
Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy

 , ,
, , 
Simple Summary
Abstract
1. Introduction
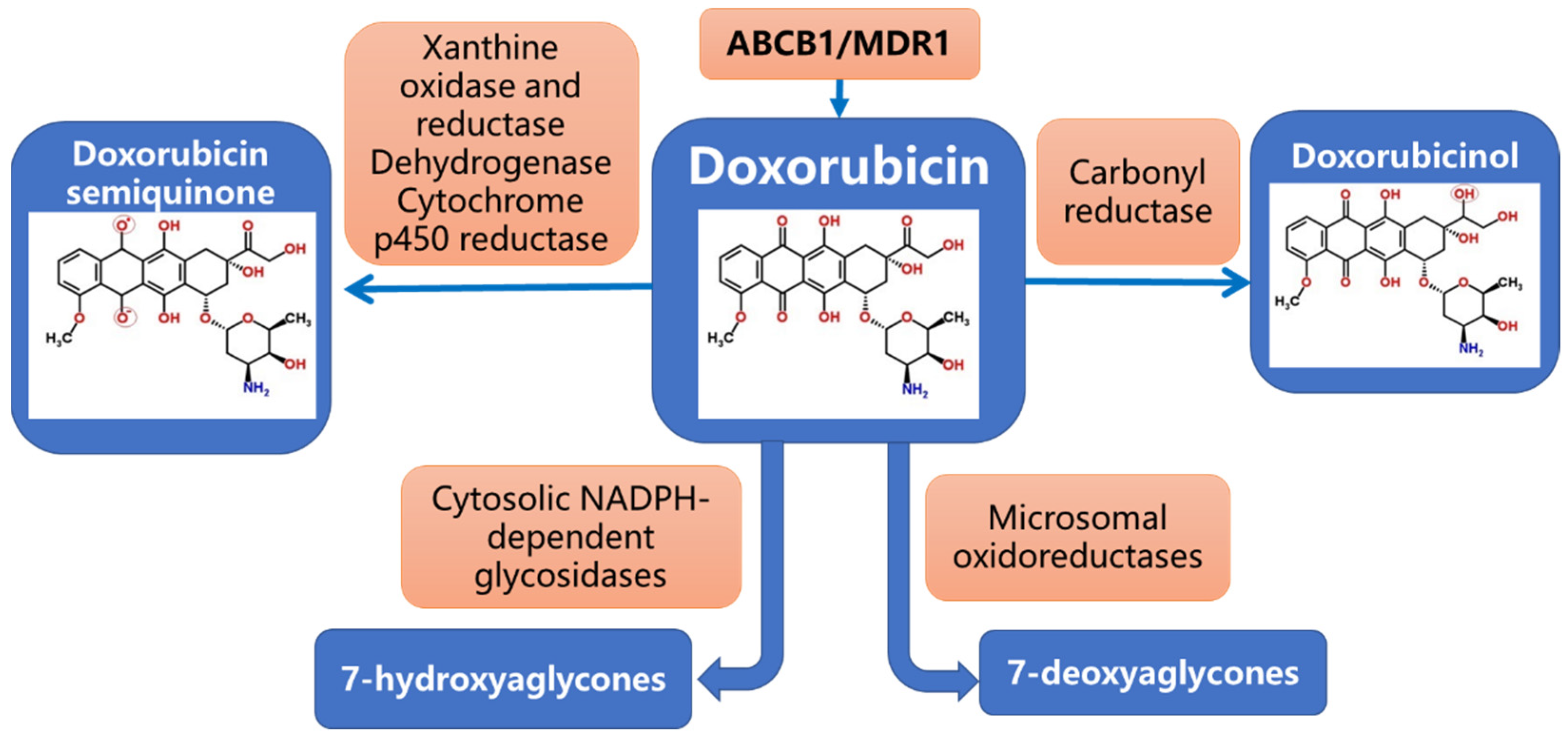
2. Pharmacogenetics of Dox Metabolism
2.1. CYP3A4 Polymorphism
2.2. CYP2D6 Polymorphism
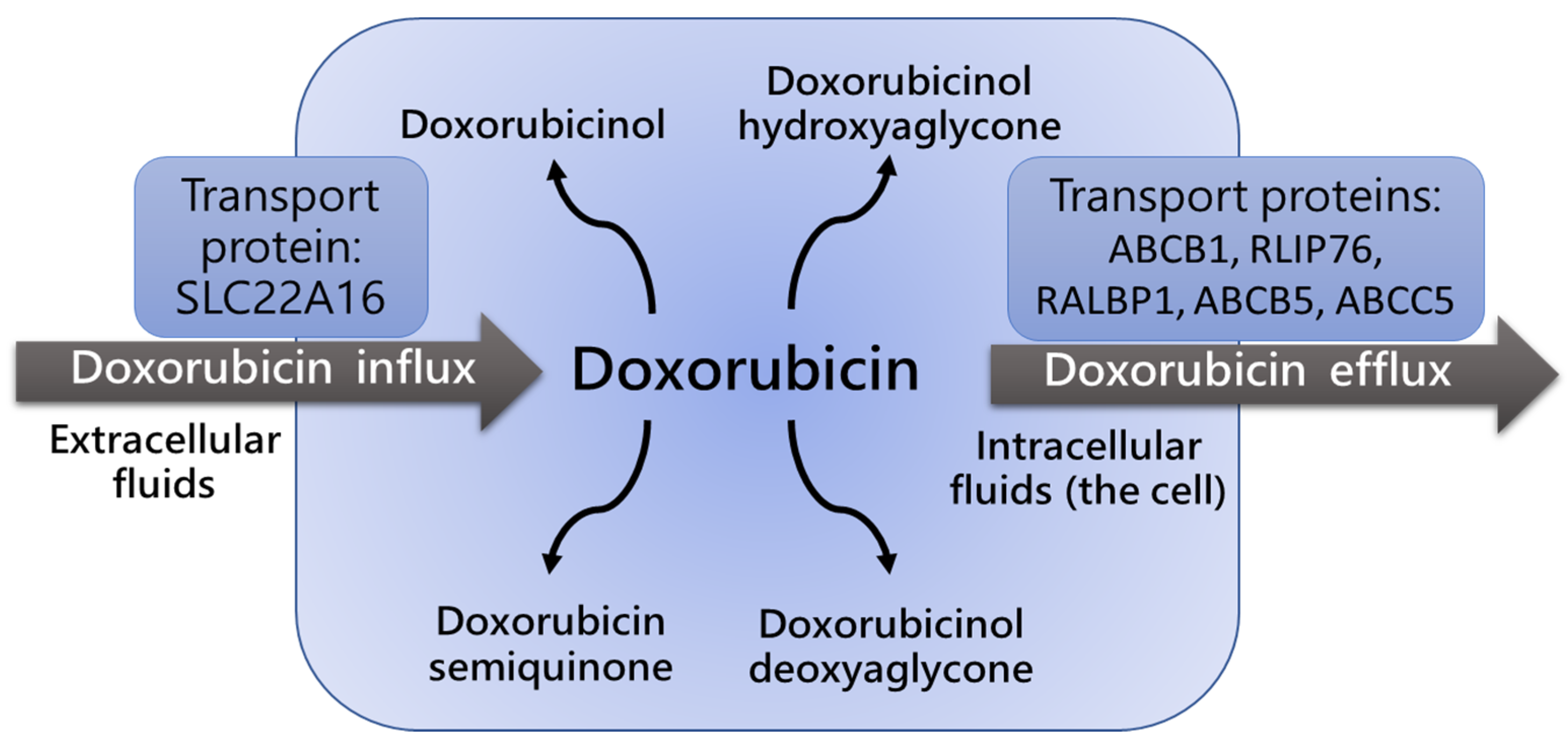
2.3. P-Glycoprotein Polymorphism and Dox Blood Concentration and Clearance
3. Genetic Polymorphisms of Detoxifying Enzymes and Drug Resistance
4. Genetic Polymorphisms and Cardiotoxicity in Dox-Treated Patients
5. Genetic Polymorphisms Associated with Dox-Induced Hematological, Nephrological, and Gastrointestinal (GI) Toxicities
5.1. Role of Gene Polymorphism in Dox-Associated Hematotoxicity
5.2. Role of Gene Polymorphism in Dox-Associated Nephrotoxicity and GI Toxicity
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Health Estimates 2016: Disease Burden by Cause, Age, Sex, by Country and by Region, 2000–2016; World Health Organization: Geneva, Switzerland, 2018; Available online: https://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html (accessed on 9 July 2019).
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.F.; Anderson, B.O.; Buzaid, A.C.; Jazieh, A.R.; Niessen, L.W.; Blauvelt, B.M.; Buchanan, D.R. Identifying important breast cancer control strategies in Asia, Latin America and the Middle East/North Africa. BMC Health Serv. Res. 2011, 11, 227. [Google Scholar] [CrossRef]
- Sheikh, A.; Hussain, S.A.; Ghori, Q.; Naeem, N.; Fazil, A.; Giri, S.; Sathian, B.; Mainali, P.; Al Tamimi, D.M. The Spectrum of Genetic Mutations in Breast Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 2177–2185. [Google Scholar] [CrossRef]
- Manahan, E.R.; Kuerer, H.M.; Sebastian, M.; Hughes, K.S.; Boughey, J.C.; Euhus, D.M.; Boolbol, S.K.; Taylor, W.A. Consensus Guidelines on Genetic’ Testing for Hereditary Breast Cancer from the American Society of Breast Surgeons. Ann. Surg. Oncol. 2019, 26, 3025–3031. [Google Scholar] [CrossRef]
- Jiang, Y.; Tian, T.; Yu, C.; Zhou, W.; Yang, J.; Wang, Y.; Wen, Y.; Chen, J.; Dai, J.; Jin, G.; et al. Identification of Recurrent Variants in BRCA1 and BRCA2 across Multiple Cancers in the Chinese Population. BioMed Res. Int. 2020, 2020, 6739823. [Google Scholar] [CrossRef]
- Chen, J.; Bae, E.; Zhang, L.; Hughes, K.; Parmigiani, G.; Braun, D.; Rebbeck, T.R. Penetrance of Breast and Ovarian Cancer in Women Who Carry a BRCA1/2 Mutation and Do Not Use Risk-Reducing Salpingo-Oophorectomy: An Updated Meta-Analysis. JNCI Cancer Spectr. 2020, 4, pkaa029. [Google Scholar] [CrossRef]
- Tan, M.-H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime Cancer Risks in Individuals with Germline PTEN Mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Welch, J.S. Patterns of mutations in TP53 mutated AML. Best Pract. Res. Clin. Haematol. 2018, 31, 379–383. [Google Scholar] [CrossRef]
- Figueiredo, J.; Melo, S.; Carneiro, P.; Moreira, A.M.; Fernandes, M.S.; Ribeiro, A.S.; Guilford, P.; Paredes, J.; Seruca, R. Clinical spectrum and pleiotropic nature of CDH1 germline mutations. J. Med. Genet. 2019, 56, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Heestand, G.M.; Kurzrock, R. Molecular landscape of pancreatic cancer: Implications for current clinical trials. Oncotarget 2015, 6, 4553–4561. [Google Scholar] [CrossRef] [PubMed]
- Balmaña, J.; Díez, O.; Rubio, I.T.; Cardoso, F.; ESMO Guidelines Working Group. BRCA in breast cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. 2011, 22 (Suppl. S6), vi31–vi34. [Google Scholar] [CrossRef]
- Niell, B.L.; Freer, P.E.; Weinfurtner, R.J.; Arleo, E.K.; Drukteinis, J.S. Screening for Breast Cancer. Radiol. Clin. N. Am. 2017, 55, 1145–1162. [Google Scholar] [CrossRef]
