

The Role of Fusobacterium nucleatum in Colorectal Cancer Cell Proliferation and Migration


Abstract
Simple Summary
Abstract
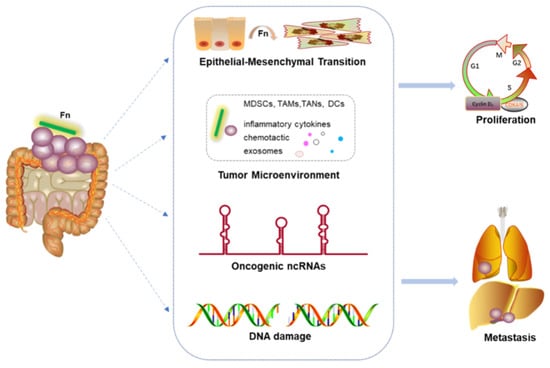
1. Introduction
2. Potential Mechanisms by Which Fn May Promote CRC Proliferation and Migration
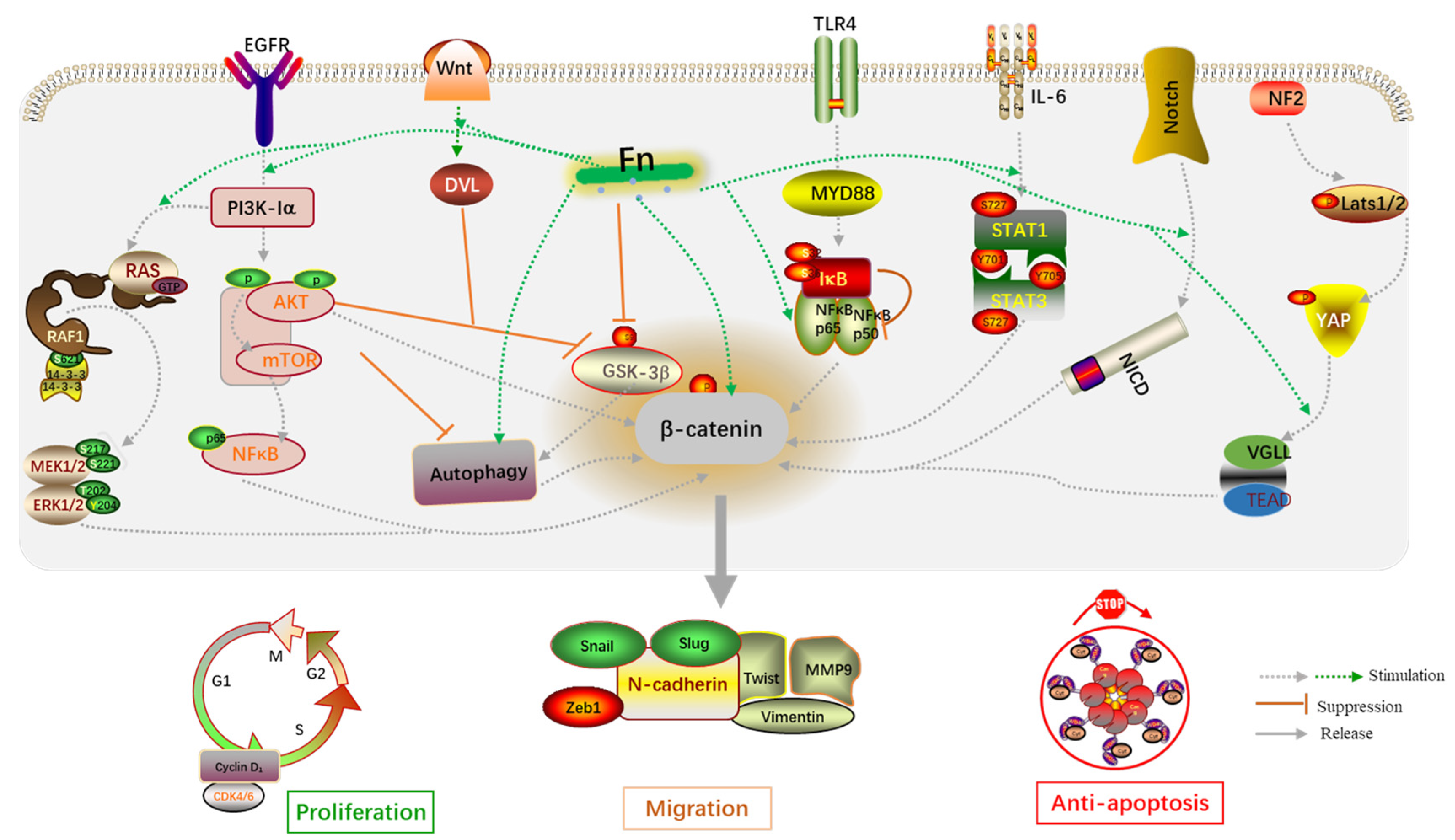
2.1. Proliferative Effects on Primary Tumor
2.2. Induction of Epithelial–Mesenchymal Transition
2.2.1. E-Cadherin/β-Catenin Complex
2.2.2. Cancer Stem-Like Cells
2.2.3. Autophagy Signaling
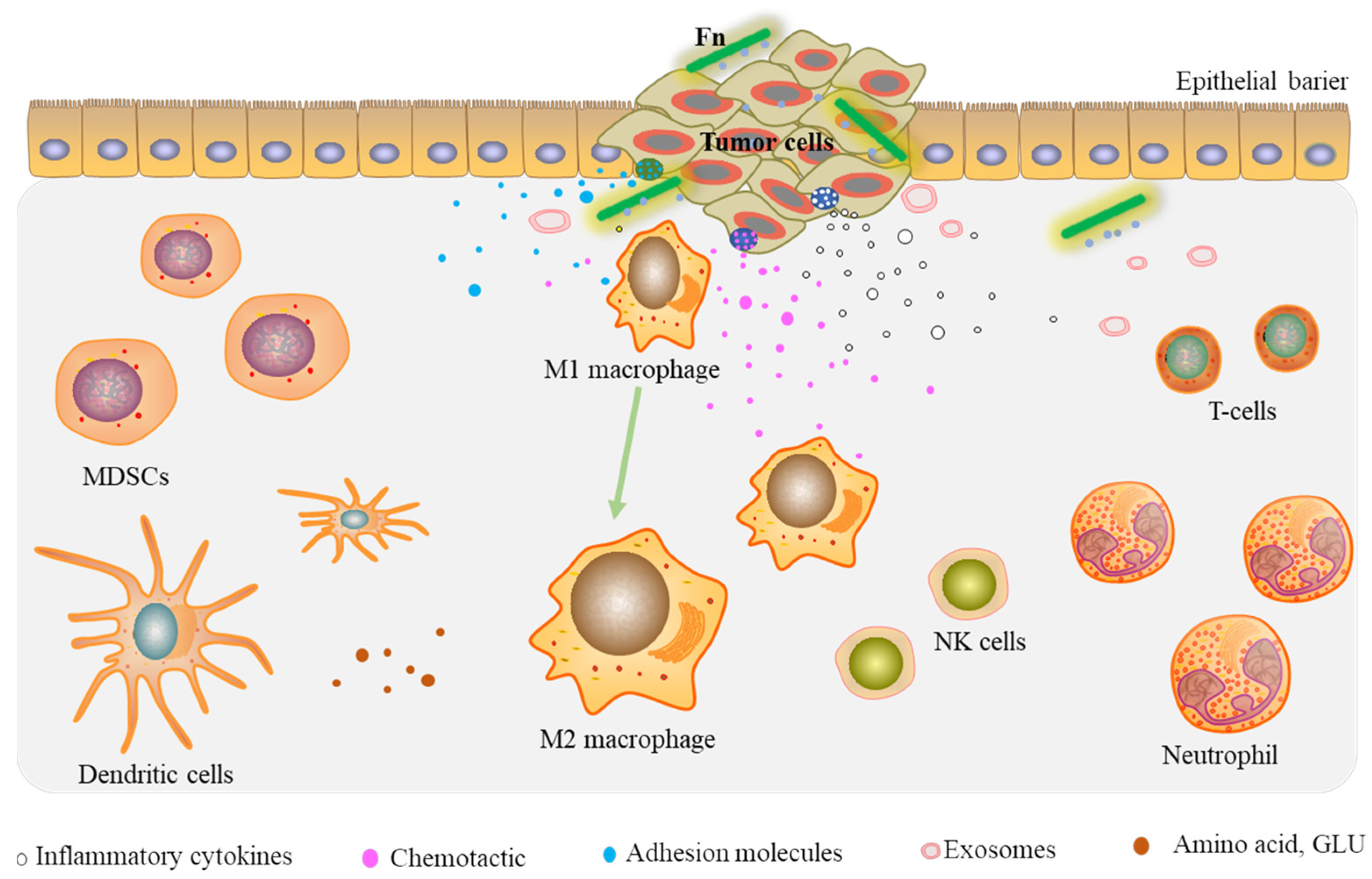
2.3. Reshaping the Tumor Microenvironment
2.3.1. Tumor-Infiltrating Immune Cells
2.3.2. Prometastatic Cytokines
2.3.3. Tumor Metabolism
2.3.4. Tumor-Associated Microbiota
2.4. Oncogenic ncRNAs
2.4.1. LncRNAs
2.4.2. miRNAs
2.5. DNA Damage
3. Discussion
3.1. Current Status
3.2. Future Directions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thanikachalam, K.; Khan, G. Colorectal Cancer and Nutrition. Nutrients 2019, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Wu, N.; Feng, Y.Q.; Lyu, N.; Wang, D.; Yu, W.D.; Hu, Y.F. Fusobacterium nucleatum promotes colon cancer progression by changing the mucosal microbiota and colon transcriptome in a mouse model. World J. Gastroenterol. 2022, 28, 1981–1995. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Y.; Li, J.; Zhao, L.; Yan, W.; Lin, B.; Guo, X.; Wei, Y. Fusobacterium nucleatum Acts as a Pro-carcinogenic Bacterium in Colorectal Cancer: From Association to Causality. Front. Cell. Dev. Biol. 2021, 9, 710165. [Google Scholar] [CrossRef]
- Zhang, N.; Ng, A.S.; Cai, S.; Li, Q.; Yang, L.; Kerr, D. Novel therapeutic strategies: Targeting epithelial–mesenchymal transition in colorectal cancer. Lancet Oncol. 2021, 22, e358–e368. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—Symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.H.; Morell-Perez, C.; Wyche, T.P.; Sana, T.R.; Lieberman, L.A.; Hett, E.C. Colorectal cancer-associated anaerobic bacteria proliferate in tumor spheroids and alter the microenvironment. Sci. Rep. 2020, 10, 5321. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, D.L.; Marchetti, S.; Pignatelli, P.; Piattelli, A.; Curia, M.C. The Oncobiome in Gastroenteric and Genitourinary Cancers. Int. J. Mol. Sci. 2022, 23, 9664. [Google Scholar] [CrossRef]
- Pignatelli, P.; Iezzi, L.; Pennese, M.; Raimondi, P.; Cichella, A.; Bondi, D.; Grande, R.; Cotellese, R.; Di Bartolomeo, N.; Innocenti, P.; et al. The Potential of Colonic Tumor Tissue Fusobacterium nucleatum to Predict Staging and Its Interplay with Oral Abundance in Colon Cancer Patients. Cancers 2021, 13, 1032. [Google Scholar] [CrossRef]
- Henstra, C.; van Praagh, J.; Olinga, P.; Nagelkerke, A. The gastrointestinal microbiota in colorectal cancer cell migration and invasion. Clin. Exp. Metastasis 2021, 38, 495–510. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/beta-catenin modulator Annexin A1. EMBO Rep. 2019, 20, e47638. [Google Scholar] [CrossRef]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-kappaB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866 e24. [Google Scholar] [CrossRef]
- Dadashi, M.; Hajikhani, B.; Faghihloo, E.; Owlia, P.; Yaslianifard, S.; Goudarzi, M.; Nasiri, M.J.; Fallah, F. Proliferative Effect of FadA Recombinant Protein from Fusobacterium nucleatum on SW480 Colorectal Cancer Cell Line. Infect. Disord. Drug Targets 2021, 21, 623–628. [Google Scholar] [CrossRef]
