
Adrenomedullin Secreted by Melanoma Cells Promotes Melanoma Tumor Growth through Angiogenesis and Lymphangiogenesis

 , and
, and 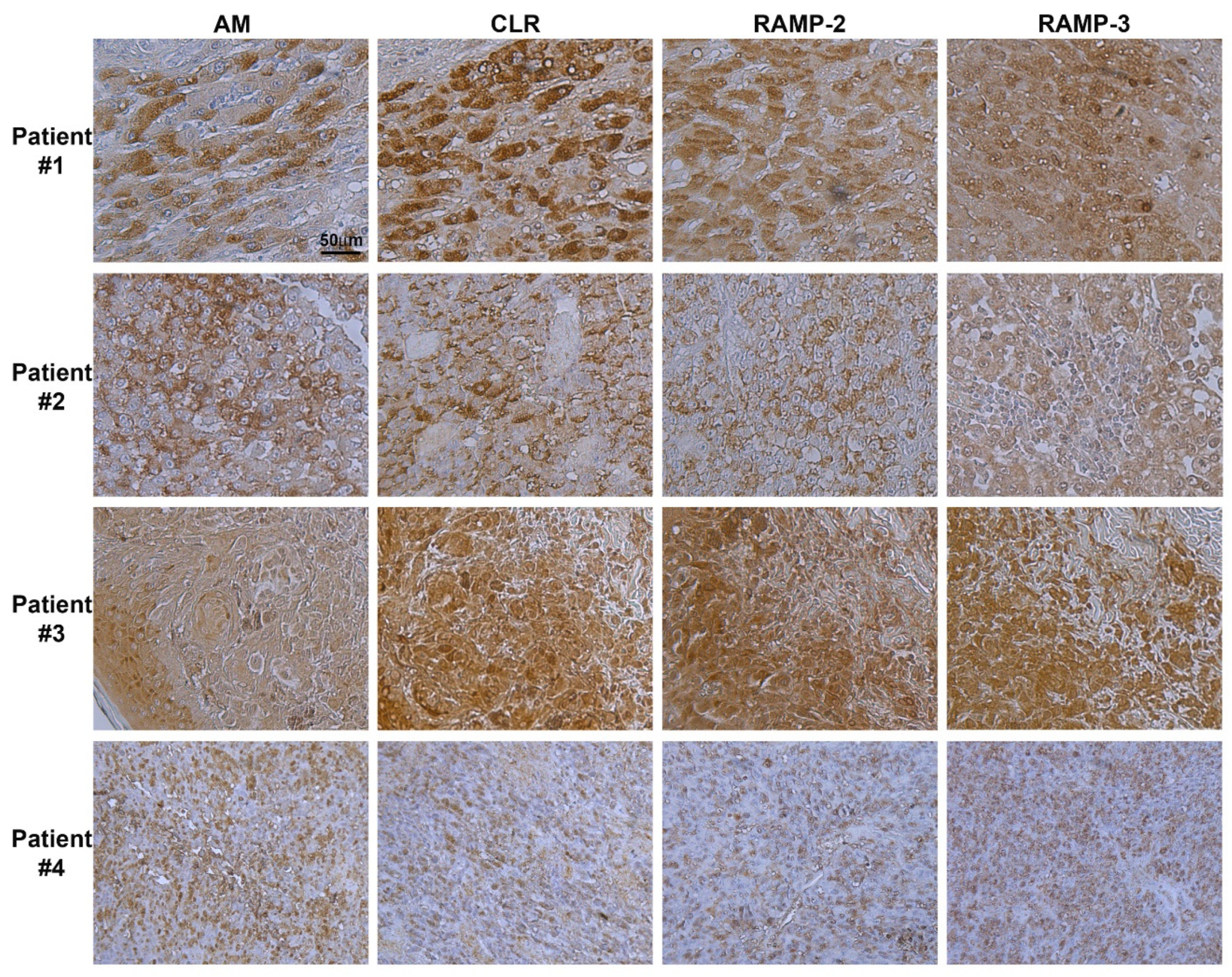{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Melanoma Tissues
2.2. Immunohistochemistry of the AM System in Human Melanoma
2.3. Cell Lines and Hypoxic Treatment
2.4. RNA Preparation and Real-Time Quantitative RT-PCR
2.5. Immunostaining of the Melanoma Cells
2.6. Cell Proliferation Assay
2.7. Cell Migration and Invasion Assays
2.8. In Vivo Matrigel Plug Studies
2.9. In Vivo Tumor Growth
2.10. Immunohistochemical Staining
2.11. Statistical Analysis
3. Results
3.1. Immunohistochemistry of AM, CLR, RAMP2, and RAMP3 Proteins in Human Melanoma
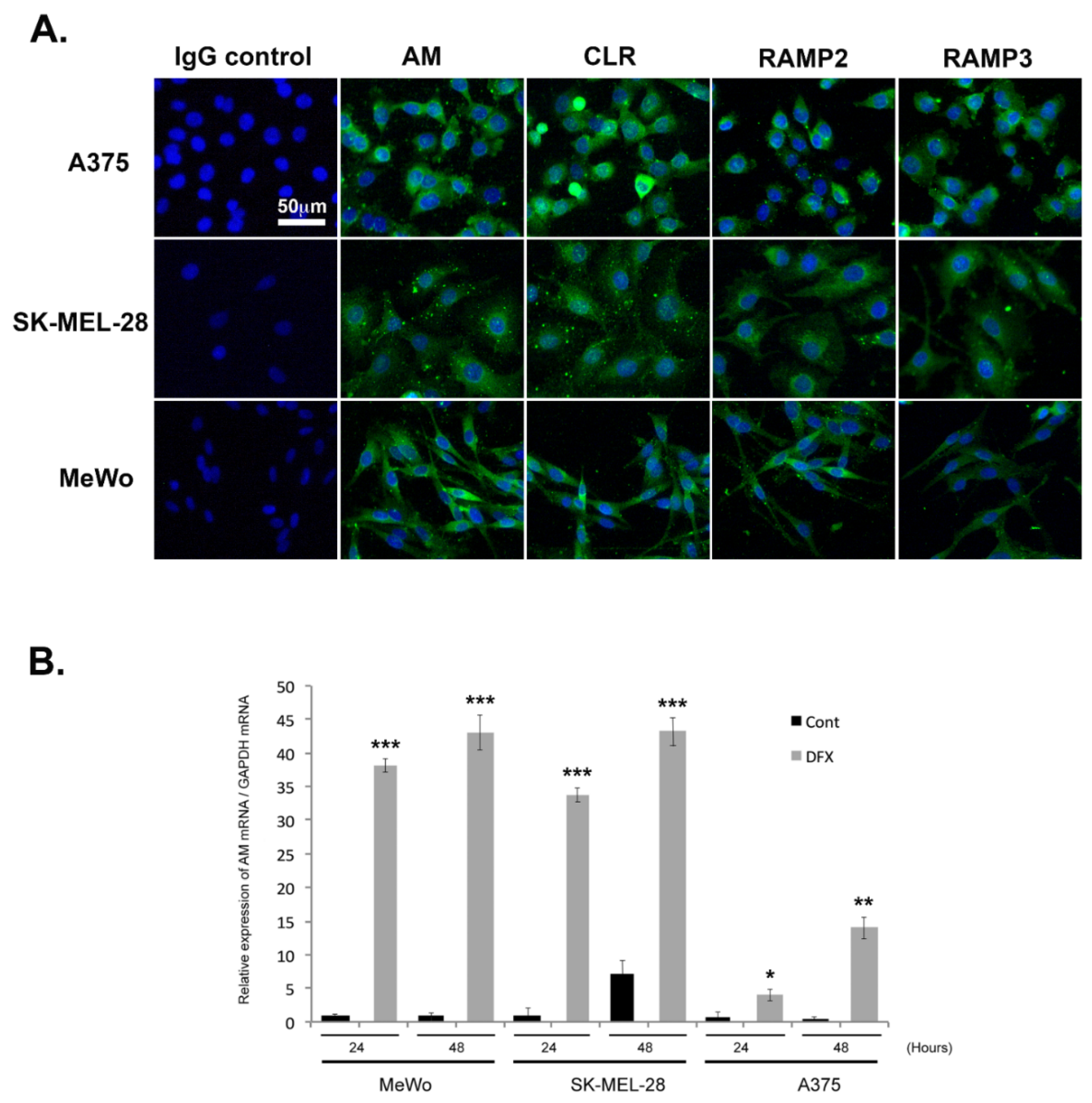
3.2. Expression of AM and AM Receptors in Melanoma Cells
3.3. Regulation of AM Expression by Hypoxia
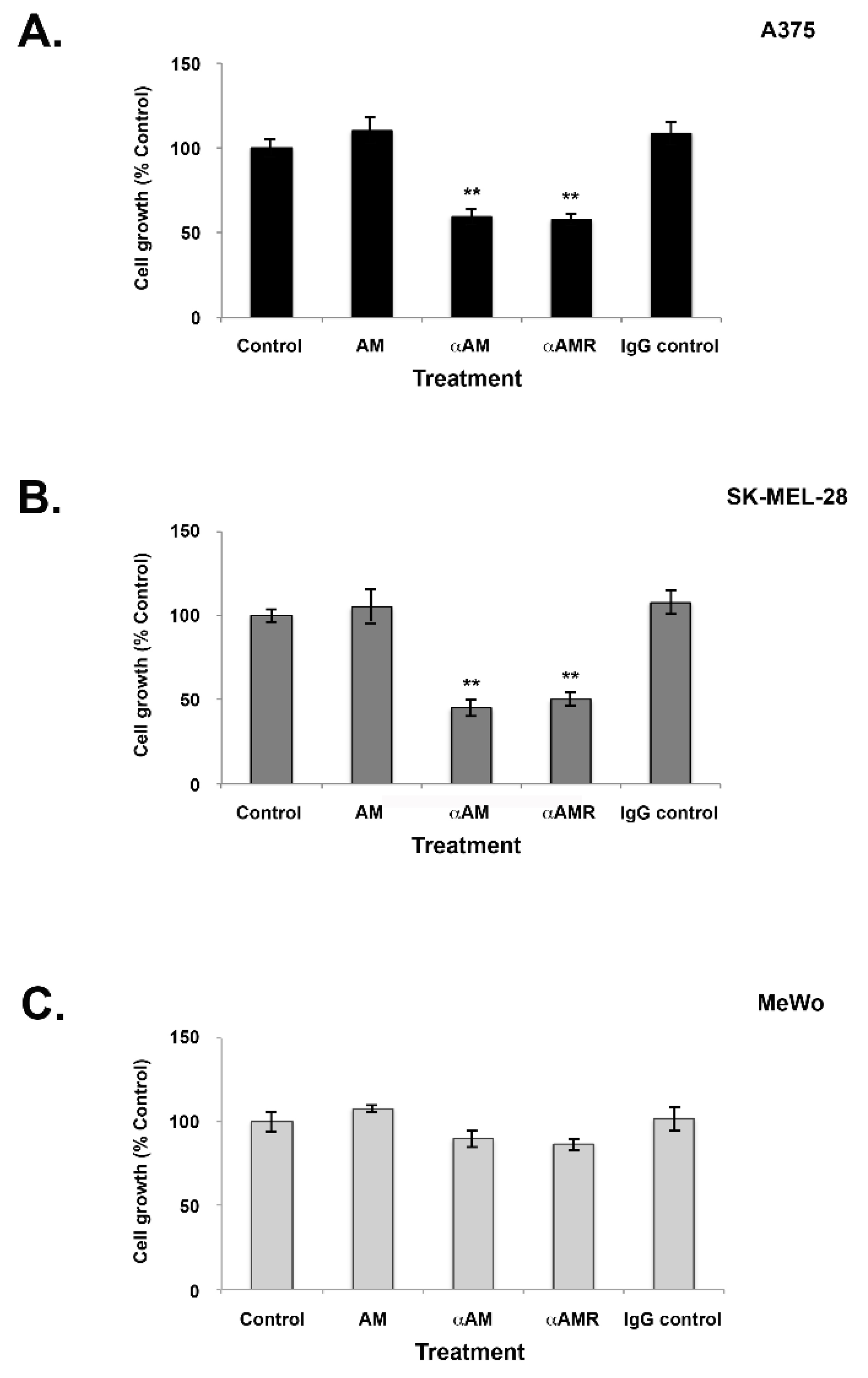
3.4. Effects of AM and AM Blockade on Melanoma Cell Proliferation
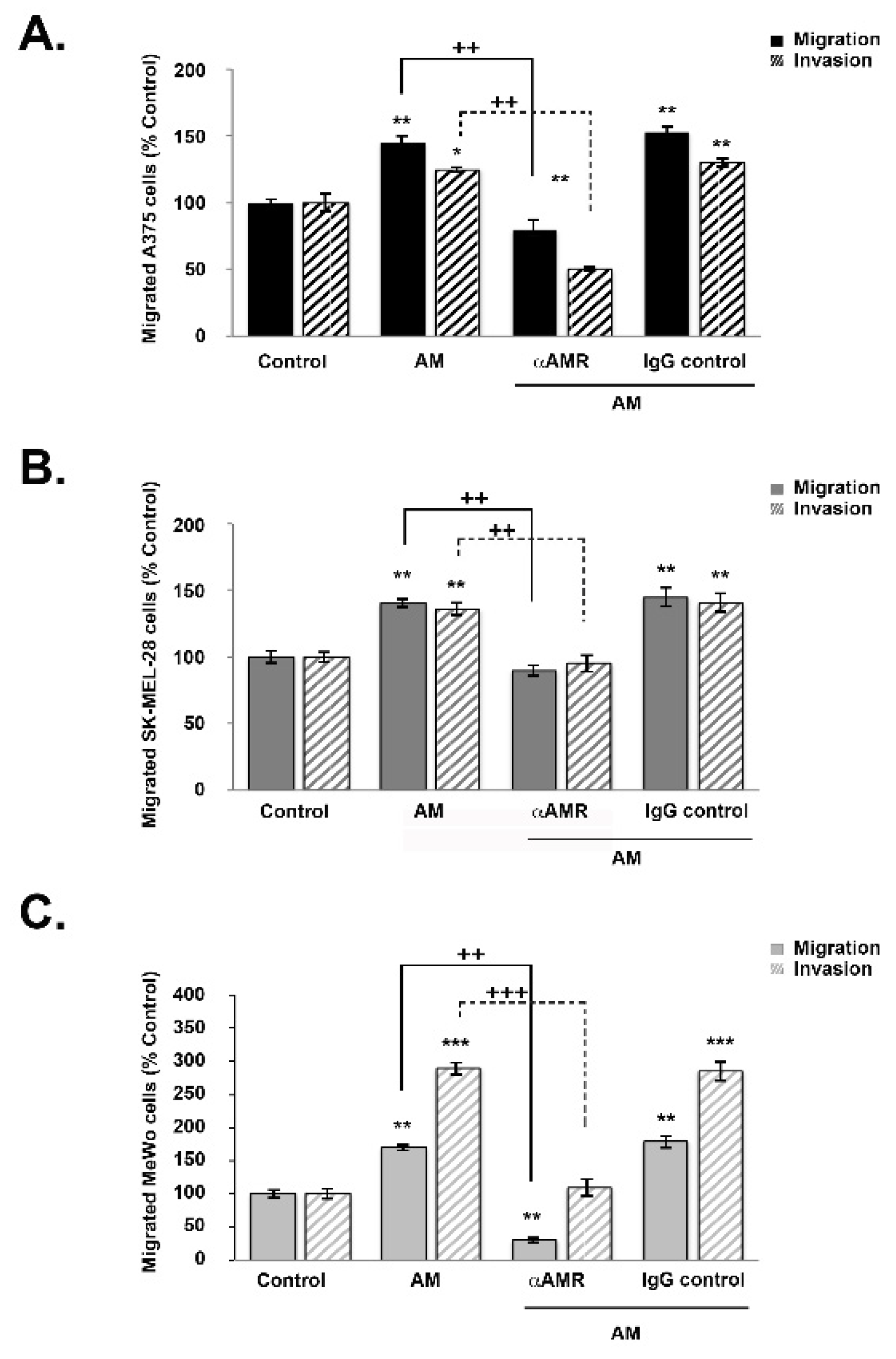
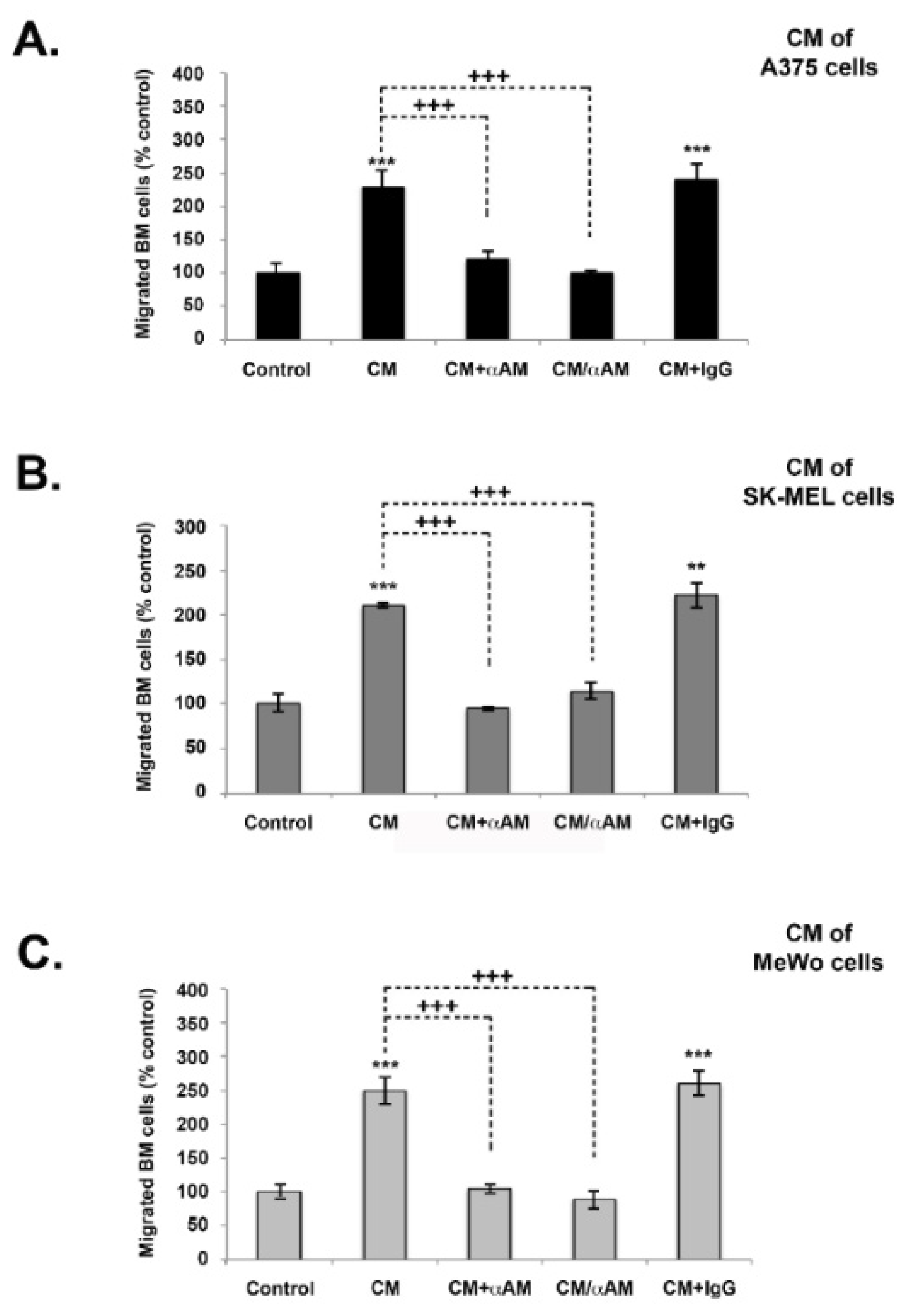
3.5. AM Induces Melanoma Cell Migration and Invasion In Vitro
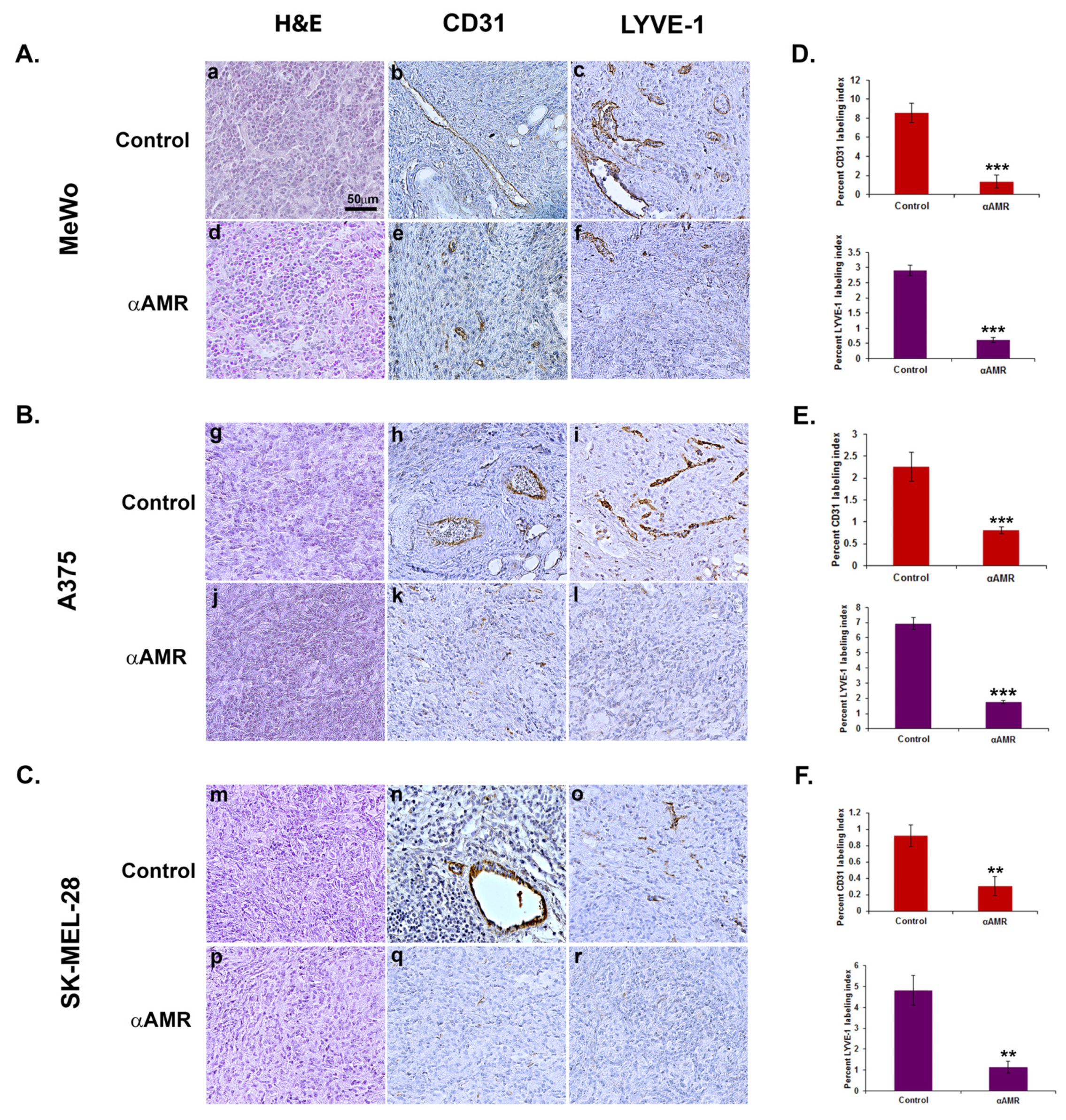
3.6. AM Released by Melanoma Cells Contributes to Angiogenesis and Lymphangiogenesis in Matrigel Plug Bioassays
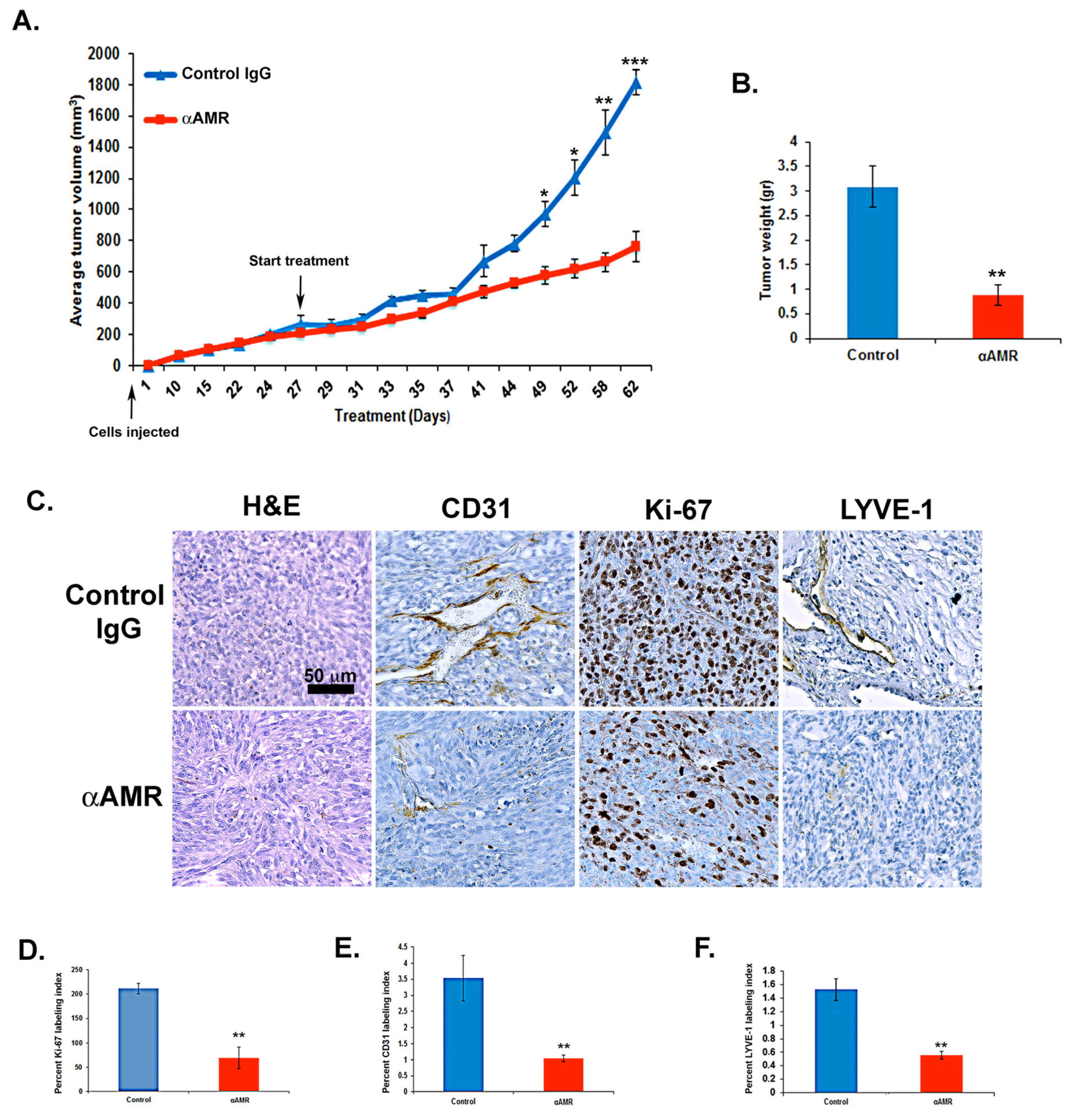
3.7. AM Blockade Inhibits the Growth of MeWo Tumor Xenografts In Vivo
3.8. AM Blockade Decreases Tumor Cell Proliferation and Impairs Tumor Angiogenesis and Lymphangiogenesis In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Streit, M.; Detmar, M. Angiogenesis, lymphangiogenesis, and melanoma metastasis. Oncogene 2003, 22, 3172–3179. [Google Scholar] [CrossRef]
- Greig, R.; Dunnington, D.; Murthy, U.; Anzano, M. Growth Factors as Novel Therapeutic Targets in Neoplastic Disease. Cancer Surv. 1988, 7, 653–674. [Google Scholar]
- Fernandez-Sauze, S.; Delfino, C.; Mabrouk, K.; Dussert, C.; Chinot, O.; Martin, P.-M.; Grisoli, F.; Ouafik, L. Effects of adrenomedullin on endothelial cells in the multistep process of angiogenesis: Involvement of CRLR/RAMP2 and CRLR/RAMP3 receptors. Int. J. Cancer 2003, 108, 797–804. [Google Scholar] [CrossRef] [PubMed]
