
The Anti-Vascular Endothelial Growth Factor Receptor 1 (VEGFR-1) D16F7 Monoclonal Antibody Inhibits Melanoma Adhesion to Soluble VEGFR-1 and Tissue Invasion in Response to Placenta Growth Factor
 ,
,  , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Cell Adhesion Assay
2.3. Transendothelial Cell Migration Assay
2.4. Invasion Assay
2.5. Quantification of PlGF-2 in B16F10 Cell Culture Conditioned Media by ELISA
2.6. Immunoblot Analysis
2.7. In Vivo Studies
2.8. Statistical Analysis
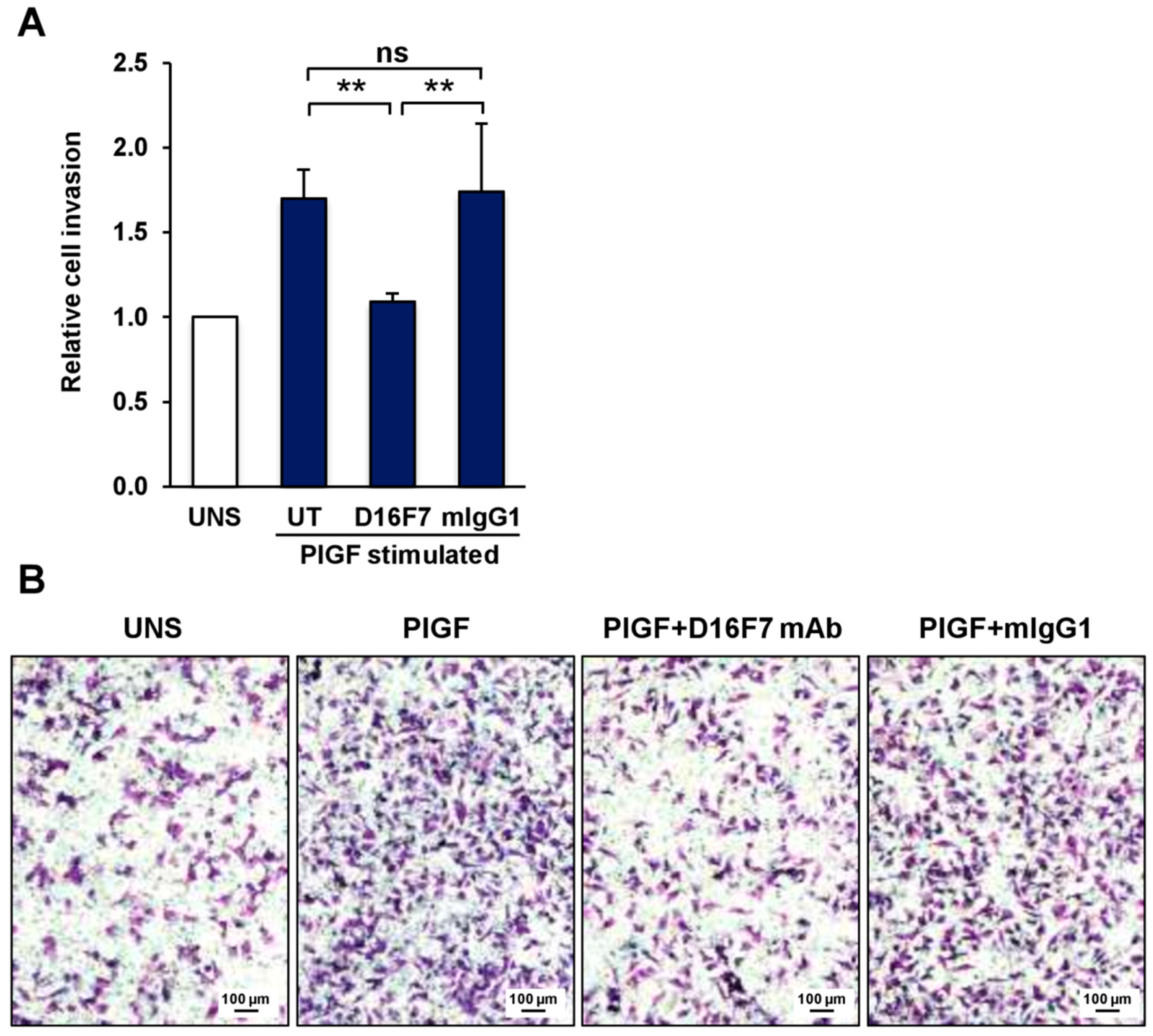
3. Results
3.1. Involvement of VEGFR-1 in Melanoma Cell Invasiveness and Adhesion to ECM
3.2. The Role of PlGF in an In Vivo Murine Melanoma Model
3.3. Effects of D16F7 mAb on Human Melanoma and Endothelial Cell Adhesion to sVEGFR-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewerchin, M.; Carmeliet, P. Placental growth factor in cancer. Expert Opin. Ther. Targets 2014, 18, 1339–1354. [Google Scholar] [CrossRef]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef]
- Chau, K.; Hennessy, A.; Makris, A. Placental growth factor and pre-eclampsia. J. Hum. Hypertens. 2017, 31, 782–786. [Google Scholar] [CrossRef]
- DiPalma, T.; Tucci, M.; Russo, G.; Maglione, D.; Lago, C.T.; Romano, A.; Saccone, S.; Della Valle, G.; De Gregorio, L.; Dragani, T.A.; et al. The placenta growth factor gene of the mouse. Mamm. Genome 1996, 7, 6–12. [Google Scholar] [CrossRef]
- Dewerchi, M.; Carmeliet, P. PlGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef]
- Lacal, P.M.; Graziani, G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol. Res. 2018, 136, 97–107. [Google Scholar] [CrossRef]
- Albonici, L.; Giganti, M.G.; Modesti, A.; Manzari, V.; Bei, R. Multifaceted Role of the Placental Growth Factor (PlGF) in the Antitumor Immune Response and Cancer Progression. Int. J. Mol. Sci. 2019, 20, 2970. [Google Scholar] [CrossRef]
- Kong, X.; Bu, J.; Chen, J.; Ni, B.; Fu, B.; Zhou, F.; Pang, S.; Zhang, J.; Xu, S.; He, C. PIGF and Flt-1 on the surface of macrophages induces the production of TGF-β1 by polarized tumor-associated macrophages to promote lung cancer angiogenesis. Eur. J. Pharmacol. 2021, 912, 174550. [Google Scholar] [CrossRef]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; De Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef]
- Li, T.; Zhu, Y.; Han, L.; Ren, W.; Liu, H.; Qin, C. VEGFR-1 activation-induced MMP-9-dependent invasion in hepatocellular carcinoma. Future Oncol. 2015, 11, 3143–3157. [Google Scholar] [CrossRef]
- Lacal, P.M.; Failla, C.M.; Pagani, E.; Odorisio, T.; Schietroma, C.; Falcinelli, S.; Zambruno, G.; D’Atri, S. Human melanoma cells secrete and respond to placenta growth factor and vascular endothelial growth factor. J. Investig. Dermatol. 2000, 115, 1000–1007. [Google Scholar] [CrossRef]