- Xu, Q.; Lu, X.; Li, J.; Feng, Y.; Tang, J.; Zhang, T.; Mao, Y.; Lan, Y.; Luo, H.; Zeng, L.; et al. Fusobacterium nucleatum induces excess methyltransferase-like 3-mediated microRNA-4717-3p maturation to promote colorectal cancer cell proliferation. Cancer Sci. 2022. [Google Scholar] [CrossRef]
- Feng, Y.Y.; Zeng, D.Z.; Tong, Y.N.; Lu, X.X.; Dun, G.D.; Tang, B.; Zhang, Z.J.; Ye, X.L.; Li, Q.; Xie, J.P.; et al. Alteration of microRNA-4474/4717 expression and CREB-binding protein in human colorectal cancer tissues infected with Fusobacterium nucleatum. PLoS ONE 2019, 14, e0215088. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, L.; Li, M.; Zhang, Y.; Sun, M.; Wang, L.; Lin, J.; Cui, Y.; Chen, Q.; Jin, C.; et al. Fusobacterium nucleatum reduces METTL3-mediated m(6)A modification and contributes to colorectal cancer metastasis. Nat. Commun. 2022, 13, 1248. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Yoo, S.Y.; Oh, H.J.; Jeong, S.; Cho, N.Y.; Kang, G.H.; Kim, J.H. Differential immune microenvironmental features of microsatellite-unstable colorectal cancers according to Fusobacterium nucleatum status. Cancer Immunol. Immunother. 2021, 70, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, Y.; Yu, J.; Chen, T.; Wu, Y.; Shi, L.; Li, Q.; Wu, J.; Fu, X. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 2017, 8, 31802–31814. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, J.; Yu, T.; Fang, X.; Lou, L.; Xin, S.; Ji, L.; Jiang, F.; Lou, Y. Fusobacterium nucleatum Promotes the Progression of Colorectal Cancer Through Cdk5-Activated Wnt/beta-Catenin Signaling. Front. Microbiol. 2020, 11, 545251. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Tian, Z.; Kong, X.; Yang, L.; Shan, X.; Dong, B.; Ding, X.; Jing, X.; Jiang, C.; Jiang, N.; et al. FadA promotes DNA damage and progression of Fusobacterium nucleatum-induced colorectal cancer through up-regulation of chk2. J. Exp. Clin. Cancer Res. 2020, 39, 202. [Google Scholar] [CrossRef]
- Ma, C.T.; Luo, H.S.; Gao, F.; Tang, Q.C.; Chen, W. Fusobacterium nucleatum promotes the progression of colorectal cancer by interacting with E-cadherin. Oncol. Lett. 2018, 16, 2606–2612. [Google Scholar] [CrossRef]
- Morrissey, S.M.; Zhang, F.; Ding, C.; Montoya-Durango, D.E.; Hu, X.; Yang, C.; Wang, Z.; Yuan, F.; Fox, M.; Zhang, H.G.; et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021, 33, 2040–2058.e10. [Google Scholar] [CrossRef]
- Guo, S.; Chen, J.; Chen, F.; Zeng, Q.; Liu, W.L.; Zhang, G. Exosomes derived from Fusobacterium nucleatum-infected colorectal cancer cells facilitate tumour metastasis by selectively carrying miR-1246/92b-3p/27a-3p and CXCL16. Gut 2020, 70, 1507–1519. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Ternes, D.; Tsenkova, M.; Pozdeev, V.I.; Meyers, M.; Koncina, E.; Atatri, S.; Schmitz, M.; Karta, J.; Schmoetten, M.; Heinken, A.; et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat. Metab. 2022, 4, 458–475. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xu, Q.; Tong, Y.; Zhang, Z.; Dun, G.; Feng, Y.; Tang, J.; Han, D.; Mao, Y.; Deng, L.; et al. Long non-coding RNA EVADR induced by Fusobacterium nucleatum infection promotes colorectal cancer metastasis. Cell Rep. 2022, 40, 111127. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Babaei, G.; Aziz, S.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharm. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Yan, X.; Liu, L.; Li, H.; Qin, H.; Sun, Z. Clinical significance of Fusobacterium nucleatum, epithelial-mesenchymal transition, and cancer stem cell markers in stage III/IV colorectal cancer patients. Onco. Targets Ther. 2017, 10, 5031–5046. [Google Scholar] [CrossRef]