- Wang, M.; Hou, L.; Chen, M.; Zhou, Y.; Liang, Y.; Wang, S.; Jiang, J.; Zhang, Y. Neoadjuvant Chemotherapy Creates Surgery Opportunities for Inoperable Locally Advanced Breast Cancer. Sci. Rep. 2017, 7, 44673. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Kaufmann, M.; von Minckwitz, G.; Mamounas, E.P.; Cameron, D.; Carey, L.A.; Cristofanilli, M.; Denkert, C.; Eiermann, W.; Gnant, M.; Harris, J.R.; et al. Recommendations from an International Consensus Conference on the Current Status and Future of Neoadjuvant Systemic Therapy in Primary Breast Cancer. Ann. Surg. Oncol. 2011, 19, 1508–1516. [Google Scholar] [CrossRef]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal drug delivery systems and anticancer drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef]
- Butowska, K.; Woziwodzka, A.; Borowik, A.; Piosik, J. Polymeric Nanocarriers: A Transformation in Doxorubicin Therapies. Materials 2021, 14, 2135. [Google Scholar] [CrossRef]
- Speth, P.A.J.; Van Hoesel, Q.G.C.M.; Haanen, C. Clinical Pharmacokinetics of Doxorubicin. Clin. Pharmacokinet. 1988, 15, 15–31. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Tecza, K.; Pamula-Pilat, J.; Lanuszewska, J.; Butkiewicz, D.; Grzybowska, E. Pharmacogenetics of toxicity of 5-fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget 2018, 9, 9114–9136. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Rizwan, A.; Jiang, L.; Bhujwalla, Z.M.; Glunde, K. Molecular Effects of Doxorubicin on Choline Metabolism in Breast Cancer. Neoplasia 2017, 19, 617–627. [Google Scholar] [CrossRef]
- Reis-Mendes, A.; Carvalho, F.; Remião, F.; Sousa, E.; Bastos, M.D.L.; Costa, V.M. The Main Metabolites of Fluorouracil + Adriamycin + Cyclophosphamide (FAC) Are Not Major Contributors to FAC Toxicity in H9c2 Cardiac Differentiated Cells. Biomolecules 2019, 9, 98. [Google Scholar] [CrossRef]
- Mordente, A.; Meucci, E.; Silvestrini, A.; Martorana, G.; Giardina, B. New Developments in Anthracycline-Induced Cardiotoxicity. Curr. Med. Chem. 2009, 16, 1656–1672. [Google Scholar] [CrossRef] [PubMed]
- Siebel, C.; Lanvers-Kaminsky, C.; Würthwein, G.; Hempel, G.; Boos, J. Bioanalysis of doxorubicin aglycone metabolites in human plasma samples–implications for doxorubicin drug monitoring. Sci. Rep. 2020, 10, 18562. [Google Scholar] [CrossRef]
- Rendic, S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev. 2002, 34, 83–448. [Google Scholar] [CrossRef]
- Chang, V.Y.; Wang, J.J. Pharmacogenetics of Chemotherapy-Induced Cardiotoxicity. Curr. Oncol. Rep. 2018, 20, 52. [Google Scholar] [CrossRef]
- Tavira, B.; Coto, E.; Diaz-Corte, C.; Alvarez, V.; López-Larrea, C.; Ortega, F. A search for new CYP3A4 variants as determinants of tacrolimus dose requirements in renal-transplanted patients. Pharm. Genom. 2013, 23, 445–448. [Google Scholar] [CrossRef]
- Werk, A.N.; Cascorbi, I. Functional Gene Variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef]
- Andreu, F.; Colom, H.; Elens, L.; van Gelder, T.; van Schaik, R.H.N.; Hesselink, D.A.; Bestard, O.; Torras, J.; Cruzado, J.M.; Grinyó, J.M.; et al. A New CYP3A5*3 and CYP3A4*22 Cluster Influencing Tacrolimus Target Concentrations: A Population Approach. Clin. Pharmacokinet. 2017, 56, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Sandanaraj, E.; Lal, S.; Selvarajan, V.; Ooi, L.L.; Wong, Z.W.; Wong, N.S.; Ang, P.C.S.; Lee, E.J.; Chowbay, B. PXR Pharmacogenetics: Association of Haplotypes with Hepatic CYP3A4 and ABCB1 Messenger RNA Expression and Doxorubicin Clearance in Asian Breast Cancer Patients. Clin. Cancer Res. 2008, 14, 7116–7126. [Google Scholar] [CrossRef] [PubMed]
- Lum, D.W.K.; Perel, P.; Hingorani, A.D.; Holmes, M.V. CYP2D6 Genotype and Tamoxifen Response for Breast Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e76648. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Im, S.; Keam, B.; Ham, H.S.; Lee, K.H.; Kim, T.Y.; Kim, Y.J.; Oh, D.; Kim, J.H.; Han, W.; et al. ABCB 1 polymorphism as prognostic factor in breast cancer patients treated with docetaxel and doxorubicin neoadjuvant chemotherapy. Cancer Sci. 2014, 106, 86–93. [Google Scholar] [CrossRef]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.-Q. Pharmacogenomics of Drug Metabolizing Enzymes and Transporters: Relevance to Precision Medicine. Genom. Proteom. Bioinform. 2016, 14, 298–313. [Google Scholar] [CrossRef]