- De Angeli, S.; Di Liddo, R.; Buoro, S.; Toniolo, L.; Conconi, M.T.; Belloni, A.S.; Parnigotto, P.P.; Nussdorfer, G.G. New immortalized human stromal cell lines enhancing in vitro expansion of cord blood hematopoietic stem cells. Int. J. Mol. Med. 2004, 13, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.L.; Walker, C.S.; Poyner, D.R. Adrenomedullin and calcitonin gene-related peptide receptors in endocrine-related cancers: Opportunities and challenges. Endocr. Relat. Cancer 2011, 18, C1–C14. [Google Scholar] [CrossRef]
- Hinson, J.P.; Kapas, S.; Smith, D.M. Adrenomedullin, a Multifunctional Regulatory Peptide*. Endocr. Rev. 2000, 21, 138–167. [Google Scholar] [CrossRef]
- Zhao, Y.; Hague, S.; Manek, S.; Zhang, L.; Bicknell, R.; Rees, M.C. PCR display identifies tamoxifen induction of the novel angiogenic factor adrenomedullin by a non estrogenic mechanism in the human endometrium. Oncogene 1998, 16, 409–415. [Google Scholar] [CrossRef] [PubMed]
- McLatchie, L.M.; Fraser, N.J.; Main, M.J.; Wise, A.; Brown, J.; Thompson, N.; Solari, R.; Lee, M.G.; Foord, S.M. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 1998, 393, 333–339. [Google Scholar] [CrossRef]
- Poyner, D.R.; Poyner, D.R.; Sexton, P.M.; Marshall, I.; Smith, D.M.; Quirion, R.; Born, W.; Muff, R.; Fischer, J.A.; Foord, S.M. International Union of Pharmacology. XXXII. The Mammalian Calcitonin Gene-Related Peptides, Adrenomedullin, Amylin, and Calcitonin Receptors. Pharmacol. Rev. 2002, 54, 233–246. [Google Scholar] [CrossRef]
- Zudaire, E.; Martínez, A.; Cuttitta, F. Adrenomedullin and cancer. Regul. Pept. 2003, 112, 175–183. [Google Scholar] [CrossRef]
- Vázquez, R.; Riveiro, M.E.; Berenguer-Daizé, C.; O’Kane, A.; Gormley, J.; Touzelet, O.; Rezai, K.; Bekradda, M.; Ouafik, L. Targeting Adrenomedullin in Oncology: A Feasible Strategy with Potential as Much More than an Alternative Anti-Angiogenic Therapy. Front. Oncol. 2020, 10, 589218. