandrey, J.; Prost-Fingerle, K. Ways into Understanding HIF Inhibition. Cancers 2021, 13, 159. [Google Scholar] [CrossRef]
- Kallio, P.J.; Pongratz, I.; Gradin, K.; McGuire, J.; Poellinger, L. Activation of hypoxia-inducible factor 1α: Posttranscriptional regulation and conformational change by recruitment of the Arnt transcription factor. Proc. Natl. Acad. Sci. USA 1997, 94, 5667–5672. [Google Scholar] [CrossRef]
- Scholz, C.C.; Taylor, C.T. Targeting the HIF pathway in inflammation and immunity. Curr. Opin. Pharmacol. 2013, 13, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Grimshaw, M.J. Endothelins and hypoxia-inducible factor in cancer. Endocr.-Relat. Cancer 2007, 14, 233–244. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A., 3rd; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Naik, R.; Ban, H.S.; Jang, K.; Kim, I.; Xu, X.; Harmalkar, D.; Shin, S.A.; Kim, M.; Kim, B.K.; Park, J.; et al. Methyl 3-(3-(4-(2,4,4-Trimethylpentan-2-yl)phenoxy)-propanamido)benzoate as a Novel and Dual Malate Dehydrogenase (MDH) 1/2 Inhibitor Targeting Cancer Metabolism. J. Med. Chem. 2017, 60, 8631–8646. [Google Scholar] [CrossRef]
- Ren, B.F.; Deng, L.F.; Wang, J.; Zhu, Y.P.; Wei, L.; Zhou, Q. Hypoxia regulation of facilitated glucose transporter-1 and glucose transporter-3 in mouse chondrocytes mediated by HIF-1α. Jt. Bone Spine 2008, 75, 176–181. [Google Scholar] [CrossRef]
- Won, M.; Ban, H.S.; Lee, K.; Lee, H.; Kim, B.-K.; Kim, H.; Naik, R.; Park, S.-K.; Park, J.-T.; Kim, I.; et al. Abstract 1164: A novel hypoxia-inducible factor-1 inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cancer Res. 2017, 77, 1164. [Google Scholar] [CrossRef]
- Kim, I.; Kim, M.; Park, M.K.; Naik, R.; Park, J.H.; Kim, B.-K.; Choi, Y.; Chang, K.Y.; Won, M.; Ban, H.S.; et al. The disubstituted adamantyl derivative LW1564 inhibits the growth of cancer cells by targeting mitochondrial respiration and reducing hypoxia-inducible factor (HIF)-1α accumulation. Exp. Mol. Med. 2020, 52, 1845–1856. [Google Scholar] [CrossRef]
- Flinck, M.; Kramer, S.H.; Pedersen, S.F. Roles of pH in control of cell proliferation. Acta Physiol. 2018, 223, e13068. [Google Scholar] [CrossRef]
- Ivanov, S.V.; Kuzmin, I.; Wei, M.H.; Pack, S.; Geil, L.; Johnson, B.E.; Stanbridge, E.J.; Lerman, M.I. Down-regulation of transmembrane carbonic anhydrases in renal cell carcinoma cell lines by wild-type von Hippel-Lindau transgenes. Proc. Natl. Acad. Sci. USA 1998, 95, 12596–12601. [Google Scholar] [CrossRef] [PubMed]
- Silagi, E.S.; Schoepflin, Z.R.; Seifert, E.L.; Merceron, C.; Schipani, E.; Shapiro, I.M.; Risbud, M.V. Bicarbonate Recycling by HIF-1-Dependent Carbonic Anhydrase Isoforms 9 and 12 Is Critical in Maintaining Intracellular pH and Viability of Nucleus Pulposus Cells. J. Bone Miner. Res. 2018, 33, 338–355. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients with Advanced Solid Tumors. Am. J. Clin. Oncol. 2020, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.L.; Elkamhawy, A.; Choi, Y.H.; Lee, C.H.; Lee, K.; Cho, J. Suppression of Tumor Growth and Cell Migration by Indole-Based Benzenesulfonamides and Their Synergistic Effects in Combination with Doxorubicin. Int. J. Mol. Sci. 2022, 23, 9903. [Google Scholar] [CrossRef]
- Hipfner, D.R.; Cohen, S.M. Connecting proliferation and apoptosis in development and disease. Nat. Rev. Mol. Cell Biol. 2004, 5, 805–815. [Google Scholar] [CrossRef]
- Sasabe, E.; Tatemoto, Y.; Li, D.; Yamamoto, T.; Osaki, T. Mechanism of HIF-1α-dependent suppression of hypoxia-induced apoptosis in squamous cell carcinoma cells. Cancer Sci. 2005, 96, 394–402. [Google Scholar] [CrossRef]
- Piret, J.P.; Mottet, D.; Raes, M.; Michiels, C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann. N. Y. Acad. Sci. 2002, 973, 443–447. [Google Scholar] [CrossRef]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Wang, P.; Long, M.; Zhang, S.; Cheng, Z.; Zhao, X.; He, F.; Liu, H.; Ming, L. Hypoxia inducible factor-1α regulates autophagy via the p27-E2F1 signaling pathway. Mol. Med. Rep. 2017, 16, 2107–2112. [Google Scholar] [CrossRef]
- Farrall, A.L.; Whitelaw, M.L. The HIF1α-inducible pro-cell death gene BNIP3 is a novel target of SIM2s repression through cross-talk on the hypoxia response element. Oncogene 2009, 28, 3671–3680. [Google Scholar] [CrossRef]
- Ma, J.; Weng, L.; Jia, Y.; Liu, B.; Wu, S.; Xue, L.; Yin, X.; Mao, A.; Wang, Z.; Shang, M. PTBP3 promotes malignancy and hypoxia-induced chemoresistance in pancreatic cancer cells by ATG12 up-regulation. J. Cell. Mol. Med. 2020, 24, 2917–2930. [Google Scholar] [CrossRef]