- Yu, M.R.; Kim, H.J.; Park, H.R. Fusobacterium nucleatum Accelerates the Progression of Colitis-Associated Colorectal Cancer by Promoting EMT. Cancers 2020, 12, 2728. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Du, J.; Chao, S.; Li, S.; Cai, H.; Zhang, H.; Chen, G.; Liu, P.; Bu, P. Fusobacterium nucleatum Promotes Colorectal Cancer Cell to Acquire Stem Cell-Like Features by Manipulating Lipid Droplet-Mediated Numb Degradation. Adv. Sci. (Weinh.) 2022, 9, e2105222. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Zhang, J.; Cao, P.; Su, W.; Deng, Y.; Zhan, N.; Fu, X.; Huang, Y.; Dong, W. Fusobacterium nucleatum Promotes Metastasis in Colorectal Cancer by Activating Autophagy Signaling via the Upregulation of CARD3 Expression. Theranostics 2020, 10, 323–339. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563 e16. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Mima, K.; Ishimoto, T.; Ogata, Y.; Imai, K.; Miyamoto, Y.; Akiyama, T.; Daitoku, N.; Hiyoshi, Y.; Iwatsuki, M.; et al. Relationship between Fusobacterium nucleatum and antitumor immunity in colorectal cancer liver metastasis. Cancer Sci. 2021, 112, 4470–4477. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, Q.; Wu, J.; Wu, Y.; Peng, W.; Li, H.; Wang, J.; Tang, X.; Peng, Y.; Fu, X. Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours via a TLR4-dependent mechanism. Cancer Immunol. Immunother. 2018, 67, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J. Hematol. Oncol. 2019, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Xu, C.; Fan, L.; Lin, Y.; Shen, W.; Qi, Y.; Zhang, Y.; Chen, Z.; Wang, L.; Long, Y.; Hou, T.; et al. Fusobacterium nucleatum promotes colorectal cancer metastasis through miR-1322/CCL20 axis and M2 polarization. Gut Microbes 2021, 13, 1980347. [Google Scholar] [CrossRef]
- Hu, L.; Liu, Y.; Kong, X.; Wu, R.; Peng, Q.; Zhang, Y.; Zhou, L.; Duan, L. Fusobacterium nucleatum Facilitates M2 Macrophage Polarization and Colorectal Carcinoma Progression by Activating TLR4/NF-kappaB/S100A9 Cascade. Front. Immunol. 2021, 12, 658681. [Google Scholar] [CrossRef]
- Lamprinaki, D.; Garcia-Vello, P.; Marchetti, R.; Hellmich, C.; McCord, K.A.; Bowles, K.M.; Macauley, M.S.; Silipo, A.; De Castro, C.; Crocker, P.R.; et al. Siglec-7 Mediates Immunomodulation by Colorectal Cancer-Associated Fusobacterium nucleatum ssp. animalis. Front. Immunol. 2021, 12, 744184. [Google Scholar] [CrossRef]
- Casasanta, M.A.; Yoo, C.C.; Udayasuryan, B.; Sanders, B.E.; Umana, A.; Zhang, Y.; Peng, H.; Duncan, A.J.; Wang, Y.; Li, L.; et al. Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci. Signal. 2020, 13, eaba9157. [Google Scholar] [CrossRef]