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Elsasser, T.H.; Muro-Cacho, C.; Moody, T.W.; Miller, M.J.; Macri, C.J.; Cuttitta, F. Expression of Adrenomedullin and Its Receptor in Normal and Malignant Human Skin: A Potential Pluripotent Role in the Integument. Endocrinology 1997, 138, 5597–5604. [Google Scholar] [CrossRef]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-Associated Macrophages Promote Angiogenesis and Melanoma Growth via Adrenomedullin in a Paracrine and Autocrine Manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [PubMed]
- Deville, J.-L.; Bartoli, C.; Berenguer, C.; Fernandez-Sauze, S.; Kaafarani, I.; Delfino, C.; Fina, F.; Salas, S.; Muracciole, X.; Mancini, J.; et al. Expression and role of adrenomedullin in renal tumors and value of its mRNA levels as prognostic factor in clear-cell renal carcinoma. Int. J. Cancer 2009, 125, 2307–2315. [Google Scholar] [CrossRef]
- Kocemba, K.; Van Andel, H.; De Haan-Kramer, A.; Mahtouk, K.; Versteeg, R.; Kersten, M.J.; Spaargaren, M.; Pals, S.T. The hypoxia target adrenomedullin is aberrantly expressed in multiple myeloma and promotes angiogenesis. Leukemia 2013, 27, 1729–1737. [Google Scholar] [CrossRef]
- Shichiri, M.; Hirata, Y. Regulation of Cell Growth and Apoptosis by Adrenomedullin. Hypertens. Res. 2003, 26, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Belloni, A.S.; Trejter, M.; Malendowicz, L.K.; Nussdorfer, G.G. Adrenomedullin stimulates proliferation and inhibits apoptosis of immature rat thymocytes cultured in vitro. Peptides 2003, 24, 295–300. [Google Scholar] [CrossRef]
- Uzan, B.; Ea, H.-K.; Launay, J.-M.; Garel, J.-M.; Champy, R.; Cressent, M.; Lioté, F. A Critical Role for Adrenomedullin-Calcitonin Receptor-Like Receptor in Regulating Rheumatoid Fibroblast-Like Synoviocyte Apoptosis. J. Immunol. 2006, 176, 5548–5558. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Vos, M.; Guédez, L.; Kaur, G.; Chen, Z.; Garayoa, M.; Pio, R.; Moody, T.; Stetler-Stevenson, W.; Kleinman, H.K.; et al. The Effects of Adrenomedullin Overexpression in Breast Tumor Cells. Gynecol. Oncol. 