- Qureshi-Baig, K.; Kuhn, D.; Viry, E.; Pozdeev, V.I.; Schmitz, M.; Rodriguez, F.; Ullmann, P.; Koncina, E.; Nurmik, M.; Frasquilho, S.; et al. Hypoxia-induced autophagy drives colorectal cancer initiation and progression by activating the PRKC/PKC-EZR (ezrin) pathway. Autophagy 2020, 16, 1436–1452. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Gruber, M.; Simon, M.C. Hypoxia-inducible factors, hypoxia, and tumor angiogenesis. Curr. Opin. Hematol. 2006, 13, 169–174. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Kim, B.; Lee, K.; Jung, H.; Bhattarai, D.; Kwon, H.J. HIF-1α suppressing small molecule, LW6, inhibits cancer cell growth by binding to calcineurin b homologous protein 1. Biochem. Biophys. Res. Commun. 2015, 458, 14–20. [Google Scholar] [CrossRef]
- Naik, R.; Han, S.; Lee, K. Chemical biology approach for the development of hypoxia inducible factor (HIF) inhibitor LW6 as a potential anticancer agent. Arch. Pharm. Res. 2015, 38, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Condorelli, F.; Park, J.; Ferrara, N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef]
- Nilsson, I.; Shibuya, M.; Wennstrom, S. Differential activation of vascular genes by hypoxia in primary endothelial cells. Exp. Cell Res. 2004, 299, 476–485. [Google Scholar] [CrossRef]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Y.J.; Yu, Q. Angiopoietin-3 inhibits pulmonary metastasis by inhibiting tumor angiogenesis. Cancer Res. 2004, 64, 6119–6126. [Google Scholar] [CrossRef]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ. Res. 2003, 93, 664–673. [Google Scholar] [CrossRef]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; De Marco, P.; Cirillo, F.; Cappello, A.R.; Dolce, V.; Belfiore, A.; et al. Copper activates HIF-1α/GPER/VEGF signalling in cancer cells. Oncotarget 2015, 6, 34158–34177. [Google Scholar] [CrossRef]
- Lolmede, K.; Durand de Saint Front, V.; Galitzky, J.; Lafontan, M.; Bouloumie, A. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int. J. Obes. 2003, 27, 1187–1195. [Google Scholar] [CrossRef]
- Kietzmann, T.; Samoylenko, A.; Roth, U.; Jungermann, K. Hypoxia-inducible factor-1 and hypoxia response elements mediate the induction of plasminogen activator inhibitor-1 gene expression by insulin in primary rat hepatocytes. Blood 2003, 101, 907–914. [Google Scholar] [CrossRef]
- Jung, F.; Palmer, L.A.; Zhou, N.; Johns, R.A. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ. Res. 2000, 86, 319–325. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Chang, J.; Erler, J. Hypoxia-mediated metastasis. Adv. Exp. Med. Biol. 2014, 772, 55–81. [Google Scholar] [CrossRef]
- Imai, T.; Horiuchi, A.; Wang, C.; Oka, K.; Ohira, S.; Nikaido, T.; Konishi, I. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am. J. Pathol. 2003, 163, 1437–1447. [Google Scholar] [CrossRef]
- Hugo, H.J.; Pereira, L.; Suryadinata, R.; Drabsch, Y.; Gonda, T.J.; Gunasinghe, N.P.; Pinto, C.; Soo, E.T.; van Denderen, B.J.; Hill, P.; et al. Direct repression of MYB by ZEB1 suppresses proliferation and epithelial gene expression during epithelial-to-mesenchymal transition of breast cancer cells. Breast Cancer Res. 2013, 15, R113. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Zagzag, D.; Nagasawa, H.; Rainey, K.; Okuyama, H.; Baek, J.H.; Semenza, G.L. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006, 66, 2725–2731. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Calorini, L.; Chiarugi, P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid. Redox Signal. 2011, 14, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; De Marco, M.A.; Graziani, I.; Gazdar, A.F.; Strack, P.R.; Miele, L.; Bocchetta, M. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007, 67, 7954–7959. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.H.; Forsdike, J.; Fitzgerald, C.J.; Macdonald-Goodfellow, S. Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int. J. Cancer 1999, 80, 617–623. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Y.; Qian, Y.; Zhang, H.; Guo, S.; Sunagawa, M.; Hisamitsu, T.; Liu, Y. Interleukin-17A promotes rheumatoid arthritis synoviocytes migration and invasion under hypoxia by increasing MMP2 and MMP9 expression through NF-κB/HIF-1α pathway. Mol. Immunol. 2013, 53, 227–236. [Google Scholar] [CrossRef]
- Rohwer, N.; Welzel, M.; Daskalow, K.; Pfander, D.; Wiedenmann, B.; Detjen, K.; Cramer, T. Hypoxia-inducible factor 1α mediates anoikis resistance via suppression of α5 integrin. Cancer Res. 2008, 68, 10113–10120. [Google Scholar] [CrossRef]
- Zhang, H.; Wong, C.C.; Wei, H.; Gilkes, D.M.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P.T., Jr.; et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012, 31, 1757–1770. [Google Scholar] [CrossRef]
- Jin, F.; Brockmeier, U.; Otterbach, F.; Metzen, E. New insight into the SDF-1/CXCR4 axis in a breast carcinoma model: Hypoxia-induced endothelial SDF-1 and tumor cell CXCR4 are required for tumor cell intravasation. Mol. Cancer Res. 2012, 10, 1021–1031. [Google Scholar] [CrossRef]