- Yin, H.; Miao, Z.; Wang, L.; Su, B.; Liu, C.; Jin, Y.; Wu, B.; Han, H.; Yuan, X. Fusobacterium nucleatum promotes liver metastasis in colorectal cancer by regulating the hepatic immune niche and altering gut microbiota. Aging (Albany NY) 2022, 14, 1941–1958. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Zheng, S.; Li, M.; Xu, C.; Jia, D.; Qi, Y.; Hou, T.; Wang, L.; Wang, B.; et al. Fusobacterium nucleatum promotes colorectal cancer cells adhesion to endothelial cells and facilitates extravasation and metastasis by inducing ALPK1/NF-kappaB/ICAM1 axis. Gut Microbes 2022, 14, 2038852. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Ji, L.; Lou, L.; Ye, S.; Fang, X.; Li, C.; Jiang, F.; Gao, H.; Lou, Y.; Li, X. Fusobacterium nucleatum Affects Cell Apoptosis by Regulating Intestinal Flora and Metabolites to Promote the Development of Colorectal Cancer. Front. Microbiol. 2022, 13, 841157. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Liu, R.; Zhou, C.; Yu, H.; Luo, W.; Zhu, J.; Liu, J.; Zhang, Z.; Xie, N.; Peng, X.; et al. ANGPTL4-Mediated Promotion of Glycolysis Facilitates the Colonization of Fusobacterium nucleatum in Colorectal Cancer. Cancer Res. 2021, 81, 6157–6170. [Google Scholar] [CrossRef]
- Hong, J.; Guo, F.; Lu, S.Y.; Shen, C.; Ma, D.; Zhang, X.; Xie, Y.; Yan, T.; Yu, T.; Sun, T.; et al. F. nucleatum targets lncRNA ENO1-IT1 to promote glycolysis and oncogenesis in colorectal cancer. Gut 2021, 70, 2123–2137. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Zhuo, W.; Liu, Y.; Li, S.; Guo, D.; Sun, Q.; Jin, J.; Rao, X.; Li, M.; Sun, M.; Jiang, M.; et al. Long Noncoding RNA GMAN, Up-regulated in Gastric Cancer Tissues, Is Associated with Metastasis in Patients and Promotes Translation of Ephrin A1 by Competitively Binding GMAN-AS. Gastroenterology 2019, 156, 676–691 e11. [Google Scholar] [CrossRef]
- Liang, Y.; Song, X.; Li, Y.; Chen, B.; Zhao, W.; Wang, L.; Zhang, H.; Liu, Y.; Han, D.; Zhang, N.; et al. LncRNA BCRT1 promotes breast cancer progression by targeting miR-1303/PTBP3 axis. Mol. Cancer 2020, 19, 85. [Google Scholar] [CrossRef]
- Chen, S.; Su, T.; Zhang, Y.; Lee, A.; He, J.; Ge, Q.; Wang, L.; Si, J.; Zhuo, W.; Wang, L. Fusobacterium nucleatum promotes colorectal cancer metastasis by modulating KRT7-AS/KRT7. Gut Microbes 2020, 11, 511–525. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, Z.; Lin, Q.; Luo, Q.; Cen, Y.; Li, J.; Fang, X.; Gong, C. MiRNAs and Cancer: Key Link in Diagnosis and Therapy. Genes 2021, 12, 1289. [Google Scholar] [CrossRef] [PubMed]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Chakraborty, A.; Abd El-Hafeez, A.A.; Sharma, A.; Sahan, A.Z.; Huang, W.J.M.; Sahoo, D.; Ghosh, P.; Hazra, T.K.; Das, S. The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells. Cells 2020, 9, 1980. [Google Scholar] [CrossRef]
- Parhi, L.; Alon-Maimon, T.; Sol, A.; Nejman, D.; Shhadeh, A.; Fainsod-Levi, T.; Yajuk, O.; Isaacson, B.; Abed, J.; Maalouf, N.; et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat. Commun. 2020, 11, 3259. [Google Scholar] [CrossRef]
- McIlvanna, E.; Linden, G.J.; Craig, S.G.; Lundy, F.T.; James, J.A. Fusobacterium nucleatum and oral cancer: A critical review. BMC Cancer 2021, 21, 1212. [Google Scholar] [CrossRef]
- Gkountela, S.; Aceto, N. Stem-like features of cancer cells on their way to metastasis. Biol. Direct 2016, 11, 33. [Google Scholar] [CrossRef]