- Fung, K.L.; Gottesman, M.M. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2009, 1794, 860–871. [Google Scholar] [CrossRef]
- Singhal, S.S.; Singhal, J.; Nair, M.P.; Lacko, A.G.; Awasthi, Y.C.; Awasthi, S. Doxorubicin transport by RALBP1 and ABCG2 in lung and breast cancer. Int. J. Oncol. 2007, 30, 717–725. [Google Scholar] [CrossRef]
- Lal, S.; Sutiman, N.; Ooi, L.L.; Wong, Z.W.; Wong, N.S.; Ang, P.C.S.; Chowbay, B. Pharmacogenetics of ABCB5, ABCC5 and RLIP76 and doxorubicin pharmacokinetics in Asian breast cancer patients. Pharm. J. 2016, 17, 337–343. [Google Scholar] [CrossRef]
- Li, Z.; Chen, C.; Chen, L.; Hu, D.; Yang, X.; Zhuo, W.; Chen, Y.; Yang, J.; Zhou, Y.; Mao, M.; et al. STAT5a Confers Doxorubicin Resistance to Breast Cancer by Regulating ABCB1. Front. Oncol. 2021, 11, 697950. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Orive, G. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 2021, 27, 436–455. [Google Scholar] [CrossRef] [PubMed]
- Sparreboom, A.; Planting, A.S.; Jewell, R.C.; El Van Der Burg, M.; Van Der Gaast, A.; De Bruijn, P.; Loos, W.J.; Nooter, K.; Chandler, L.H.; Paul, E.M.; et al. Clinical pharmacokinetics of doxorubicin in combination with GF120918, a potent inhibitor of MDR1 P-glycoprotein. Anti-Cancer Drugs 1999, 10, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Stearns, V.; Davidson, N.E.; Flockhart, D.A. Pharmacogenetics in the treatment of breast cancer. Pharm. J. 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Pulford, D.J. The Glut athione S-Transferase Supergene Family: Regulation of GST and the Contribution of the lsoenzymes to Cancer Chemoprotection and Drug Resistance Part I. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–520. [Google Scholar] [CrossRef]
- Stewart, D.J. Tumor and host factors that may limit efficacy of chemotherapy in non-small cell and small cell lung cancer. Crit. Rev. Oncol. 2010, 75, 173–234. [Google Scholar] [CrossRef]
- Zhao, E.A.J.; Zhang, H.; Lei, T.; Liu, J.; Zhang, S.; Wu, N.; Sun, B.; Wang, M. Drug resistance gene expression and chemotherapy sensitivity detection in Chinese women with different molecular subtypes of breast cancer. Cancer Biol. Med. 2020, 17, 1014–1025. [Google Scholar] [CrossRef]
- Jeong, H.; Herskowitz, I.; Kroetz, D.L.; Rine, J. Function-Altering SNPs in the Human Multidrug Transporter Gene ABCB1 Identified Using a Saccharomyces-Based Assay. PLoS Genet. 2007, 3, e39. [Google Scholar] [CrossRef]
- Mittal, B.; Tulsyan, S.; Mittal, R. The effect of ABCB1 polymorphisms on the outcome of breast cancer treatment. Pharm. Pers. Med. 2016, 9, 47–58. [Google Scholar] [CrossRef]
- Madrid-Paredes, A.; Cañadas-Garre, M.; Sánchez-Pozo, A.; Expósito-Ruiz, M.; Calleja-Hernández, M. ABCB1 gene polymorphisms and response to chemotherapy in breast cancer patients: A meta-analysis. Surg. Oncol. 2017, 26, 473–482. [Google Scholar] [CrossRef]
- Ruiz-Pinto, S.; Martin, M.; Pita, G.; Caronia, D.; de la Torre-Montero, J.C.; Moreno, L.T.; Moreno, F.; García-Sáenz, J.; Benítez, J.; González-Neira, A. Pharmacogenetic variants and response to neoadjuvant single-agent doxorubicin or docetaxel: A study in locally advanced breast cancer patients participating in the NCT00123929 phase 2 randomized trial. Pharm. Genom. 2018, 28, 245–250. [Google Scholar] [CrossRef]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.-M.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early Detection of Anthracycline Cardiotoxicity and Improvement With Heart Failure Therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Saleh, Y.; Abdelkarim, O.; Herzallah, K.; Abela, G.S. Anthracycline-induced cardiotoxicity: Mechanisms of action, incidence, risk factors, prevention, and treatment. Heart Fail. Rev. 2020, 26, 1159–1173. [Google Scholar] [CrossRef]