- Wendel, C.; Hemping-Bovenkerk, A.; Krasnyanska, J.; Mees, S.T.; Kochetkova, M.; Stoeppeler, S.; Haier, J. CXCR4/CXCL12 participate in extravasation of metastasizing breast cancer cells within the liver in a rat model. PLoS ONE 2012, 7, e30046. [Google Scholar] [CrossRef]
- Gassmann, P.; Haier, J.; Schluter, K.; Domikowsky, B.; Wendel, C.; Wiesner, U.; Kubitza, R.; Engers, R.; Schneider, S.W.; Homey, B.; et al. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia 2009, 11, 651–661. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef]
- Sion, A.M.; Figg, W.D. Lysyl oxidase (LOX) and hypoxia-induced metastases. Cancer Biol. Ther. 2006, 5, 909–911. [Google Scholar] [CrossRef]
- Wong, C.C.-L.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.-M.; Khoo, U.-S.; Ng, I.O.-L.; et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef]
- Fang, Y.; Sullivan, R.; Graham, C.H. Confluence-dependent resistance to doxorubicin in human MDA-MB-231 breast carcinoma cells requires hypoxia-inducible factor-1 activity. Exp. Cell Res. 2007, 313, 867–877. [Google Scholar] [CrossRef]
- Huang, L.; Ao, Q.; Zhang, Q.; Yang, X.; Xing, H.; Li, F.; Chen, G.; Zhou, J.; Wang, S.; Xu, G.; et al. Hypoxia induced paclitaxel resistance in human ovarian cancers via hypoxia-inducible factor 1α. J. Cancer Res. Clin. Oncol. 2010, 136, 447–456. [Google Scholar] [CrossRef]
- Jia, X.; Hong, Q.; Lei, L.; Li, D.; Li, J.; Mo, M.; Wang, Y.; Shao, Z.; Shen, Z.; Cheng, J.; et al. Basal and therapy-driven hypoxia-inducible factor-1α confers resistance to endocrine therapy in estrogen receptor-positive breast cancer. Oncotarget 2015, 6, 8648–8662. [Google Scholar] [CrossRef]
- Chen, J.; Ding, Z.; Peng, Y.; Pan, F.; Li, J.; Zou, L.; Zhang, Y.; Liang, H. HIF-1α inhibition reverses multidrug resistance in colon cancer cells via downregulation of MDR1/P-glycoprotein. PLoS ONE 2014, 9, e98882. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Creemers, S.G.; van Koetsveld, P.M.; De Herder, W.W.; Dogan, F.; Franssen, G.J.H.; Feelders, R.A.; Hofland, L.J. MDR1 inhibition increases sensitivity to doxorubicin and etoposide in adrenocortical cancer. Endocr.-Relat. Cancer 2019, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Doublier, S.; Belisario, D.C.; Polimeni, M.; Annaratone, L.; Riganti, C.; Allia, E.; Ghigo, D.; Bosia, A.; Sapino, A. HIF-1 activation induces doxorubicin resistance in MCF7 3-D spheroids via P-glycoprotein expression: A potential model of the chemo-resistance of invasive micropapillary carcinoma of the breast. BMC Cancer 2012, 12, 4. [Google Scholar] [CrossRef]
- Sun, Y.; Guan, Z.; Liang, L.; Cheng, Y.; Zhou, J.; Li, J.; Xu, Y. HIF-1α/MDR1 pathway confers chemoresistance to cisplatin in bladder cancer. Oncol. Rep. 2016, 35, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Deeley, R.G.; Cole, S.P.C. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS Lett. 2006, 580, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. OncoTargets Ther. 2015, 8, 1941–1948. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Pinzón-Daza, M.L.; Cuellar-Saenz, Y.; Nualart, F.; Ondo-Mendez, A.; Del Riesgo, L.; Castillo-Rivera, F.; Garzón, R. Oxidative Stress Promotes Doxorubicin-Induced Pgp and BCRP Expression in Colon Cancer Cells Under Hypoxic Conditions. J. Cell. Biochem. 2017, 118, 1868–1878. [Google Scholar] [CrossRef]
- Bergandi, L.; Canosa, S.; Pittatore, G.; Silvagno, F.; Doublier, S.; Gennarelli, G.; Benedetto, C.; Revelli, A. Human recombinant FSH induces chemoresistance in human breast cancer cells via HIF-1α activation. Biol. Reprod. 2019, 100, 1521–1535. [Google Scholar] [CrossRef]
- Yang, S.F.; Chang, C.W.; Wei, R.J.; Shiue, Y.L.; Wang, S.N.; Yeh, Y.T. Involvement of DNA damage response pathways in hepatocellular carcinoma. BioMed Res. Int. 2014, 2014, 153867. [Google Scholar] [CrossRef]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Rezaeian, A.H.; Wang, Y.H.; Lin, H.K. DNA damage players are linked to HIF-1α/hypoxia signaling. Cell Cycle 2017, 16, 725–726. [Google Scholar] [CrossRef][Green Version]
- Shenoy, N.; Dronca, R.; Quevedo, F.; Boorjian, S.A.; Cheville, J.; Costello, B.; Kohli, M.; Witzig, T.; Pagliaro, L. Low hypoxia inducible factor-1α (HIF-1α) expression in testicular germ cell tumors—A major reason for enhanced chemosensitivity? Chin. J. Cancer Res. 2017, 29, 374–378. [Google Scholar] [CrossRef][Green Version]
- Sugrue, T.; Lowndes, N.F.; Ceredig, R. Hypoxia enhances the radioresistance of mouse mesenchymal stromal cells. Stem Cells 2014, 32, 2188–2200. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wang, J.; Xu, S.; Xiao, L.; Chen, G.; Zhang, W.; Li, J. Radioprotective effect on HepG2 cells of low concentrations of cobalt chloride: Induction of hypoxia-inducible factor-1 alpha and clearance of reactive oxygen species. J. Radiat. Res. 2013, 54, 203–209. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, S.C.; Chien, C.W.; Lin, S.C.; Lee, C.T.; Lin, B.W.; Lee, J.C.; Tsai, S.J. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am. J. Pathol. 2011, 179, 1405–1414. [Google Scholar] [CrossRef]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef]