2002, 94, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Ouafik, L.; Sauze, S.; Boudouresque, F.; Chinot, O.; Delfino, C.; Fina, F.; Vuaroqueaux, V.; Dussert, C.; Palmari, J.; Dufour, H.; et al. Neutralization of Adrenomedullin Inhibits the Growth of Human Glioblastoma Cell Lines in Vitro and Suppresses Tumor Xenograft Growth in Vivo. Am. J. Pathol. 2002, 160, 1279–1292. [Google Scholar] [CrossRef]
- Kaafarani, I.; Fernandez-Sauze, S.; Berenguer, C.; Chinot, O.; Delfino, C.; Dussert, C.; Metellus, P.; Boudouresque, F.; Mabrouk, K.; Grisoli, F.; et al. Targeting adrenomedullin receptors with systemic delivery of neutralizing antibodies inhibits tumor angiogenesis and suppresses growth of human tumor xenografts in mice. FASEB J. 2009, 23, 3424–3435. [Google Scholar] [CrossRef] [PubMed]
- Berenguer-Daizé, C.; Boudouresque, F.; Bastide, C.; Tounsi, A.; Benyahia, Z.; Acunzo, J.; Dussault, N.; Delfino, C.; Baeza, N.; Daniel, L.; et al. Adrenomedullin Blockade Suppresses Growth of Human Hormone–Independent Prostate Tumor Xenograft in Mice. Clin. Cancer Res. 2013, 19, 6138–6150. [Google Scholar] [CrossRef] [PubMed]
- Oehler, M.K.; Hague, S.; Rees, M.C.; Bicknell, R. Adrenomedullin promotes formation of xenografted endometrial tumors by stimulation of autocrine growth and angiogenesis. Oncogene 2002, 21, 2815–2821. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karpinich, N.O.; Kechele, D.O.; Espenschied, S.T.; Willcockson, H.H.; Fedoriw, Y.; Caron, K.M. Adrenomedullin gene dosage correlates with tumor and lymph node lymphangiogenesis. FASEB J. 2013, 27, 590–600. [Google Scholar] [CrossRef]
- Ishikawa, T.; Chen, J.; Wang, J.; Okada, F.; Sugiyama, T.; Kobayashi, T.; Shindo, M.; Higashino, F.; Katoh, H.; Asaka, M.; et al. Adrenomedullin antagonist suppresses in vivo growth of human pancreatic cancer cells in SCID mice by suppressing angiogenesis. Oncogene 2003, 22, 1238–1242. [Google Scholar] [CrossRef]
- Ramachandran, V.; Arumugam, T.; Hwang, R.F.; Greenson, J.K.; Simeone, D.M.; Logsdon, C.D. Adrenomedullin Is Expressed in Pancreatic Cancer and Stimulates Cell Proliferation and Invasion in an Autocrine Manner via the Adrenomedullin Receptor, ADMR. Cancer Res. 2007, 67, 2666–2675. [Google Scholar] [CrossRef]