- Fernandez-Chas, M.; Curtis, M.J.; Niederer, S.A. Mechanism of doxorubicin cardiotoxicity evaluated by integrating multiple molecular effects into a biophysical model. J. Cereb. Blood Flow Metab. 2017, 175, 763–781. [Google Scholar] [CrossRef]
- Aminkeng, F.; Ross, C.J.D.; Rassekh, S.R.; Hwang, S.; Rieder, M.J.; Bhavsar, A.P.; Smith, A.; Sanatani, S.; Gelmon, K.A.; Bernstein, D.; et al. Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br. J. Clin. Pharmacol. 2016, 82, 683–695. [Google Scholar] [CrossRef]
- Bhagat, A.; Kleinerman, E.S. Anthracycline-Induced Cardiotoxicity: Causes, Mechanisms, and Prevention. Adv. Exp. Med. Biol. 2020, 1257, 181–192. [Google Scholar] [CrossRef]
- Ravi, D.; Das, K.C. Redox-cycling of anthracyclines by thioredoxin system: Increased superoxide generation and DNA damage. Cancer Chemother. Pharmacol. 2004, 54, 449–458. [Google Scholar] [CrossRef]
- Osataphan, N.; Phrommintikul, A.; Chattipakorn, S.C.; Chattipakorn, N. Effects of doxorubicin-induced cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial function: Insights for future interventions. J. Cell. Mol. Med. 2020, 24, 6534–6557. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.-I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Norton, N.; Weil, R.M.; Advani, P.P. Inter-Individual Variation and Cardioprotection in Anthracycline-Induced Heart Failure. J. Clin. Med. 2021, 10, 4079. [Google Scholar] [CrossRef] [PubMed]
- Timm, K.N.; Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, J.; Teng, H.; Peng, J. Os Efeitos da Doxorrubicina na Biossíntese e no Metabolismo do Heme em Cardiomiócitos. Arq. Bras. Cardiol. 2021, 116, 315–322. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, X.; Song, P.; Yuan, Y.-P.; Kong, C.-Y.; Wu, H.-M.; Xu, S.-C.; Ma, Z.-G.; Tang, Q.-Z. Meteorin-like protein attenuates doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1 pathway. Redox Biol. 2020, 37, 101747. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity Through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, e108–e125. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2019, 29, 101394. [Google Scholar] [CrossRef]
- Tan, T.C.; Neilan, T.G.; Francis, S.; Plana, J.C.; Scherrer-Crosbie, M. Anthracycline-Induced Cardiomyopathy in Adults. Compr. Physiol. 2015, 5, 1517–1540. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Raj, S.; Franco, V.I.; Lipshultz, S.E. Anthracycline-Induced Cardiotoxicity: A Review of Pathophysiology, Diagnosis, and Treatment. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 315. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, C.-L.; Quiñones-Lombraña, A.; Singh, P.; Landier, W.; Hageman, L.; Mather, M.; Rotter, J.I.; Taylor, K.D.; Chen, Y.-D.I.; et al. CELF4 Variant and Anthracycline-Related Cardiomyopathy: A Children’s Oncology Group Genome-Wide Association Study. J. Clin. Oncol. 2016, 34, 863–870. [Google Scholar] [CrossRef]
- Krajinovic, M.; Elbared, J.; Drouin, S.; Bertout, L.; Rezgui, A.; Ansari, M.; Raboisson, M.-J.; Lipshultz, S.E.; Silverman, L.B.; Sallan, S.E.; et al. Polymorphisms of ABCC5 and NOS3 genes influence doxorubicin cardiotoxicity in survivors of childhood acute lymphoblastic leukemia. Pharm. J. 2015, 16, 530–535. [Google Scholar] [CrossRef]
- Wojnowski, L.; Kulle, B.; Schirmer, M.; Schlüter, G.; Schmidt, A.; Rosenberger, A.; Vonhof, S.; Bickeböller, H.; Toliat, M.R.; Suk, E.-K.; et al. NAD(P)H Oxidase and Multidrug Resistance Protein Genetic Polymorphisms Are Associated With Doxorubicin-Induced Cardiotoxicity. Circulation 2005, 112, 3754–3762. [Google Scholar] [CrossRef]