- Mitani, T.; Ito, Y.; Harada, N.; Nakano, Y.; Inui, H.; Ashida, H.; Yamaji, R. Resveratrol reduces the hypoxia-induced resistance to doxorubicin in breast cancer cells. J. Nutr. Sci. Vitaminol. 2014, 60, 122–128. [Google Scholar] [CrossRef]
- Hickman, J.A. Apoptosis and chemotherapy resistance. Eur. J. Cancer 1996, 32, 921–926. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Tan, B.B.; Li, Y.; Fan, L.Q.; Yang, P.G.; Tian, Y. Enhancement of Drug Sensitivity by Knockdown of HIF-1α in Gastric Carcinoma Cells. Oncol. Res. 2016, 23, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Dame, C.; Haugstetter, A.; Wiedenmann, B.; Detjen, K.; Schmitt, C.A.; Cramer, T. Hypoxia-inducible factor 1α determines gastric cancer chemosensitivity via modulation of p53 and NF-κB. PLoS ONE 2010, 5, e12038. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.T.; Wu, C.W.; Lin, W.W. Hypoxia-induced decoy receptor 2 gene expression is regulated via a hypoxia-inducible factor 1α-mediated mechanism. Biochem. Biophys. Res. Commun. 2010, 391, 1274–1279. [Google Scholar] [CrossRef]
- Minassian, L.M.; Cotechini, T.; Huitema, E.; Graham, C.H. Hypoxia-Induced Resistance to Chemotherapy in Cancer. Adv. Exp. Med. Biol. 2019, 1136, 123–139. [Google Scholar] [CrossRef]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updat. 2011, 14, 191–201. [Google Scholar] [CrossRef]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and doxorubicin resistance in cancer. Anti-Cancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Wu, H.M.; Jiang, Z.F.; Ding, P.S.; Shao, L.J.; Liu, R.Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef]
- Long, F.; Liu, W.; Jia, P.; Wang, H.; Jiang, G.; Wang, T. HIF-1α-induced autophagy contributes to cisplatin resistance in ovarian cancer cells. Pharmazie 2018, 73, 533–536. [Google Scholar] [CrossRef]
- Yang, X.; Yin, H.; Zhang, Y.; Li, X.; Tong, H.; Zeng, Y.; Wang, Q.; He, W. Hypoxia-induced autophagy promotes gemcitabine resistance in human bladder cancer cells through hypoxia-inducible factor 1α activation. Int. J. Oncol. 2018, 53, 215–224. [Google Scholar] [CrossRef]
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67–73. [Google Scholar] [CrossRef]
- Zou, Y.M.; Hu, G.Y.; Zhao, X.Q.; Lu, T.; Zhu, F.; Yu, S.Y.; Xiong, H. Hypoxia-induced autophagy contributes to radioresistance via c-Jun-mediated Beclin1 expression in lung cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 761–767. [Google Scholar] [CrossRef]
- Song, J.G.; Lee, Y.S.; Park, J.A.; Lee, E.H.; Lim, S.J.; Yang, S.J.; Zhao, M.; Lee, K.; Han, H.K. Discovery of LW6 as a new potent inhibitor of breast cancer resistance protein. Cancer Chemother. Pharmacol. 2016, 78, 735–744. [Google Scholar] [CrossRef]
- Han, S.; Kim, E.-S.; You, B.; Chae, H.-S.; Liu, Q.; Chin, Y.-W.; Ahn, H.-C.; Chung, S.-J.; Lee, K.; Choi, Y.H. Effect of treatment period with LC478, a disubstituted adamantayl derivative, on P-glycoprotein inhibition: Its application to increase docetaxel absorption in rats. Xenobiotica 2020, 50, 863–874. [Google Scholar] [CrossRef]
- Zander, S.A.; Sol, W.; Greenberger, L.; Zhang, Y.; van Tellingen, O.; Jonkers, J.; Borst, P.; Rottenberg, S. EZN-2208 (PEG-SN38) overcomes ABCG2-mediated topotecan resistance in BRCA1-deficient mouse mammary tumors. PLoS ONE 2012, 7, e45248. [Google Scholar] [CrossRef]
- Onnis, B.; Rapisarda, A.; Melillo, G. Development of HIF-1 inhibitors for cancer therapy. J. Cell. Mol. Med. 2009, 13, 2780–2786. [Google Scholar] [CrossRef]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1α inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10, 605. [Google Scholar] [CrossRef]
- Zhao, T.; Ren, H.; Jia, L.; Chen, J.; Xin, W.; Yan, F.; Li, J.; Wang, X.; Gao, S.; Qian, D.; et al. Inhibition of HIF-1α by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 2250–2262. [Google Scholar] [CrossRef]
- Schwartz, D.L.; Bankson, J.A.; Lemos, R., Jr.; Lai, S.Y.; Thittai, A.K.; He, Y.; Hostetter, G.; Demeure, M.J.; Von Hoff, D.D.; Powis, G. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 2057–2067. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, R.; Molteni, R.; Gariboldi, M.B.; Marras, E.; Perletti, G.; Monti, E. Effect of HIF-1 modulation on the response of two- and three-dimensional cultures of human colon cancer cells to 5-fluorouracil. Eur. J. Cancer 2009, 45, 890–898. [Google Scholar] [CrossRef]