- Marcellini, M.; De Luca, N.; Riccioni, T.; Ciucci, A.; Orecchia, A.; Lacal, P.M.; Ruffini, F.; Pesce, M.; Cianfarani, F.; Zambruno, G.; et al. Increased melanoma growth and metastasis spreading in mice overexpressing placenta growth factor. Am. J. Pathol. 2006, 169, 643–654. [Google Scholar] [CrossRef]
- Aoki, S.; Inoue, K.; Klein, S.; Halvorsen, S.; Chen, J.; Matsui, A.; Nikmaneshi, M.R.; Kitahara, S.; Hato, T.; Chen, X.; et al. Placental growth factor promotes tumour desmoplasia and treatment resistance in intrahepatic cholangiocarcinoma. Gut 2022, 71, 185–193. [Google Scholar] [CrossRef]
- Nixon, A.B.; Sibley, A.B.; Liu, Y.; Hatch, A.J.; Jiang, C.; Mulkey, F.; Starr, M.D.; Brady, J.C.; Niedzwiecki, D.; Venook, A.P.; et al. Plasma Protein Biomarkers in Advanced or Metastatic Colorectal Cancer Patients Receiving Chemotherapy With Bevacizumab or Cetuximab: Results from CALGB 80405 (Alliance). Clin. Cancer. Res. 2022, 28, 2779–2788. [Google Scholar] [CrossRef]
- Pagani, E.; Ruffini, F.; Antonini Cappellini, G.C.; Scoppola, A.; Fortes, C.; Marchetti, P.; Graziani, G.; D’Atri, S.; Lacal, P.M. Placenta growth factor and neuropilin-1 collaborate in promoting melanoma aggressiveness. Int. J. Oncol. 2016, 48, 1581–1589. [Google Scholar] [CrossRef]
- Graziani, G.; Ruffini, F.; Tentori, L.; Scimeca, M.; Dorio, A.S.; Atzori, M.G.; Failla, C.M.; Morea, V.; Bonanno, E.; D’Atri, S.; et al. Antitumor activity of a novel anti-vascular endothelial growth factor receptor-1 monoclonal antibody that does not interfere with ligand binding. Oncotarget 2016, 7, 72868–72885. [Google Scholar] [CrossRef]
- Atzori, M.G.; Tentori, L.; Ruffini, F.; Ceci, C.; Lisi, L.; Bonanno, E.; Scimeca, M.; Eskilsson, E.; Daubon, T.; Miletic, H.; et al. The anti-vascular endothelial growth factor receptor-1 monoclonal antibody D16F7 inhibits invasiveness of human glioblastoma and glioblastoma stem cells. J. Exp. Clin. Cancer Res. 2017, 36, 106. [Google Scholar] [CrossRef]
- Atzori, M.G.; Tentori, L.; Ruffini, F.; Ceci, C.; Bonanno, E.; Scimeca, M.; Lacal, P.M.; Graziani, G. The Anti-Vascular Endothelial Growth Factor Receptor-1 Monoclonal Antibody D16F7 Inhibits Glioma Growth and Angiogenesis In Vivo. J. Pharmacol. Exp. Ther. 2018, 364, 77–86. [Google Scholar] [CrossRef]
- Lacal, P.M.; Atzori, M.G.; Ruffini, F.; Scimeca, M.; Bonanno, E.; Cicconi, R.; Mattei, M.; Bernardini, R.; D’Atri, S.; Tentori, L.; et al. Targeting the vascular endothelial growth factor receptor-1 by the monoclonal antibody D16F7 to increase the activity of immune checkpoint inhibitors against cutaneous melanoma. Pharmacol. Res. 2020, 159, 104957. [Google Scholar] [CrossRef]