- Berenguer, C.; Boudouresque, F.; Dussert, C.; Daniel, L.; Muracciole, X.; Grino, M.; Rossi, D.; Mabrouk, K.; Figarella-Branger, D.; Martin, P.-M.; et al. Adrenomedullin, an autocrine/paracrine factor induced by androgen withdrawal, stimulates ‘neuroendocrine phenotype’ in LNCaP prostate tumor cells. Oncogene 2008, 27, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Benyahia, Z.; Dussault, N.; Cayol, M.; Sigaud, R.; Berenguer-Daizé, C.; Delfino, C.; Tounsi, A.; Garcia, S.; Martin, P.-M.; Mabrouk, K.; et al. Stromal fibroblasts present in breast carcinomas promote tumor growth and angiogenesis through adrenomedullin secretion. Oncotarget 2017, 8, 15744–15762. [Google Scholar] [CrossRef]
- Nouguerède, E.; Berenguer, C.; Garcia, S.; Bennani, B.; Delfino, C.; Nanni, I.; Dahan, L.; Gasmi, M.; Seitz, J.; Martin, P.; et al. Expression of adrenomedullin in human colorectal tumors and its role in cell growth and invasion in vitro and in xenograft growth in vivo. Cancer Med. 2013, 2, 196–207. [Google Scholar] [CrossRef]
- Greillier, L.; Tounsi, A.; Berenguer-Daizé, C.; Dussault, N.; Delfino, C.; Benyahia, Z.; Cayol, M.; Mabrouk, K.; Garcia, S.; Martin, P.-M.; et al. Functional Analysis of the Adrenomedullin Pathway in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2016, 11, 94–107. [Google Scholar] [CrossRef]
- Michaylira, C.Z.; Nakagawa, H. Hypoxic microenvironment as a cradle for melanoma development and progression. Cancer Biol. Ther. 2006, 5, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Giaccia, A.J. The Unique Physiology of Solid Tumors: Opportunities (and Problems) for Cancer Therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar] [PubMed]
- Metellus, P.; Voutsinos-Porche, B.; Nanni-Metellus, I.; Colin, C.; Fina, F.; Berenguer, C.; Dussault, N.; Boudouresque, F.; Loundou, A.; Intagliata, D.; et al. Adrenomedullin expression and regulation in human glioblastoma, cultured human glioblastoma cell lines and pilocytic astrocytoma. Eur. J. Cancer 2011, 47, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Kangawa, K.; Kawamoto, M.; Ichiki, Y.; Nakamura, S.; Matsuo, H.; Eto, T. Adrenomedullin: A Novel Hypotensive Peptide Isolated from Human Pheochromocytoma. Biochem. Biophys. Res. Commun. 1993, 192, 553–560. [Google Scholar] [CrossRef]