- Wu, Y.; Dong, L.; Bao, S.; Wang, M.; Yun, Y.; Zhu, R. FK228 augmented temozolomide sensitivity in human glioma cells by blocking PI3K/AKT/mTOR signal pathways. Biomed. Pharmacother. 2016, 84, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Namgung, Y.; Kim, S.Y.; Kim, I. Down-regulation of Survivin by BIX-01294 Pretreatment Overcomes Resistance of Hepatocellular Carcinoma Cells to TRAIL. Anticancer Res. 2019, 39, 3571–3578. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Lin, H.; Moh, J.S.; Chen, K.D.; Wang, I.W.; Ou, Y.C.; You, Y.S.; Lung, C.C. Low-dose LBH589 increases the sensitivity of cisplatin to cisplatin-resistant ovarian cancer cells. Taiwan J. Obstet. Gynecol. 2011, 50, 165–171. [Google Scholar] [CrossRef][Green Version]
- Maiso, P.; Carvajal-Vergara, X.; Ocio, E.M.; Lopez-Perez, R.; Mateo, G.; Gutierrez, N.; Atadja, P.; Pandiella, A.; San Miguel, J.F. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006, 66, 5781–5789. [Google Scholar] [CrossRef]
- Zang, H.; Qian, G.; Zong, D.; Fan, S.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.Y. Overcoming acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib by combining osimertinib with the histone deacetylase inhibitor panobinostat (LBH589). Cancer 2020, 126, 2024–2033. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, K.; Zhu, Q.; Shen, Q.; Zhang, Q.; Yu, J.; Chen, Y.; Lu, W. Synthesis and biological evaluation of paclitaxel and vorinostat co-prodrugs for overcoming drug resistance in cancer therapy in vitro. Bioorg. Med. Chem. 2019, 27, 1405–1413. [Google Scholar] [CrossRef]
- Lautz, T.B.; Jie, C.; Clark, S.; Naiditch, J.A.; Jafari, N.; Qiu, Y.Y.; Zheng, X.; Chu, F.; Madonna, M.B. The effect of vorinostat on the development of resistance to doxorubicin in neuroblastoma. PLoS ONE 2012, 7, e40816. [Google Scholar] [CrossRef]
- Barbone, D.; Cheung, P.; Battula, S.; Busacca, S.; Gray, S.G.; Longley, D.B.; Bueno, R.; Sugarbaker, D.J.; Fennell, D.A.; Broaddus, V.C. Vorinostat eliminates multicellular resistance of mesothelioma 3D spheroids via restoration of Noxa expression. PLoS ONE 2012, 7, e52753. [Google Scholar] [CrossRef]
- Adamski, J.; Price, A.; Dive, C.; Makin, G. Hypoxia-induced cytotoxic drug resistance in osteosarcoma is independent of HIF-1Alpha. PLoS ONE 2013, 8, e65304. [Google Scholar] [CrossRef]
- Lee, M.R.; Lin, C.; Lu, C.C.; Kuo, S.C.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Lee, F.Y.; Yang, J.S.; Tsai, F.J. YC-1 induces G0/G1 phase arrest and mitochondria-dependent apoptosis in cisplatin-resistant human oral cancer CAR cells. BioMedicine 2017, 7, 12. [Google Scholar] [CrossRef]
- Hu, H.; Miao, X.K.; Li, J.Y.; Zhang, X.W.; Xu, J.J.; Zhang, J.Y.; Zhou, T.X.; Hu, M.N.; Yang, W.L.; Mou, L.Y. YC-1 potentiates the antitumor activity of gefitinib by inhibiting HIF-1α and promoting the endocytic trafficking and degradation of EGFR in gefitinib-resistant non-small-cell lung cancer cells. Eur. J. Pharmacol. 2020, 874, 172961. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, Z.-A.; Liu, Q.-H.; Kong, Q.-Y.; Liu, L.; Cui, T.; Wu, Y.-P. RAD001 can reverse drug resistance of SGC7901/DDP cells. Tumour Biol. 2014, 35, 9171–9177. [Google Scholar] [CrossRef]
- Kim, E.S.; Kies, M.S.; Fossella, F.V.; Glisson, B.S.; Zaknoen, S.; Statkevich, P.; Munden, R.F.; Summey, C.; Pisters, K.M.; Papadimitrakopoulou, V.; et al. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer 2005, 104, 561–569. [Google Scholar] [CrossRef]
- Smalley, K.S.; Eisen, T.G. Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int. J. Cancer 2003, 105, 165–175. [Google Scholar] [CrossRef]
- Wang, E.J.; Johnson, W.W. The farnesyl protein transferase inhibitor lonafarnib (SCH66336) is an inhibitor of multidrug resistance proteins 1 and 2. Chemotherapy 2003, 49, 303–308. [Google Scholar] [CrossRef]
- Shevrin, D.H.; Lad, T.E.; Guinan, P.; Kilton, L.J.; Greenburg, A.; Johnson, P.; Blough, R.R.; Hoyer, H. Phase II trial of echinomycin in advanced hormone-resistant prostate cancer. An Illinois Cancer Council study. Investig. New Drugs 1994, 12, 65–66. [Google Scholar] [CrossRef]
- Evenepoel, M.; Haenen, V.; De Baerdemaecker, T.; Meeus, M.; Devoogdt, N.; Dams, L.; Van Dijck, S.; Van der Gucht, E.; De Groef, A. Pain Prevalence During Cancer Treatment: A Systematic Review and Meta-Analysis. J. Pain Symptom Manag. 2022, 63, e317–e335. [Google Scholar] [CrossRef]
- Neufeld, N.J.; Elnahal, S.M.; Alvarez, R.H. Cancer pain: A review of epidemiology, clinical quality and value impact. Future Oncol. 2017, 13, 833–841. [Google Scholar] [CrossRef]
- Esin, E.; Yalcin, S. Neuropathic cancer pain: What we are dealing with? How to manage it? OncoTargets Ther. 2014, 7, 599–618. [Google Scholar] [CrossRef]
- Howard, P. Hypoxia, almitrine, and peripheral neuropathy. Thorax 1989, 44, 247–250. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boyette-Davis, J.A.; Walters, E.T.; Dougherty, P.M. Mechanisms involved in the development of chemotherapy-induced neuropathy. Pain Manag. 2015, 5, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Ludman, T.; Melemedjian, O.K. Bortezomib-induced aerobic glycolysis contributes to chemotherapy-induced painful peripheral neuropathy. Mol. Pain 2019, 15, 1744806919837429. [Google Scholar] [CrossRef] [PubMed]