- Failla, C.M.; Carbo, M.; Morea, V. Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1. Int. J. Mol. Sci. 2018, 19, 1306. [Google Scholar] [CrossRef]
- Orecchia, A.; Lacal, P.M.; Schietroma, C.; Morea, V.; Zambruno, G.; Failla, C.M. Vascular endothelial growth factor receptor-1 is deposited in the extracellular matrix by endothelial cells and is a ligand for the alpha 5 beta 1 integrin. J. Cell Sci. 2003, 116, 3479–3489. [Google Scholar] [CrossRef]
- Orecchia, A.; Mettouchi, A.; Uva, P.; Simon, G.C.; Arcelli, D.; Avitabile, S.; Ragone, G.; Meneguzzi, G.; Pfenninger, K.H.; Zambruno, G.; et al. Endothelial cell adhesion to soluble vascular endothelial growth factor receptor-1 triggers a cell dynamic and angiogenic phenotype. FASEB J. 2014, 28, 692–704. [Google Scholar] [CrossRef]
- Colotti, G.; Failla, C.M.; Lacal, P.M.; Ungarelli, M.; Ruffini, F.; Di Micco, P.; Orecchia, A.; Morea, V. Neuropilin-1 is required for endothelial cell adhesion to soluble vascular endothelial growth factor receptor 1. FEBS J. 2022, 289, 183–198. [Google Scholar] [CrossRef]
- Ruffini, F.; Failla, C.M.; Orecchia, A.; Bani, M.R.; Dorio, A.S.; Fortes, C.; Zambruno, G.; Graziani, G.; Giavazzi, R.; D’Atri, S.; et al. Expression of the soluble vascular endothelial growth factor receptor-1 in cutaneous melanoma: Role in tumour progression. Br. J. Dermatol. 2011, 164, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Vergati, M.; Muzi, A.; Levati, L.; Ruffini, F.; Forini, O.; Vernole, P.; Lacal, P.M.; Graziani, G. Generation of an immortalized human endothelial cell line as a model of neovascular proliferating endothelial cells to assess chemosensitivity to anticancer drugs. Int. J. Oncol. 2005, 27, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Lacal, P.M.; Petrillo, M.G.; Ruffini, F.; Muzi, A.; Bianchini, R.; Ronchetti, S.; Migliorati, G.; Riccardi, C.; Graziani, G.; Nocentini, G. Glucocorticoid-induced tumor necrosis factor receptor family-related ligand triggering upregulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 and promotes leukocyte adhesion. J. Pharmacol. Exp. Ther. 2013, 347, 164–172. [Google Scholar] [CrossRef]
- Lacal, P.M.; Ruffini, F.; Pagani, E.; D’Atri, S. An autocrine loop directed by the vascular endothelial growth factor promotes invasiveness of human melanoma cells. Int. J. Oncol. 2005, 27, 1625–1632. [Google Scholar]
- Sissaoui, S.; Egginton, S.; Ting, L.; Ahmed, A.; Hewett, P.W. Hyperglycemia up-regulates placental growth factor (PlGF) expression and secretion in endothelial cells via suppression of PI3 kinase-Akt signaling and activation of FOXO1. Sci. Rep. 2021, 11, 16344. [Google Scholar] [CrossRef]