- Bao, Y.; Ding, Z.; Zhao, P.; Li, J.; Chen, P.; Zheng, J.; Qian, Z. Autophagy inhibition potentiates the anti-EMT effects of alteronol through TGF-beta/Smad3 signaling in melanoma cells. Cell Death Dis. 2020, 11, 223. [Google Scholar] [CrossRef]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef]
- Saw, P.E.; Xu, X.; Chen, J.; Song, E.W. Non-coding RNAs: The new central dogma of cancer biology. Sci. China Life Sci. 2021, 64, 22–50. [Google Scholar] [CrossRef]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Chen, H.; Song, J.; Chen, X.; Lin, C.; Zhang, X.; Hou, N.; Pan, J.; Zhou, Z.; Wang, L.; et al. MiR-454-3p-Mediated Wnt/beta-catenin Signaling Antagonists Suppression Promotes Breast Cancer Metastasis. Theranostics 2019, 9, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lai, Q.; He, C.; Fang, Y.; Yan, Q.; Zhang, Y.; Wang, X.; Gu, C.; Wang, Y.; Ye, L.; et al. RUNX1 promotes tumour metastasis by activating the Wnt/beta-catenin signalling pathway and EMT in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 334. [Google Scholar] [CrossRef]
- Wang, J.; Cai, H.; Liu, Q.; Xia, Y.; Xing, L.; Zuo, Q.; Zhang, Y.; Chen, C.; Xu, K.; Yin, P.; et al. Cinobufacini Inhibits Colon Cancer Invasion and Metastasis via Suppressing Wnt/beta-Catenin Signaling Pathway and EMT. Am. J. Chin. Med. 2020, 48, 703–718. [Google Scholar] [CrossRef]
- Wang, R.; Ma, Y.; Zhan, S.; Zhang, G.; Cao, L.; Zhang, X.; Shi, T.; Chen, W. B7-H3 promotes colorectal cancer angiogenesis through activating the NF-kappaB pathway to induce VEGFA expression. Cell Death Dis. 2020, 11, 55. [Google Scholar] [CrossRef]
- Gao, Y.; Nan, X.; Shi, X.; Mu, X.; Liu, B.; Zhu, H.; Yao, B.; Liu, X.; Yang, T.; Hu, Y.; et al. SREBP1 promotes the invasion of colorectal cancer accompanied upregulation of MMP7 expression and NF-kappaB pathway activation. BMC Cancer 2019, 19, 685. [Google Scholar] [CrossRef]
- Zhong, B.; Cheng, B.; Huang, X.; Xiao, Q.; Niu, Z.; Chen, Y.F.; Yu, Q.; Wang, W.; Wu, X.J. Colorectal cancer-associated fibroblasts promote metastasis by up-regulating LRG1 through stromal IL-6/STAT3 signaling. Cell Death Dis. 2021, 13, 16. [Google Scholar] [CrossRef]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. Onco. Targets Ther. 2020, 13, 13023–13032. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef]
- Garreta, E.; Kamm, R.D.; Chuva de Sousa Lopes, S.M.; Lancaster, M.A.; Weiss, R.; Trepat, X.; Hyun, I.; Montserrat, N. Rethinking organoid technology through bioengineering. Nat. Mater. 2021, 20, 145–155. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Tammela, T.; Cetinbas, N.M.; Akkad, A.; Roghanian, A.; Rickelt, S.; Almeqdadi, M.; Wu, K.; Oberli, M.A.; Sanchez-Rivera, F.J.; et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 2017, 35, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Fong, E.L.S.; Zhu, C.; Lin, Q.X.X.; Xiong, M.; Li, A.; Li, T.; Benoukraf, T.; Yu, H.; Liu, S. Hydrogel-based colorectal cancer organoid co-culture models. Acta Biomater. 2021, 132, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Mao, Y.; Wang, W.; Zhou, X.; Wang, W.; Gao, S.; Li, J.; Wen, L.; Fu, W.; Tang, F. Systematic evaluation of colorectal cancer organoid system by single-cell RNA-Seq analysis. Genome Biol. 2022, 23, 106. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Gui, X.; Zhang, Y.; Zhang, Z.; Chen, W.; Zhang, X.; Wang, Y.; Zhang, M.; Shang, Z.; et al. Salivary Fusobacterium nucleatum serves as a potential biomarker for colorectal cancer. iScience 2022, 25, 104203. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviations | Full Name | Abbreviations | Full Name |