- Ludman, T.; Melemedjian, O.K. Bortezomib and metformin opposingly regulate the expression of hypoxia-inducible factor alpha and the consequent development of chemotherapy-induced painful peripheral neuropathy. Mol. Pain 2019, 15, 1744806919850043. [Google Scholar] [CrossRef]
- Richardson, P.G.; Chanan-Khan, A.A.; Lonial, S.; Krishnan, A.Y.; Carroll, M.P.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: Results of a phase 1/2 study. Br. J. Haematol. 2011, 153, 729–740. [Google Scholar] [CrossRef]
- Zhang, G.; Feng, G.-Y.; Guo, Y.-R.; Liang, D.-Q.; Yuan, Y.; Wang, H.-L. Correlation between liver cancer pain and the HIF-1 and VEGF expression levels. Oncol. Lett. 2017, 13, 77–80. [Google Scholar] [CrossRef][Green Version]
- Zhang, Z.; Deng, M.; Huang, J.; Wu, J.; Li, Z.; Xing, M.; Wang, J.; Guo, Q.; Zou, W. Microglial annexin A3 downregulation alleviates bone cancer-induced pain through inhibiting the Hif-1α/vascular endothelial growth factor signaling pathway. Pain 2020, 161, 2750–2762. [Google Scholar] [CrossRef]
- Stosser, S.; Schweizerhof, M.; Kuner, R. Hematopoietic colony-stimulating factors: New players in tumor-nerve interactions. J. Mol. Med. 2011, 89, 321–329. [Google Scholar] [CrossRef]
- Schweizerhof, M.; Stosser, S.; Kurejova, M.; Njoo, C.; Gangadharan, V.; Agarwal, N.; Schmelz, M.; Bali, K.K.; Michalski, C.W.; Brugger, S.; et al. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat. Med. 2009, 15, 802–807. [Google Scholar] [CrossRef]
- Mroczko, B.; Szmitkowski, M.; Wereszczynska-Siemiatkowska, U.; Jurkowska, G. Hematopoietic cytokines in the sera of patients with pancreatic cancer. Clin. Chem. Lab. Med. 2005, 43, 146–150. [Google Scholar] [CrossRef]
- Wang, H.; Jia, R.; Zhao, T.; Li, X.; Lang, M.; Lan, C.; Wang, H.; Li, Z.; Zhou, B.; Wu, L.; et al. HIF-1α mediates tumor-nerve interactions through the up-regulation of GM-CSF in pancreatic ductal adenocarcinoma. Cancer Lett. 2019, 453, 10–20. [Google Scholar] [CrossRef]
- Kanngiesser, M.; Mair, N.; Lim, H.-Y.; Zschiebsch, K.; Blees, J.; Häußler, A.; Brüne, B.; Ferreiros Bouzas, N.; Kress, M.; Tegeder, I. Hypoxia-Inducible Factor 1 Regulates Heat and Cold Pain Sensitivity and Persistence. Antioxid. Redox Signal. 2014, 20, 2555–2571. [Google Scholar] [CrossRef]
- Qiu, G.Z.; Jin, M.Z.; Dai, J.X.; Sun, W.; Feng, J.H.; Jin, W.L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef]
- Garje, R.; An, J.J.; Sanchez, K.; Greco, A.; Stolwijk, J.; Devor, E.; Rustum, Y.; Zakharia, Y. Current Landscape and the Potential Role of Hypoxia-Inducible Factors and Selenium in Clear Cell Renal Cell Carcinoma Treatment. Int. J. Mol. Sci. 2018, 19, 3834. [Google Scholar] [CrossRef]
- Bhattarai, D.; Xu, X.; Lee, K. Hypoxia-inducible factor-1 (HIF-1) inhibitors from the last decade (2007 to 2016): A “structure-activity relationship” perspective. Med. Res. Rev. 2018, 38, 1404–1442. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Soung, N.-K.; Kim, H.-M.; Asami, Y.; Kim, D.H.; Cho, Y.; Naik, R.; Jang, Y.; Jang, K.; Han, H.J.; Ganipisetti, S.R.; et al. Mechanism of the natural product moracin-O derived MO-460 and its targeting protein hnRNPA2B1 on HIF-1α inhibition. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Jeong, W.; Park, S.R.; Rapisarda, A.; Fer, N.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; et al. Weekly EZN-2208 (PEGylated SN-38) in combination with bevacizumab in patients with refractory solid tumors. Investig. New Drugs 2014, 32, 340–346. [Google Scholar] [CrossRef]
- Norris, R.E.; Shusterman, S.; Gore, L.; Muscal, J.A.; Macy, M.E.; Fox, E.; Berkowitz, N.; Buchbinder, A.; Bagatell, R. Phase 1 evaluation of EZN-2208, a polyethylene glycol conjugate of SN38, in children adolescents and young adults with relapsed or refractory solid tumors. Pediatr. Blood Cancer 2014, 61, 1792–1797. [Google Scholar] [CrossRef]
- Garrett, C.R.; Bekaii-Saab, T.S.; Ryan, T.; Fisher, G.A.; Clive, S.; Kavan, P.; Shacham-Shmueli, E.; Buchbinder, A.; Goldberg, R.M. Randomized phase 2 study of pegylated SN-38 (EZN-2208) or irinotecan plus cetuximab in patients with advanced colorectal cancer. Cancer 2013, 119, 4223–4230. [Google Scholar] [CrossRef] [PubMed]
- Borsi, E.; Perrone, G.; Terragna, C.; Martello, M.; Dico, A.F.; Solaini, G.; Baracca, A.; Sgarbi, G.; Pasquinelli, G.; Valente, S.; et al. Hypoxia inducible factor-1 alpha as a therapeutic target in multiple myeloma. Oncotarget 2014, 5, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Mie Lee, Y.; Kim, S.H.; Kim, H.S.; Jin Son, M.; Nakajima, H.; Jeong Kwon, H.; Kim, K.W. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1α activity. Biochem. Biophys. Res. Commun. 2003, 300, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Sasakawa, Y.; Naoe, Y.; Noto, T.; Inoue, T.; Sasakawa, T.; Matsuo, M.; Manda, T.; Mutoh, S. Antitumor efficacy of FK228, a novel histone deacetylase inhibitor, depends on the effect on expression of angiogenesis factors. Biochem. Pharmacol. 2003, 66, 897–906. [Google Scholar] [CrossRef]