- D’Agostino, R.B. Tests for Normal Distribution in Goodness-Of-Fit Techniques; D’Agostino, R.B., Stephens, M.A., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1986. [Google Scholar]
- Das, K.R.; Rahmatullah Imon, A.H.M. A Brief Review of Tests for Normality. Am. J. Theor. App. Stat. 2016, 5, 5–12. [Google Scholar]
- Kim, K.J.; Cho, C.S.; Kim, W.U. Role of placenta growth factor in cancer and inflammation. Exp. Mol. Med. 2012, 44, 10–19. [Google Scholar] [CrossRef]
- Taylor, A.P.; Goldenberg, D.M. Role of placenta growth factor in malignancy and evidence that an antagonistic PlGF/Flt-1 peptide inhibits the growth and metastasis of human breast cancer xenografts. Mol. Cancer Ther. 2007, 6, 524–531. [Google Scholar] [CrossRef]
- Martinsson-Niskanen, T.; Riisbro, R.; Larsson, L.; Winstedt, L.; Stenberg, Y.; Pakola, S.; Stassen, J.M.; Glazer, S. Monoclonal antibody TB-403: A first-in-human, Phase I, double-blind, dose escalation study directed against placental growth factor in healthy male subjects. Clin. Ther 2011, 33, 1142–1149. [Google Scholar] [CrossRef]
- Lassen, U.; Nielsen, D.L.; Sørensen, M.; Winstedt, L.; Niskanen, T.; Stenberg, Y.; Pakola, S.; Stassen, J.M.; Glazer, S. A phase I, dose-escalation study of TB-403, a monoclonal antibody directed against PlGF, in patients with advanced solid tumours. Br. J. Cancer 2012, 106, 678–684. [Google Scholar] [CrossRef]
- Lassen, U.; Chinot, O.L.; McBain, C.; Mau-Sørensen, M.; Larsen, V.A.; Barrie, M.; Roth, P.; Krieter, O.; Wang, K.; Habben, K.; et al. Phase 1 dose-escalation study of the antiplacental growth factor monoclonal antibody RO5323441 combined with bevacizumab in patients with recurrent glioblastoma. Neuro. Oncol. 2015, 17, 1007–1015. [Google Scholar] [CrossRef][Green Version]
- Saulnier Sholler, G.; Duda, D.G.; Bergendahl, G.; Ebb, D.; Snuderl, M.; Laetsch, T.W.; Michlitsch, J.; Hanson, D.; Isakoff, M.S.; Bielamowicz, K.; et al. A Phase I Trial of TB-403 in Relapsed Medulloblastoma, Neuroblastoma, Ewing Sarcoma, and Alveolar Rhabdomyosarcoma. Clin. Cancer Res. 2022, 28, 3950–3957. [Google Scholar] [CrossRef]
- Abou Faycal, C.; Brambilla, E.; Agorreta, J.; Lepeltier, N.; Jacquet, T.; Lemaître, N.; Emadali, A.; Lucas, A.; Lacal, P.M.; Montuenga, L.; et al. The sVEGFR1-i13 splice variant regulates a β1 integrin/VEGFR autocrine loop involved in the progression and the response to anti-angiogenic therapies of squamous cell lung carcinoma. Br. J. Cancer 2018, 118, 1596–1608. [Google Scholar] [CrossRef]