|---|---|---|---|
| APC | adenomatous polyposis coli | lncRNAs | long noncoding RNAs |
| ATCC | American Type Culture Collection | MDSCs | myeloid-derived suppressor cells |
| CCSCs | colorectal cancer stem-like cells | miRNAs | microRNAs (small ncRNAs) |
| CDX | cell-derived xenograft | ncRNAs | noncoding RNAs |
| CSCs | cancer stem-like cells | PDX | patient-derived xenograft |
| ECM | extracellular matrix | TAMs | tumor-associated macrophages |
| EMT | epithelial–mesenchymal transition | TANs | tumor-associated neutrophils |
| FadA | Fusobacterium nucleatum adhesin A | TEXs | tumor-derived exosomes |
| Fn | Fusobacterium nucleatum | TME | tumor microenvironment |
| Mechanisms | References | Fn Strain | Models | Findings | Experiment | Metastatic Sites | ||
|---|---|---|---|---|---|---|---|---|
| Signaling | Upregulates | Downregulates | ||||||
| Induced EMT | ||||||||
| E-cadherin/β-catenin | [24] | F01 ATCC 10953 | CRC tissue SW480, Caco-2 | Activates β-catenin signaling via a TLR4/p-PAK1/β-catenin S675 cascade | β-catenin TLR4 PAK1 c-Myc cyclin D1 | In vitro | ||
| [25] | ATCC 25586 | CRC tissue DLD-1, SW480 | Activates Wnt/β-catenin and IL-6/STAT3 signaling via upregulation of Cdk5 | CDK5 β-catenin c-Myc cyclin D1 p-STAT3 IL-6,8 COX-2 TNF-β | In vitro | |||
| [3] | ATCC 25586 | AOM/DSS mice | Impairs the function of the intestinal barrier and the aberrant activation of EMT | Ki-67 NETO2 ANGPTL4 PCK1 | E-cadherin | In vivo | ||
| [26] | ATCC 25586 (FadA) | CRC tissue HCT29, HT116 CDX mice, APCMin/+ mice | Activates the E-cadherin/β-catenin pathway, leading to the upregulation of chk2 | chk2 β-catenin Ki-67 PCNA | E-cadherin | In vitro In vivo | ||
| [27] | ATCC 25586 | NCM460 | Interacts with E-cadherin and enhances the malignant phenotype of CRC cells | NF-κB p65 IL-6 IL-1β MMP-13 TNF-α | In vitro | |||
| [29] | Fn-Ex (TEXs) | HCT116, SW480 CDX mice | Activates the Wnt/β-catenin pathway | β-catenin vimentin c-Myc cyclin D1 | E-cadherin | In vitro In vivo | Liver lung | |
| [30] | ATCC 12230 US1U SF81 (FadA) | CRC tissue HCT116, DLD1, HT29, SW480, RKO CDX mice | Regulates the inflammatory and oncogenic responses via E-cadherin/β-catenin | β-catenin NF-κB IL-6,8,18 LEF/TCF c-Myc cyclin D1 | E-cadherin | In vitro In vivo | ||
| [17] | WAL12230 ATCC 25586 (FadA) | CRC tissue 10C, HCT116, DLD1, RKO, SW480, HT29 CDX mice, APCMin/+ mice | Induces the Wnt/β-catenin modulator Annexin A1 | Annexin A1 β-catenin cyclin D1 | E-cadherin | In vitro In vivo | ||
| [31] | ATCC 23726 ATCC 25586 (formate) | CRC tissue HCT116, Caco-2 CDX mice | Induces cancer stemness and thereby metastatic dissemination by activation of the AhR-Wnt/β-catenin pathway | AhR β-catenin IL8 Th17 MAPK ERK MEK-p38 NF-κB p65 | In vitro In vivo | lung | ||
| [32] | CRC tissue HCT116, LoVo, SW480, SW620, HT29 CDX mice | Enhances the translation of EMT-related factors by the lncRNA EVADR-YBX1 axis | N-cadherin vimentin Snail Slug Zeb1 | E-cadherin | In vitro In vivo | liver lung | ||
| CSCs | [35] | CRC tissue | Potential involvement of Fn in EMT–CSC crosstalk | N-cadherin Nanog Oct-4 Sox-2 | E-cadherin | In vitro | lung, liver ovary node | |