- Armenian, S.; Bhatia, S. Predicting and Preventing Anthracycline-Related Cardiotoxicity. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.K.; Karthikeyan, B.; Quiñones-Lombraña, A.; Blair, R.H.; Early, A.P.; Levine, E.G.; Sharma, U.C.; Blanco, J.G.; O’Connor, T. CBR3 V244M is associated with LVEF reduction in breast cancer patients treated with doxorubicin. Cardio-Oncology 2021, 7, 17. [Google Scholar] [CrossRef]
- Reichwagen, A.; Ziepert, M.; Kreuz, M.; Gödtel-Armbrust, U.; Rixecker, T.; Poeschel, V.; Toliat, M.R.; Nürnberg, P.; Tzvetkov, M.; Deng, S.; et al. Association of NADPH oxidase polymorphisms with anthracycline-induced cardiotoxicity in the RICOVER-60 trial of patients with aggressive CD20+ B-cell lymphoma. Pharmacogenomics 2015, 16, 361–372. [Google Scholar] [CrossRef]
- Oliveira, A.; Rodrigues, F.; Santos, R.; Aoki, T.; Rocha, M.; Longui, C.; Melo, M. GSTT1, GSTM1, and GSTP1 polymorphisms and chemotherapy response in locally advanced breast cancer. Genet. Mol. Res. 2010, 9, 1045–1053. [Google Scholar] [CrossRef]
- Visscher, H.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; Carleton, B.C.; et al. Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 2015, 16, 1065–1076. [Google Scholar] [CrossRef]
- Hart, S.N.; Zhong, X.-B. P450 oxidoreductase: Genetic polymorphisms and implications for drug metabolism and toxicity. Expert Opin. Drug Metab. Toxicol. 2008, 4, 439–452. [Google Scholar] [CrossRef]
- Vaitiekus, D.; Muckiene, G.; Vaitiekiene, A.; Sereikaite, L.; Inciuraite, R.; Insodaite, R.; Cepuliene, D.; Kupcinskas, J.; Ugenskiene, R.; Jurkevicius, R.; et al. HFE Gene Variants’ Impact on Anthracycline-Based Chemotherapy-Induced Subclinical Cardiotoxicity. Cardiovasc. Toxicol. 2020, 21, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Magdy, T.; Jiang, Z.; Jouni, M.; Fonoudi, H.; Lyra-Leite, D.; Jung, G.; Romero-Tejeda, M.; Kuo, H.-H.; Fetterman, K.A.; Gharib, M.; et al. RARG variant predictive of doxorubicin-induced cardiotoxicity identifies a cardioprotective therapy. Cell Stem Cell 2021, 28, 2076–2089.e7. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Caram, M.V.; Kidwell, K.M.; Thibert, J.N.; Gersch, C.; Seewald, N.J.; Smerage, J.; Rubenfire, M.; Henry, N.L.; A Cooney, K.; et al. Evidence for association of SNPs in ABCB1 and CBR3, but not RAC2, NCF4, SLC28A3 or TOP2B, with chronic cardiotoxicity in a cohort of breast cancer patients treated with anthracyclines. Pharmacogenomics 2016, 17, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.; Sludden, J.; Griffin, M.J.; Cole, M.; Verrill, M.; Jamieson, D.; Boddy, A.V. Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br. J. Cancer 2010, 102, 1003–1009. [Google Scholar] [CrossRef]
- Yang, X.; Li, G.; Yang, T.; Guan, M.; An, N.; Yang, F.; Dai, Q.; Zhong, C.; Luo, C.; Gao, Y.; et al. Possible Susceptibility Genes for Intervention against Chemotherapy-Induced Cardiotoxicity. Oxidative Med. Cell. Longev. 2020, 2020, 4894625. [Google Scholar] [CrossRef]
- Ikeda, M.; Tsuji, D.; Yamamoto, K.; Kim, Y.-I.; Daimon, T.; Iwabe, Y.; Hatori, M.; Makuta, R.; Hayashi, H.; Inoue, K.; et al. Relationship between ABCB1 gene polymorphisms and severe neutropenia in patients with breast cancer treated with doxorubicin/cyclophosphamide chemotherapy. Drug Metab. Pharmacokinet. 2015, 30, 149–153. [Google Scholar] [CrossRef]
- Syarifah, S.; Hamdi, T.; Widyawati, T.; Sari, M.I.; Anggraini, D.R. Relation of polymorphism C1236T and C3435T in ABCB1 gene with bone marrow suppression in chemotherapy-treated breast cancer patients. IOP Conf. Ser. Earth Environ. Sci. 2018, 125, 012126. [Google Scholar] [CrossRef]