- Wang, K.; Stark, F.S.; Schlothauer, T.; Lahr, A.; Cosson, V.; Zhi, J.; Habben, K.; Tessier, J.; Schick, E.; Staack, R.F.; et al. An apparent clinical pharmacokinetic drug-drug interaction between bevacizumab and the anti-placental growth factor monoclonal antibody RO5323441 via a target-trapping mechanism. Cancer Chemother. Pharmacol. 2017, 79, 661–671. [Google Scholar] [CrossRef]
- Zekri, J.; Marples, M.; Taylor, D.; Kandukurti, K.; McParland, L.; Brown, J.E. Complications of bone metastases from malignant melanoma. J. Bone Oncol. 2017, 8, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Caldaria, A.; Giuffrida, R.; di Meo, N.; Massari, L.; Dianzani, C.; Cannavò, S.P.; Degrassi, F.; Casablanca, E.; Zalaudek, I.; Conforti, C. Diagnosis and treatment of melanoma bone metastasis: A multidisciplinary approach. Dermatol. Ther. 2020, 33, e14193. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Untreated Cells 1 | D16F7-Treated Cells 1 | % Decrease | p Value 2 |
|---|---|---|---|---|
| 1 | 292 ± 31 | 160 ± 27 | 45.2 | <0.05 |
| 2 | 133 ± 27 | 84 ± 15 | 36.9 | <0.05 |
| 3 | 271 ± 11 | 180 ± 11 | 33.6 | <0.05 |
| 4 | 663 ± 80 | 400 ± 109 | 39.7 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atzori, M.G.; Ceci, C.; Ruffini, F.; Scimeca, M.; Cicconi, R.; Mattei, M.; Lacal, P.M.; Graziani, G. The Anti-Vascular Endothelial Growth Factor Receptor 1 (VEGFR-1) D16F7 Monoclonal Antibody Inhibits Melanoma Adhesion to Soluble VEGFR-1 and Tissue Invasion in Response to Placenta Growth Factor. Cancers 2022, 14, 5578. https://doi.org/10.3390/cancers14225578
Atzori MG, Ceci C, Ruffini F, Scimeca M, Cicconi R, Mattei M, Lacal PM, Graziani G. The Anti-Vascular Endothelial Growth Factor Receptor 1 (VEGFR-1) D16F7 Monoclonal Antibody Inhibits Melanoma Adhesion to Soluble VEGFR-1 and Tissue Invasion in Response to Placenta Growth Factor. Cancers. 2022; 14(22):5578. https://doi.org/10.3390/cancers14225578
Chicago/Turabian StyleAtzori, Maria Grazia, Claudia Ceci, Federica Ruffini, Manuel Scimeca, Rosella Cicconi, Maurizio Mattei, Pedro Miguel Lacal, and Grazia Graziani. 2022. "The Anti-Vascular Endothelial Growth Factor Receptor 1 (VEGFR-1) D16F7 Monoclonal Antibody Inhibits Melanoma Adhesion to Soluble VEGFR-1 and Tissue Invasion in Response to Placenta Growth Factor" Cancers 14, no. 22: 5578. https://doi.org/10.3390/cancers14225578
APA StyleAtzori, M. G., Ceci, C., Ruffini, F., Scimeca, M., Cicconi, R., Mattei, M., Lacal, P. M., & Graziani, G. (2022). The Anti-Vascular Endothelial Growth Factor Receptor 1 (VEGFR-1) D16F7 Monoclonal Antibody Inhibits Melanoma Adhesion to Soluble VEGFR-1 and Tissue Invasion in Response to Placenta Growth Factor. Cancers, 14(22), 5578. https://doi.org/10.3390/cancers14225578