| [36] | LOVO, HCT8, LS174T AOM/DSS mice | Promotes the EMT through EGFR/AKT/ERK signaling and increases the stemness of CRC cells | Fibronectin N-cadherin Snail Slug CD44+ CD133+ IL-1β, 6 p-EGFR p-AKT p-ERK | E-cadherin ZO-1 | In vitro In vivo | |||
| [37] | ATCC 25586 | CRC tissue CCSCs, HEK293, HT29, HCT116 CDX mice | Promotes Numb degradation and activates Notch signaling in non-CCSCs; enhances CPT1B-mediated FAO via the TLR4/MYD88/NF-κB in CCSCs | ALDH1 CD133+ CD44+ Lgr5 Olfm4 Sox9 Aldh1 FASN NICD MDM2 CPT1B NF-κB | Numb | In vitro In vivo | ||
| Autophagy | [38] | F01 ATCC10953 | CRC tissue SW480, HCT116, CT26 xenograft mice APCMin/+ mice | Activates autophagy signaling by upregulating CARD3 | CARD3 LC3-II beclin1 vimentin ATG5,7 MAPK | E-cadherin p62 | In vitro In vivo | Lung liver node |
| Reprogramed TME | ||||||||
| MDSCs | [40] | EAVG_002 | CRC tissue APCMin/+ mice | Generates a proinflammatory microenvironment through the recruitment of tumor-infiltrating immune cells | MDSCs (CD11b+) TANs, TAMs DCs COX-2 IL1,6,8, TNF MMP3 NF-κB p65 | In vitro In vivo | ||
| [41] | CRC tissue APCMin/+ mice | The presence of Fn is associated with a lower density of CD8+ T cells and a higher density of MDSCs in CRC liver metastases | MDSCs (CD33+) TAMs (CD163+) Ki67 TIL- 6 TNF-α | CD8+ | In vitro In vivo | liver | ||
| M2 polarization | [46] | ATCC 25586 | CRC tissue HCT116, LoVo CDX mice | Promotes M2 polarization through the NF-κB pathway | F4/80+ CCR6+ CD206+ ARG1 MRC1 IL-10 TGF-β | In vitro In vivo | lung | |
| [47] | ATCC 25586 | CRC tissue HCT116, SW480, THP-1 CDX mice | Promotes M2 polarization via the TLR4/NF-κB/S100A9 pathway | CD206+ IL-10 S100A9 TLR4 NF-κB p65 PCNA N-cadherin VEGF TGF-β | CD86+ iNOS TNF-α | In vitro In vivo | ||
| [42] | ATCC10953 F01 | CRC tissue RAW 264.7 APCMin/+ mice | Promotes the M2 polarization of macrophages via the TLR4/IL-6/p-STAT3/c-Myc cascade | CD206+ IL-10 TGF-β TLR4 IL-6 p-STAT3 c-Myc | CD86+ IL-12 TGF-α | In vitro In vivo | ||
| [48] | ATCC 51,191 (LPS/OMVs) | CHO-Siglec-7-Fc, U937, moDCs, moM¢s | Promotes M2 polarization and a proinflammatory environment via the Fn–Siglec-7 interaction | Siglec-7 IL-8 I-10 IL-10 PD-L1 | CD86+ TNF-α | In vitro | ||
| Prometastatic cytokines | [49] | ATCC 23,726 (FadA, Fap2) | HCT116 | Induces the secretion of the proinflammatory and prometastatic cytokines | IL-8 CXCL1 | In vitro | ||
| [29] | Fn-Ex (TEXs) | HCT116, SW480 CDX mice | stimulates the CXCL16/RhoA/IL-8 exosomes | IL-8 CXCL16 RhoA | CXCR6 | In vitro In vivo | Liver lung | |
| [50] | ATCC 25586 | CT26-Luc mice | Affects the secretion of inflammatory cytokines and modulates the hepatic immune response | IL-6,12, 9 IL-17A CXCL1 MCP-1 TNF-α IFN-γ CD11b+ Treg | CD3+ CD4+ CD8+ NK Th17 | In vivo | liver | |
| [51] | ATCC 25586 ATCC 10953 | CRC tissue HCT116, LoVo, HUVECs CDX/PDX mice | Promotes CRC cell adhesion to endothelial cells and metastasis by activating the NF-κB/ICAM1 axis | ICAM1 ALPK1 NF-κB p65 | In vitro In vivo | Liver Lung node | ||
| Tumor metabolism | [53] | ATCC 25586 | CRC tissue SW480 AOM mice | Regulates amino acid metabolism | lactic acid aspartic acid glutamate glutathione Bcl-2 caspase 3 | propionic acid leucine isoleucine Bax | In vitro In vivo | |