- Gerber, D.E.; Boothman, D.A.; Fattah, F.J.; Dong, Y.; Zhu, H.; Skelton, R.A.; Priddy, L.L.; Vo, P.; Dowell, J.E.; Sarode, V.; et al. Phase 1 study of romidepsin plus erlotinib in advanced non-small cell lung cancer. Lung Cancer 2015, 90, 534–541. [Google Scholar] [CrossRef]
- Haigentz, M., Jr.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef]
- Fischer, C.; Leithner, K.; Wohlkoenig, C.; Quehenberger, F.; Bertsch, A.; Olschewski, A.; Olschewski, H.; Hrzenjak, A. Panobinostat reduces hypoxia-induced cisplatin resistance of non-small cell lung carcinoma cells via HIF-1α destabilization. Mol. Cancer 2015, 14, 4. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Mina, R.; Jakubowiak, A.J.; Zimmerman, T.L.; Wolf, J.J.; Lewis, C.; Gleason, C.; Sharp, C.; Martin, T.; Heffner, L.T.; et al. Combining carfilzomib and panobinostat to treat relapsed/refractory multiple myeloma: Results of a Multiple Myeloma Research Consortium Phase I Study. Blood Cancer J. 2019, 9, 3. [Google Scholar] [CrossRef]
- Wood, P.J.; Strong, R.; McArthur, G.A.; Michael, M.; Algar, E.; Muscat, A.; Rigby, L.; Ferguson, M.; Ashley, D.M. A phase I study of panobinostat in pediatric patients with refractory solid tumors, including CNS tumors. Cancer Chemother. Pharmacol. 2018, 82, 493–503. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro-Oncology 2018, 20, 546–556. [Google Scholar] [CrossRef]
- Haas, N.B.; Quirt, I.; Hotte, S.; McWhirter, E.; Polintan, R.; Litwin, S.; Adams, P.D.; McBryan, T.; Wang, L.; Martin, L.P.; et al. Phase II trial of vorinostat in advanced melanoma. Investig. New Drugs 2014, 32, 526–534. [Google Scholar] [CrossRef]
- Krug, L.M.; Kindler, H.L.; Calvert, H.; Manegold, C.; Tsao, A.S.; Fennell, D.; Ohman, R.; Plummer, R.; Eberhardt, W.E.; Fukuoka, K.; et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 2015, 16, 447–456. [Google Scholar] [CrossRef]
- Lang, S.A.; Klein, D.; Moser, C.; Gaumann, A.; Glockzin, G.; Dahlke, M.H.; Dietmaier, W.; Bolder, U.; Schlitt, H.J.; Geissler, E.K.; et al. Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastric cancer cells and reduces tumor growth and vascularization in vivo. Mol. Cancer Ther. 2007, 6, 1123–1132. [Google Scholar] [CrossRef]
- Heath, E.I.; Hillman, D.W.; Vaishampayan, U.; Sheng, S.; Sarkar, F.; Harper, F.; Gaskins, M.; Pitot, H.C.; Tan, W.; Ivy, S.P.; et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 2008, 14, 7940–7946. [Google Scholar] [CrossRef]
- Han, J.Y.; Oh, S.H.; Morgillo, F.; Myers, J.N.; Kim, E.; Hong, W.K.; Lee, H.Y. Hypoxia-inducible factor 1α and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J. Natl. Cancer Inst. 2005, 97, 1272–1286. [Google Scholar] [CrossRef]
- Hanrahan, E.O.; Kies, M.S.; Glisson, B.S.; Khuri, F.R.; Feng, L.; Tran, H.T.; Ginsberg, L.E.; Truong, M.T.; Hong, W.K.; Kim, E.S. A phase II study of Lonafarnib (SCH66336) in patients with chemorefractory, advanced squamous cell carcinoma of the head and neck. Am. J. Clin. Oncol. 2009, 32, 274–279. [Google Scholar] [CrossRef]
- Sharma, S.; Kemeny, N.; Kelsen, D.P.; Ilson, D.; O’Reilly, E.; Zaknoen, S.; Baum, C.; Statkevich, P.; Hollywood, E.; Zhu, Y.; et al. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann. Oncol. 2002, 13, 1067–1071. [Google Scholar] [CrossRef]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; He, S.; Shen, G.; Wang, Y. HIF-1 Dimerization Inhibitor Acriflavine Enhances Antitumor Activity of Sunitinib in Breast Cancer Model. Oncol. Res. 2014, 22, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.; Goodman, P.J.; Fleming, T.R.; Taylor, S.A.; Macdonald, J.S. Phase II trial of echinomycin in advanced colorectal cancer. A Southwest Oncology Group study. Investig. New Drugs 1991, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.E.; Wolf, M.K.; Crawford, E.D.; Taylor, S.; Blumenstein, B.; Flanigan, R.; Meyers, F.J.; Hynes, H.E.; Barlogie, B.; Eisenberger, M. Phase II trial of echinomycin for the treatment of advanced renal cell carcinoma. A Southwest Oncology Group study. Investig. New Drugs 1993, 11, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Muss, H.B.; Blessing, J.A.; Baker, V.V.; Barnhill, D.R.; Adelson, M.D. Echinomycin (NSC 526417) in advanced ovarian cancer. A phase II trial of the Gynecologic Oncology Group. Am. J. Clin. Oncol. 1990, 13, 299–301. [Google Scholar] [CrossRef]
- Schilsky, R.L.; Faraggi, D.; Korzun, A.; Vogelzang, N.; Ellerton, J.; Wood, W.; Henderson, I.C. Phase II study of echinomycin in patients with advanced breast cancer: A report of Cancer and Leukemia Group B protocol 8641. Investig. New Drugs 1991, 9, 269–272. [Google Scholar] [CrossRef]