- Yao, S.; Sucheston, L.E.; Zhao, H.; Barlow, W.E.; Zirpoli, G.; Liu, S.; Moore, H.C.F.; Budd, G.T.; Hershman, D.L.; Davis, W.; et al. Germline genetic variants in ABCB1, ABCC1 and ALDH1A1, and risk of hematological and gastrointestinal toxicities in a SWOG Phase III trial S0221 for breast cancer. Pharm. J. 2013, 14, 241–247. [Google Scholar] [CrossRef]
- Chen, S.; Sutiman, N.; Zhang, C.Z.; Yu, Y.; Lam, S.; Khor, C.C.; Chowbay, B. Pharmacogenetics of irinotecan, doxorubicin and docetaxel transporters in Asian and Caucasian cancer patients: A comparative review. Drug Metab. Rev. 2016, 48, 502–540. [Google Scholar] [CrossRef]
- Han, S.-A.; Kim, S.-W. BRCA and Breast Cancer-Related High-Penetrance Genes. Adv. Exp. Med. Biol. 2021, 1187, 473–490. [Google Scholar] [CrossRef]
- Li, D.; Yang, Y.; Wang, S.; He, X.; Liu, M.; Bai, B.; Tian, C.; Sun, R.; Yu, T.; Chu, X. Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol. 2021, 46, 102089. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.M.; Thomas, M.Z.; Magdy, T.; Eisenmann, E.D.; Uddin, M.E.; DiGiacomo, D.F.; Pan, A.; Keiser, M.; Otter, M.; Xia, S.H.; et al. Targeting OCT3 attenuates doxorubicin-induced cardiac injury. Proc. Natl. Acad. Sci. USA 2021, 118, e2020168118. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.V.; Svistunov, A.A.; Chubarev, V.N.; Dostdar, S.A.; Sokolov, A.V.; Brzecka, A.; Sukocheva, O.; Neganova, M.E.; Klochkov, S.G.; Somasundaram, S.G.; et al. Extracellular vesicles in cancer nanomedicine. Semin. Cancer Biol. 2021, 69, 212–225. [Google Scholar] [CrossRef]
- Kumari, H.; Huang, W.-H.; Chan, M. Review on the Role of Epigenetic Modifications in Doxorubicin-Induced Cardiotoxicity. Front. Cardiovasc. Med. 2020, 7, 56. [Google Scholar] [CrossRef]
- Vijayaraghavalu, S.; Labhasetwar, V. Nanogel-mediated delivery of a cocktail of epigenetic drugs plus doxorubicin overcomes drug resistance in breast cancer cells. Drug Deliv. Transl. Res. 2018, 8, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, J.; Skorupa, M.; Tretyn, A. MicroRNA as a Potential Therapeutic Molecule in Cancer. Cells 2022, 11, 1008. [Google Scholar] [CrossRef]
- Ruggeri, C.; Gioffré, S.; Achilli, F.; Colombo, G.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models and cancer patients. Heart Fail. Rev. 2017, 23, 109–122. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Filkowski, J.; Meservy, J.; Ilnytskyy, Y.; Tryndyak, V.P.; Chekhun, V.F.; Pogribny, I.P. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol. Cancer Ther. 2008, 7, 2152–2159. [Google Scholar] [CrossRef]
- Bao, L.; Hazari, S.; Mehra, S.; Kaushal, D.; Moroz, K.; Dash, S. Increased Expression of P-Glycoprotein and Doxorubicin Chemoresistance of Metastatic Breast Cancer Is Regulated by miR-298. Am. J. Pathol. 2012, 180, 2490–2503. [Google Scholar] [CrossRef]
- Chen, F.; Chen, C.; Yang, S.; Gong, W.; Wang, Y.; Cianflone, K.; Tang, J.; Wang, D.W. Let-7b Inhibits Human Cancer Phenotype by Targeting Cytochrome P450 Epoxygenase 2J2. PLoS ONE 2012, 7, e39197. [Google Scholar] [CrossRef]
- Gedda, M.R.; Babele, P.K.; Zahra, K.; Madhukar, P. Epigenetic Aspects of Engineered Nanomaterials: Is the Collateral Damage Inevitable? Front. Bioeng. Biotechnol. 2019, 7, 228. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-Z.; Gao, W.; Yu, A.-M. MicroRNAs Regulate CYP3A4 Expression via Direct and Indirect Targeting. Drug Metab. Dispos. 2009, 37, 2112–2117. [Google Scholar] [CrossRef]
- Sukocheva, O.A.; Lukina, E.; Friedemann, M.; Menschikowski, M.; Hagelgans, A.; Aliev, G. The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: Current findings and future perspectives. Semin. Cancer Biol. 2022, 82, 35–59. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Chen, J.; Wang, S.; Fu, F.; Zhu, X.; Wu, C.; Liu, Z.; Zhong, G.; Lin, J. A Nanodrug Consisting Of Doxorubicin And Exosome Derived From Mesenchymal Stem Cells For Osteosarcoma Treatment In Vitro. Int. J. Nanomed. 2019, 14, 8603–8610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, T.; Miao, Y.; Zhou, L.; Zhang, W. Dual-responsive doxorubicin-loaded nanomicelles for enhanced cancer therapy. J. Nanobiotechnol. 2020, 18, 136. [Google Scholar] [CrossRef]