- Kato, H.; Shichiri, M.; Marumo, F.; Hirata, Y. Adrenomedullin as an Autocrine/Paracrine Apoptosis Survival Factor for Rat Endothelial Cells*. Endocrinology 1997, 138, 2615–2620. [Google Scholar] [CrossRef]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in Cutaneous Melanoma Is Associated with RAS Activation and MEK Dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Nikitenko, L.L.; Fox, S.B.; Kehoe, S.; Rees, M.C.P.; Bicknell, R. Adrenomedullin and Tumour Angiogenesis. Br. J. Cancer 2006, 94, 1–7. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.-H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Stacker, S.A.; Achen, M.; Jussila, L.; Baldwin, M.E.; Alitalo, K. Lymphangiogenesis and cancer metastasis. Nat. Rev. Cancer 2002, 2, 573–583. [Google Scholar] [CrossRef]
- He, Y.; Kozaki, K.-I.; Karpanen, T.; Koshikawa, K.; Yla-Herttuala, S.; Takahashi, T.; Alitalo, K. Suppression of Tumor Lymphangiogenesis and Lymph Node Metastasis by Blocking Vascular Endothelial Growth Factor Receptor 3 Signaling. J. Natl. Cancer Inst. 2002, 94, 819–825. [Google Scholar] [CrossRef]
- Skobe, M.; Hawighorst, T.; Jackson, D.G.; Prevo, R.; Janes, L.; Velasco, P.; Riccardi, L.; Alitalo, K.; Claffey, K.; Detmar, M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 2001, 7, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Garayoa, M.; Martínez, A.; Lee, S.; Pío, R.; An, W.G.; Neckers, L.; Trepel, J.; Montuenga, L.M.; Ryan, H.; Johnson, R.; et al. Hypoxia-Inducible Factor-1 (HIF-1) Up-Regulates Adrenomedullin Expression in Human Tumor Cell Lines during Oxygen Deprivation: A Possible Promotion Mechanism of Carcinogenesis. Mol. Endocrinol. 2000, 14, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Six, K.L.; Dunworth, W.P.; Li, M.; Caron, K.M. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J. Clin. Invest. 2008, 118, 40–50. [Google Scholar] [CrossRef]