- Schumacher, G.; Neuhaus, P. 2-Methoxyestradiol—A new compound for cancer treatment. Dtsch. Med. Wochenschr. 2006, 131, 825–830. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Richardson, P.G.; Lacy, M.Q.; Dispenzieri, A.; Greipp, P.R.; Witzig, T.E.; Schlossman, R.; Sidor, C.F.; Anderson, K.C.; Gertz, M.A. Novel therapy with 2-methoxyestradiol for the treatment of relapsed and plateau phase multiple myeloma. Clin. Cancer Res. 2007, 13, 6162–6167. [Google Scholar] [CrossRef]
- Harrison, M.R.; Hahn, N.M.; Pili, R.; Oh, W.K.; Hammers, H.; Sweeney, C.; Kim, K.; Perlman, S.; Arnott, J.; Sidor, C.; et al. A phase II study of 2-methoxyestradiol (2ME2) NanoCrystal(R) dispersion (NCD) in patients with taxane-refractory, metastatic castrate-resistant prostate cancer (CRPC). Investig. New Drugs 2011, 29, 1465–1474. [Google Scholar] [CrossRef]
- Matei, D.; Schilder, J.; Sutton, G.; Perkins, S.; Breen, T.; Quon, C.; Sidor, C. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol. Oncol. 2009, 115, 90–96. [Google Scholar] [CrossRef]
- Kulke, M.H.; Chan, J.A.; Meyerhardt, J.A.; Zhu, A.X.; Abrams, T.A.; Blaszkowsky, L.S.; Regan, E.; Sidor, C.; Fuchs, C.S. A prospective phase II study of 2-methoxyestradiol administered in combination with bevacizumab in patients with metastatic carcinoid tumors. Cancer Chemother. Pharmacol. 2011, 68, 293–300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIF-1 Inhibitors | Anticancer Therapy | Cancer Type/ Cell Line | Mechanism for Prevention of Resistance | References |
|---|---|---|---|---|
| LW6 | Mitoxantrone Doxorubicin | Breast cancer | Drug efflux | [135] |
| LC478 | Docetaxel | Colorectal adenocarcinoma | Drug efflux | [136] |
| EZN-2208 (PEG-SN38) | Topotecan | Breast cancer | DNA damage repair | [137,138] |
| Chetomin | Radiation | Glioma | Metabolism | [139] |
| PX-478 | Radiation Gemcitabine | Pancreatic cancer | Apoptosis | [140,141] |
| PMX290 | 5-Fluorouracil | Colon adenocarcinoma | Apoptosis | [142] |
| FK228 (Romidepsin) | Temozolomide | Glioma | Apoptosis | [143] |
| BIX-01294 | TRAIL | Hepatocellular carcinoma | Apoptosis | [144] |
| LBH589 | Cisplatin Bortezomib Osimertinib | Ovarian cancer Multiple myeloma Lung cancer | Apoptosis | [145] [146] [147] |
| Vorinostat | Paclitaxel Doxorubicin Bortezomib | Breast cancer Neuroblastoma Mesothelioma | Apoptosis | [148] [149] [150] |
| NSC-134754 | Cisplatin Doxorubicin | Osteosarcoma | Apoptosis | [151] |
| YC-1 | Gefitinib Cisplatin | Lung cancer Oral cancer | Drug efflux Apoptosis | [152] [153] |
| RAD001 (Everolimus) | Cisplatin | Gastric cancer | Drug efflux Apoptosis | [154] |
| SCH66336 (Lonafarnib) | Paclitaxel Cisplatin | Lung cancer Melanoma | Drug efflux Apoptosis | [155] [156,157] |
| Echinomycin | Hormone | Prostate cancer | n.d. | [158] |
| Mechanism of HIF-1 Inhibition | HIF-1 Inhibitor | Phase | Cancer Type | Reference |
|---|---|---|---|---|
| Decreasing HIF-1α mRNA expression | EZN-2208 (PEG-SN38) | Phase I Phase II | Refractory solid tumors Metastatic colorectal cancer | [179,180] [181] |
| Decreasing HIF-1α protein synthesis | EZN-2968 | Phase I | Refractory solid tumors | [183] |
| Decreasing HIF-1α stabilization | FK228 (Romidepsin) | Phase I Phase II | NSCLC Head and neck cancer | [186] [187] |
| LBH589 (Panobinostat) | Phase I Phase I Phase III | MM Refractory solid tumors Refractory MM | [189] [190] [191] | |
| Vorinostat | Phase I/II Phase II Phase III | Glioblastoma Advanced melanoma Mesothelioma | [193] [194] [195] | |
| 17-AAG (Tanespimycin) | Phase II Phase I/II | Prostate cancer Refractory MM | [197] [166] | |
| SCH66336 (Lonafarnib) | Phase II Phase II Phase II | SCCHN Lung carcinoma Colorectal cancer | [199] [200] [155] | |
| Decreasing HIF-1/ DNA binding | Echinomycin | Phase II Phase II Phase II Phase II | Ovarian cancer Breast cancer Renal cell carcinoma Colorectal cancer | [203] [204] [205] [206] |
| Decreasing HIF-1α protein synthesis and transcriptional activity | 2ME2 | Phase II Phase II Phase II Phase II | MM Prostate cancer Ovarian cancer Carcinoid tumors | [208] [209] [210] [211] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. https://doi.org/10.3390/cancers14246054
Bui BP, Nguyen PL, Lee K, Cho J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers. 2022; 14(24):6054. https://doi.org/10.3390/cancers14246054
Chicago/Turabian StyleBui, Bich Phuong, Phuong Linh Nguyen, Kyeong Lee, and Jungsook Cho. 2022. "Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain" Cancers 14, no. 24: 6054. https://doi.org/10.3390/cancers14246054
APA StyleBui, B. P., Nguyen, P. L., Lee, K., & Cho, J. (2022). Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers, 14(24), 6054. https://doi.org/10.3390/cancers14246054