| [54] | ATCC 25586 | CRC tissue DLD1, SW480, HT-29, HCT-116 CDX mice | Promotes glycolysis via the induction of ANGPTL4 and facilitates the colonization of Fn | ANGPTL4 H3K27ac ECAR GLUT1 | HDAC | In vitro In vivo | ||
| [55] | CRC tissue HCT116, DLD1 CDX mice | Promotes CRC glycolysis via ENO1 | lactic acid ECAR | In vitro In vivo | ||||
| [3] | ATCC 25586 | AOM/DSS mice | Alters the colon mucosal microbiota | FABP1 VAV2 GalR1 | E-cadherin | In vivo | ||
| Oncogenic ncRNAs | ||||||||
| lncRNAs | [32] | CRC tissue HCT116, LoVo, HT29, SW480, SW620 CDX mice | Regulates the lncRNA EVADR-YBX1 axis | EVADR YBX1 | In vitro In vivo | Liver lung | ||
| [60] | CRC tissue HCT116, LoVo CDX mice | Upregulates KRT7-AS/KRT7 by activating the NF-κB pathway | KRT7-AS KRT7 NF-κB p65 | IκB-α | In vitro In vivo | lung | ||
| [55] | CRC tissue HCT116, DLD1 CDX mice | Targets lncRNA ENO1- IT1 to promote glycolysis and oncogenesis | ENO1-IT1 ENO1 KRT7 | In vitro In vivo | ||||
| miRNAs | [29] | Fn-Ex (TEXs) | HCT116, SW480 CDX mice | Stimulates tumor cells to generate miR-1246/92b- 3p/27a- 3p- rich | miR-1246 miR-92b-3p miR-27a-3p | GSK3β | In vitro In vivo | Liver lung |
| [21] | CRC tissue Caco-2, HEK-293 | Upregulates miR-4474/4717 by post transcriptionally regulating the target gene, CREBBP | miR-4474 miR-4717 STAT1 TP53 EWSR1 | CREBBP JUN PRKACB CAMK2B | In vitro | |||
| [18] | CRC tissue HCT116, HT29, LOVO, SW480 CDX mice, APCMin/+ mice | Regulates miR21 expression through the TLR4/MYD88/NF-κB pathway | miR-21 IL-17,21,22 MIP3a TLR4,2 MYD88 NF-κB p65 MAPK | RASA1 PDCD4 PTEN RECK SPRY1 RHOB IκB-α | In vitro In vivo | |||
| [22] | ATCC 25586 | CRC tissue HCT116, LoVo, RKO, SW620 PDX mice | Reduces m6A modifications through the Hippo-YAP/FOXD3/METTL3/KIF26B axis | KIF26B YAP NF-κB | FOXD3 METTL3 m6A NF2 KIBRA Willin\FRMD6 | In vitro In vivo | Liver bone | |
| [46] | ATCC 25586 | CRC tissue HCT116, LoVo CDX mice | Regulates miR-1322/CCL20 expression through the NF-κB pathway | CCL20 | miR-1322 | In vitro In vivo | lung | |
| DNA damage | ||||||||
| [64] | ATCC 25586 | CRC tissue EDMs APC Min/+ mice | Induces the downregulation of NEIL2 and the consequent accumulation of DNA damage | NTH1 MSH2 MSH6 Ku70 IL-8 pATM p-γH2AX | NEIL2 NEIL1 PMS2 | In vitro In vivo | ||
| [26] | ATCC 25586 (FadA) | CRC tissue HCT29, HT116 CDX mice, APC Min/+ mice | Elevates DNA damage and increases the number of CRC cells in the S phase of the cell cycle | chk2 γH2AX | In vitro In vivo | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Ma, Q.; Guo, Y.; You, F. The Role of Fusobacterium nucleatum in Colorectal Cancer Cell Proliferation and Migration. Cancers 2022, 14, 5350. https://doi.org/10.3390/cancers14215350
Wu Z, Ma Q, Guo Y, You F. The Role of Fusobacterium nucleatum in Colorectal Cancer Cell Proliferation and Migration. Cancers. 2022; 14(21):5350. https://doi.org/10.3390/cancers14215350
Chicago/Turabian StyleWu, Zihong, Qiong Ma, Ying Guo, and Fengming You. 2022. "The Role of Fusobacterium nucleatum in Colorectal Cancer Cell Proliferation and Migration" Cancers 14, no. 21: 5350. https://doi.org/10.3390/cancers14215350
APA StyleWu, Z., Ma, Q., Guo, Y., & You, F. (2022). The Role of Fusobacterium nucleatum in Colorectal Cancer Cell Proliferation and Migration. Cancers, 14(21), 5350. https://doi.org/10.3390/cancers14215350