- Li, L.; Wu, B.; Zhao, Q.; Li, J.; Han, Y.; Fan, X.; Dong, J.; Li, P. Attenuation of doxorubicin-induced cardiotoxicity by cryptotanshinone detected through association analysis of transcriptomic profiling and KEGG pathway. Aging 2020, 12, 9585–9603. [Google Scholar] [CrossRef]
- Hashemitabar, S.; Yazdian-Robati, R.; Hashemi, M.; Ramezani, M.; Abnous, K.; Kalalinia, F. ABCG2 aptamer selectively delivers doxorubicin to drug-resistant breast cancer cells. J. Biosci. 2019, 44, 39. [Google Scholar] [CrossRef]
- Schettini, F.; Giuliano, M.; Lambertini, M.; Bartsch, R.; Pinato, D.J.; Onesti, C.E.; Harbeck, N.; Lüftner, D.; Rottey, S.; van Dam, P.A.; et al. Anthracyclines Strike Back: Rediscovering Non-Pegylated Liposomal Doxorubicin in Current Therapeutic Scenarios of Breast Cancer. Cancers 2021, 13, 4421. [Google Scholar] [CrossRef] [PubMed]
- Borišev, I.; Mrđanovic, J.; Petrovic, D.; Seke, M.; Jović, D.; Srdjenovic, B.; Latinovic, N.; Djordjevic, A. Nanoformulations of doxorubicin: How far have we come and where do we go from here? Nanotechnology 2018, 29, 332002. [Google Scholar] [CrossRef]
- Bruhn, O.; Cascorbi, I. Polymorphisms of the drug transporters ABCB1, ABCG2, ABCC2 and ABCC3 and their impact on drug bioavailability and clinical relevance. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1337–1354. [Google Scholar] [CrossRef]
- Marsh, S.; Liu, G. Pharmacokinetics and pharmacogenomics in breast cancer chemotherapy. Adv. Drug Deliv. Rev. 2009, 61, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Wright, G.; Bryant, H.; Wiggins, L.; Zotto, V.D.; Schuler, M.; Malozzi, C.; Cohen, M.; Gassman, N. Cytoprotective Effect of Vitamin D on Doxorubicin-Induced Cardiac Toxicity in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 7439. [Google Scholar] [CrossRef] [PubMed]
- Gomari, H.; Moghadam, M.F.; Soleimani, M.; Ghavami, M.; Khodashenas, S. Targeted delivery of doxorubicin to HER2 positive tumor models. Int. J. Nanomed. 2019, 14, 5679–5690. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, J.; Beeraka, N.M.; Tang, C.; Babayeva, Y.V.; Sinelnikov, M.Y.; Zhang, X.; Zhang, J.; Liu, J.; Reshetov, I.V.; et al. Advances in the Prevention and Treatment of Obesity-Driven Effects in Breast Cancers. Front. Oncol. 2022, 12, 820968. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020, 83, 556–569. [Google Scholar] [CrossRef]
- Grant, M.K.; Abdelgawad, I.Y.; Lewis, C.A.; Zordoky, B.N. Sexual Dimorphism in Doxorubicin-induced Systemic Inflammation: Implications for Hepatic Cytochrome P450 Regulation. Int. J. Mol. Sci. 2020, 21, 1279. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| No-Function Alleles | Decreased-Function Alleles | Normal-Function Alleles | Increased-Function Alleles |
|---|---|---|---|
| *3, *4, *4xN, *5, *6, *7, *8, *11, *12, *36, *40, *42, and *56 | *9, *10, *17, *29, *41, *44, and *49 | *2, *35, *43, and *45 | *1xN, *2xN |
| Dox-Related Effects | Gene/Polymorphism | Reference |
|---|---|---|
| Drug clearance | ABCC5g + 7161G4A (rs1533682) expression resulted in faster Dox clearance; homozygote allele in ABCC5 g.-1679T4A locus was linked to higher Dox concentration in blood. | Lal et al. (2017) [40] |
| PXR*1B haplotype was linked to lower Dox clearance. | Sandanaraj et al. (2008) [34] | |
| C3435TT genotype was associated with higher AUC. | Kim et al. (2015) [36] | |
| Drug resistance | GSTM1*0 и GSTT1*0 presence was associated with lower risk of disease relapse/death. | Stearns et al. (2004) [44] |
| Drug resistance was detected in carriers of M89T, L662R, R669C, and S1141T polymorphisms of ABCB1 gene; lower level of drug resistance was shown in carriers of ABCB1/W1108R variant. | Jeong et al. (2007) [48] | |