- Jin, D.; Harada, K.; Ohnishi, S.; Yamahara, K.; Kangawa, K.; Nagaya, N. Adrenomedullin induces lymphangiogenesis and ameliorates secondary lymphoedema. Cardiovasc. Res. 2008, 80, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Achen, M.G.; McColl, B.K.; Stacker, S.A. Focus on lymphangiogenesis in tumor metastasis. Cancer Cell 2005, 7, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Tobler, N.E.; Detmar, M. Tumor and lymph node lymphangiogenesis-impact on cancer metastasis. J. Leukoc. Biol. 2006, 80, 691–696. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nature 2005, 438, 946–953. [Google Scholar] [CrossRef]
- Nagy, J.A.; Vasile, E.; Feng, D.; Sundberg, C.; Brown, L.F.; Detmar, M.J.; Lawitts, J.A.; Benjamin, L.; Tan, X.; Manseau, E.J.; et al. Vascular Permeability Factor/Vascular Endothelial Growth Factor Induces Lymphangiogenesis as well as Angiogenesis. J. Exp. Med. 2002, 196, 1497–1506. [Google Scholar] [CrossRef]
- Hirakawa, S.; Kodama, S.; Kunstfeld, R.; Kajiya, K.; Brown, L.F.; Detmar, M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J. Exp. Med. 2005, 201, 1089–1099. [Google Scholar] [CrossRef]
- Cao, R.; Björndahl, M.A.; Religa, P.; Clasper, S.; Garvin, S.; Galter, D.; Meister, B.; Ikomi, F.; Tritsaris, K.; Dissing, S.; et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell 2004, 6, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Koyama, T.; Sakurai, T.; Kamiyoshi, A.; Ichikawa-Shindo, Y.; Kawate, H.; Liu, T.; Xian, X.; Imai, A.; Zhai, L.; et al. The endothelial adrenomedullin-RAMP2 system regulates vascular integrity and suppresses tumour metastasis. Cardiovasc. Res. 2016, 111, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Khalfaoui-Bendriss, G.; Dussault, N.; Fernandez-Sauze, S.; Berenguer-Daizé, C.; Sigaud, R.; Delfino, C.; Cayol, M.; Metellus, P.; Chinot, O.; Mabrouk, K.; et al. Adrenomedullin blockade induces regression of tumor neovessels through interference with vascular endothelial-cadherin signalling. Oncotarget 2015, 6, 7536–7553. [Google Scholar] [CrossRef]
- Martínez, A.; Julián, M.; Bregonzio, C.; Notari, L.; Moody, T.W.; Cuttitta, F. Identification of Vasoactive Nonpeptidic Positive and Negative Modulators of Adrenomedullin Using a Neutralizing Antibody-Based Screening Strategy. Endocrinology 2004, 145, 3858–3865. [Google Scholar] [CrossRef][Green Version]
- Avgoustou, P.; Jailani, A.B.A.; Zirimwabagabo, J.-O.; Tozer, M.J.; Gibson, K.R.; Glossop, P.A.; Mills, J.E.J.; Porter, R.A.; Blaney, P.; Bungay, P.J.; et al. Discovery of a First-in-Class Potent Small Molecule Antagonist against the Adrenomedullin-2 Receptor. ACS Pharmacol. Transl. Sci. 2020, 3, 706–719. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benyahia, Z.; Gaudy-Marqueste, C.; Berenguer-Daizé, C.; Chabane, N.; Dussault, N.; Cayol, M.; Vellutini, C.; Djemli, A.; Nanni, I.; Beaufils, N.; et al. Adrenomedullin Secreted by Melanoma Cells Promotes Melanoma Tumor Growth through Angiogenesis and Lymphangiogenesis. Cancers 2022, 14, 5909. https://doi.org/10.3390/cancers14235909
Benyahia Z, Gaudy-Marqueste C, Berenguer-Daizé C, Chabane N, Dussault N, Cayol M, Vellutini C, Djemli A, Nanni I, Beaufils N, et al. Adrenomedullin Secreted by Melanoma Cells Promotes Melanoma Tumor Growth through Angiogenesis and Lymphangiogenesis. Cancers. 2022; 14(23):5909. https://doi.org/10.3390/cancers14235909
Chicago/Turabian StyleBenyahia, Zohra, Caroline Gaudy-Marqueste, Caroline Berenguer-Daizé, Norhimane Chabane, Nadège Dussault, Mylène Cayol, Christine Vellutini, Amina Djemli, Isabelle Nanni, Nathalie Beaufils, and et al. 2022. "Adrenomedullin Secreted by Melanoma Cells Promotes Melanoma Tumor Growth through Angiogenesis and Lymphangiogenesis" Cancers 14, no. 23: 5909. https://doi.org/10.3390/cancers14235909
APA StyleBenyahia, Z., Gaudy-Marqueste, C., Berenguer-Daizé, C., Chabane, N., Dussault, N., Cayol, M., Vellutini, C., Djemli, A., Nanni, I., Beaufils, N., Mabrouk, K., Grob, J.-J., & Ouafik, L. (2022). Adrenomedullin Secreted by Melanoma Cells Promotes Melanoma Tumor Growth through Angiogenesis and Lymphangiogenesis. Cancers, 14(23), 5909. https://doi.org/10.3390/cancers14235909