| MDR1 TT genotype was associated with a worse tumor response to chemotherapy. | Tulsyan et al. (2016) [49] | |
| No association was found between ABCB1/MDR1 polymorphisms and response to chemotherapy (meta-analysis study). | Madrid-Paredes et al. (2017) [50] | |
| Lower Dox efficacy was associated with expression of ABCC2/rs717620 variant. | Ruiz-Pinto et al. (2018) [51] | |
| Cardiotoxicity | Increased risk of toxicity was associated with A1629T in ABCC5 and G894T in NOS3 genes. | Krajinovic et al. (2016) [74] |
| Expression levels of Gly671Val/MRP1 and Val1188Glu-Cys1515Tyr (rs8187694-rs8187710)/MRP2 variants were linked to the increased risk of acute cardiotoxicity. | Wojnowski et al. (2005) [75] | |
| RARG variants rs2229774, SLC28A3 rs7853758, and UGT1A6 rs17863783 correlated with the increased toxicity. | Aminkeng et al. (2016) [57] | |
| ABCC5 (A-1629T, rs7627754) and ABCB4 (rs4148808) correlated with the decreased left ventricular ejection fraction. | Armenian et al. (2018) [76] | |
| Expression of rs1786814/CELF4 gene was associated with the decreased myocardial contractility. | Wang et al. (2016) [73] | |
| SLC22A16 variants A146G, T312C, and T755C correlated with the lower toxicity, while SLC22A16 variants 1226C, CYP2B6*2, and CYP2B6*5 were linked to the higher toxicity. | Bray et al. (2010) [85] | |
| Hematotoxicity | Expression of ABCB1 variant 2677G>T/A was linked to the higher risk of neutropenia. | Ikeda et al. (2015) [87] |
| No significant associations were found between ABCB1 (C1236T and C3435T) polymorphisms and myelosuppression. | Syarifah et al. (2018) [88] | |
| ABCC1 variants 809 + 54C>A (rs903880), 677 + 1391T>C (rs16967126), and 1988 + 219G>T (rs4148350) correlated with the higher toxicity. | Yao et al. (2014) [89] | |
| Allele p.Asn118 = (rs11615) in gene ERCC1, homozygote GG polymorphism p.Val417Ile (rs2273697) of ABCC2 gene, and heterozygote allele CA of variant p.Gln141Lys (rs2231142) correlated with the higher risk of anemia. G allele of p.Pro329Ala in gene ALDH3A1 (rs2228100) and homozygote CC allele of CYP2C19 c.-806C>A (rs12248560) were linked to the higher risk of leucopenia. TT allele of ABCC2 gene was associated with the higher risk of severe neutropenia. | Tecza et al. (2018) [23] | |
| GI- and nephrotoxicity | Homozygote allele CC of gene polymorphism CYP1B1, A allele of gene ATM p.Asp1853Asn (rs1801516), and A allele of gene GSTP1 p.Ile105Val were linked to the higher risk of toxicity. | Tecza et al. (2018) [23] |
| C allele of gene ERCC1 variant p.Asn118 = (rs11615) and expression of GSTT1 and GSTM1 genes was associated with the higher risk of toxicity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagdasaryan, A.A.; Chubarev, V.N.; Smolyarchuk, E.A.; Drozdov, V.N.; Krasnyuk, I.I.; Liu, J.; Fan, R.; Tse, E.; Shikh, E.V.; Sukocheva, O.A. Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy. Cancers 2022, 14, 5436. https://doi.org/10.3390/cancers14215436
Bagdasaryan AA, Chubarev VN, Smolyarchuk EA, Drozdov VN, Krasnyuk II, Liu J, Fan R, Tse E, Shikh EV, Sukocheva OA. Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy. Cancers. 2022; 14(21):5436. https://doi.org/10.3390/cancers14215436
Chicago/Turabian StyleBagdasaryan, Alina A., Vladimir N. Chubarev, Elena A. Smolyarchuk, Vladimir N. Drozdov, Ivan I. Krasnyuk, Junqi Liu, Ruitai Fan, Edmund Tse, Evgenia V. Shikh, and Olga A. Sukocheva. 2022. "Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy" Cancers 14, no. 21: 5436. https://doi.org/10.3390/cancers14215436
APA StyleBagdasaryan, A. A., Chubarev, V. N., Smolyarchuk, E. A., Drozdov, V. N., Krasnyuk, I. I., Liu, J., Fan, R., Tse, E., Shikh, E. V., & Sukocheva, O. A. (2022). Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy. Cancers, 14(21), 5436. https://doi.org/10.3390/cancers14215436