
The Predictive and Prognostic Role of RAS–RAF–MEK–ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets




Simple Summary
Abstract
1. Introduction
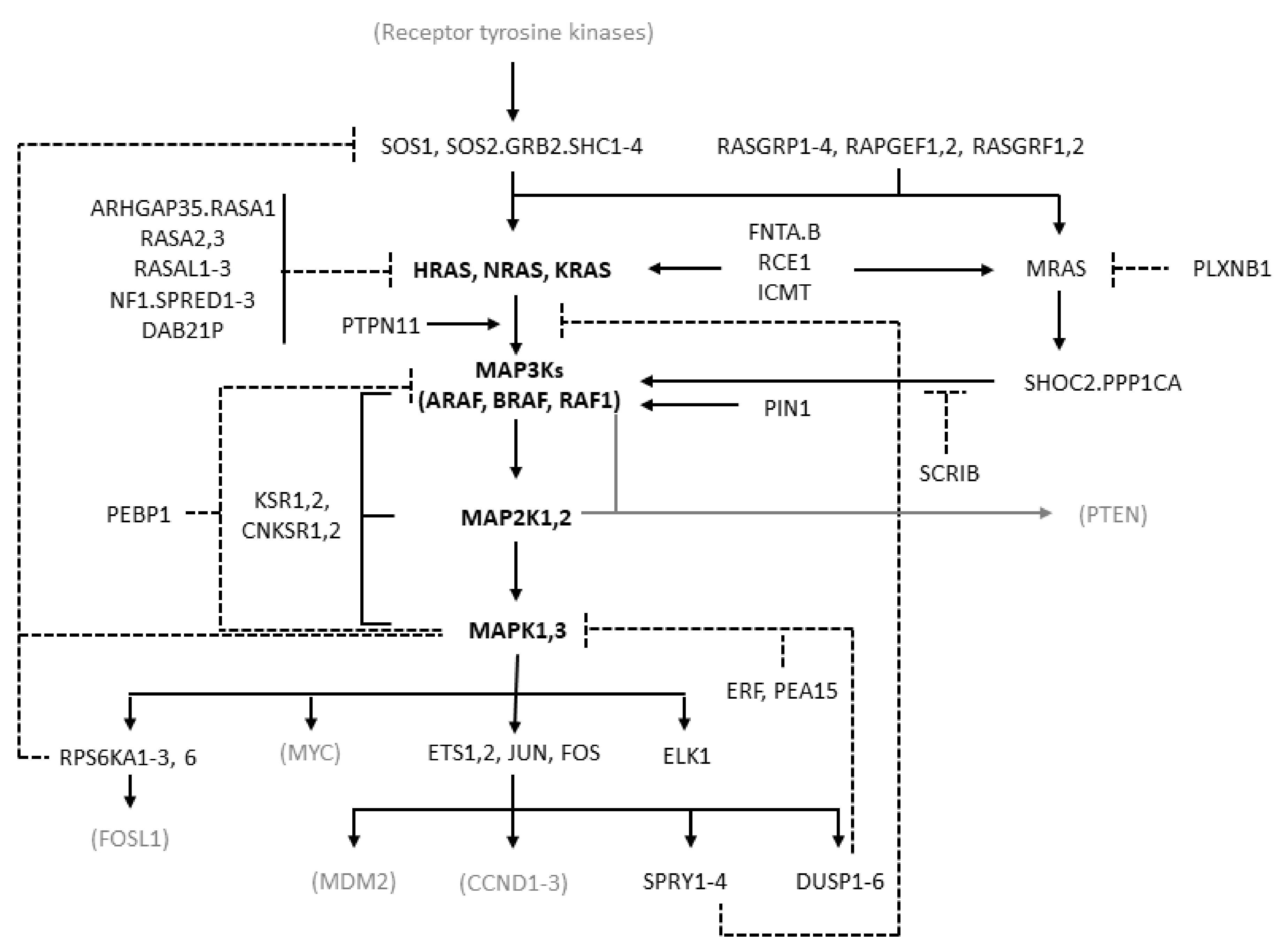
2. Pathway Description
3. The Spectrum of Genomic Alterations of the RAS/RAF/MEK/ERK Pathway in Human Tumors
4. RAS/RAF/MEK/ERK Pathway Alterations in Breast Cancer and Their Prognostic Impact
5. The Spectrum of Somatic Alterations Affecting the ERK Pathway in Breast Cancer: Findings from cBioPortal Datasets
5.1. Methods
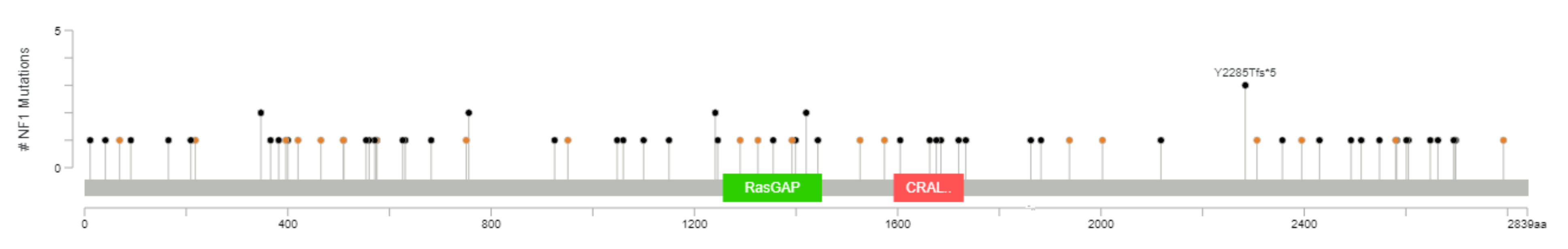
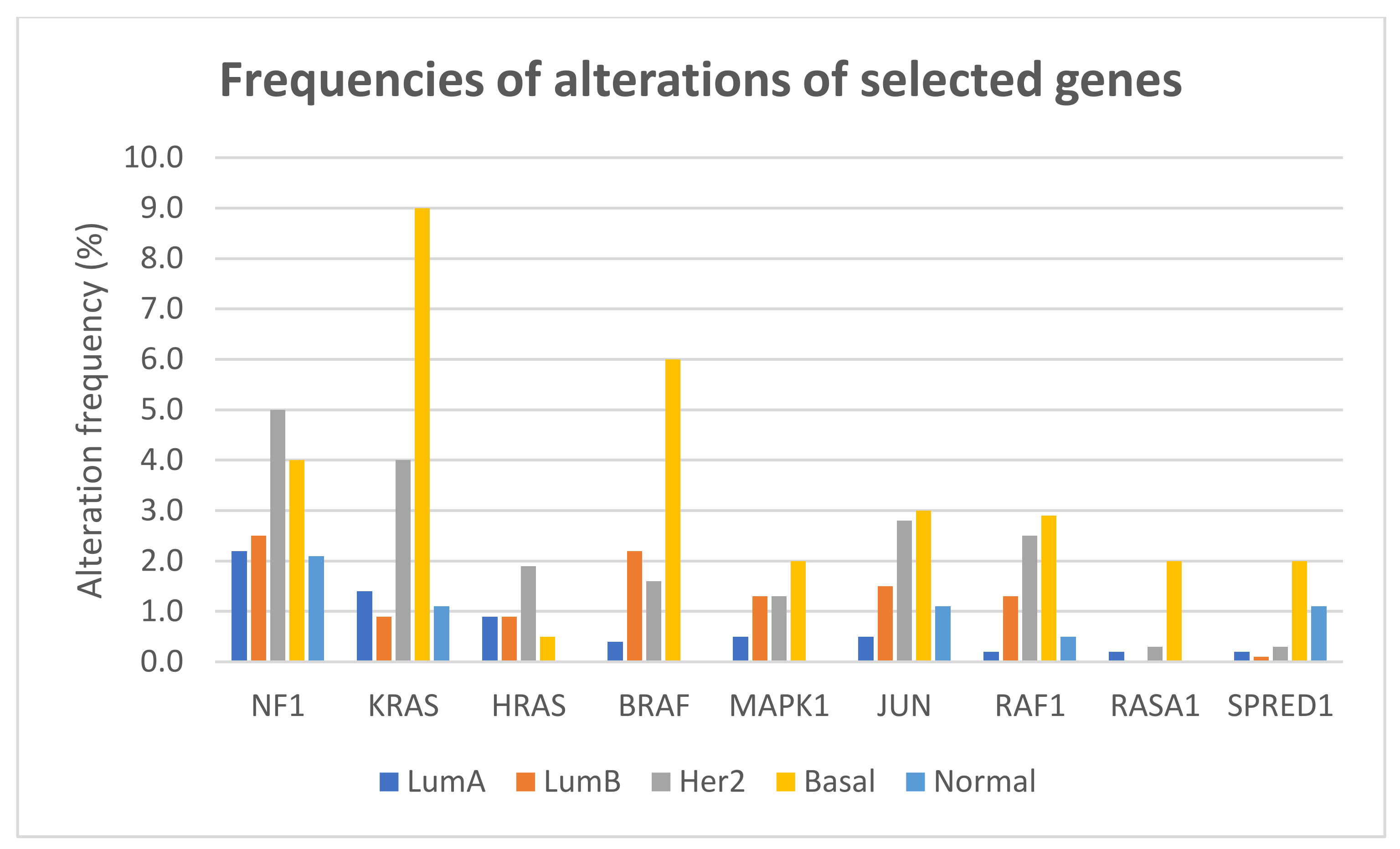
5.2. Results
6. Transcriptomic Predictors of RAS Pathway Activation in Breast Cancer
7. RAS/RAF/MEK/ERK Pathway in Luminal Cancers and Resistance to Endocrine Therapy
8. RAS/RAF/MEK/ERK Pathway in HER2-Positive Breast Cancer
9. RAS/RAF/MEK/ERK Pathway in Triple-Negative Breast Cancer and Resistance to Chemotherapy and Immunotherapy
10. Predicting the Effects of RAS/RAF/MEK/ERK Inhibitors in Breast Cancer
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Glossary
| Dabrafenib | Small-molecule, ATP-competitive inhibitor of the rapidly accelerated fibrosarcoma (RAF) kinases, especially mutant BRAF. |
| Dacomitinib | Small-molecule, irreversible inhibitor of the pan-human epidermal growth factor receptor (HER) family. |
| Doxorubicin and epirubicin | Chemotherapeutic agents belonging to the class of anthracyclines, acting by multiple mechanisms including (1) intercalating between base pairs in the DNA helix, (2) inhibiting topoisomerase 2, and (3) forming oxygen free radicals. |
| Everolimus | Allosteric inhibitor of the mammalian target of rapamycin (mTOR), inhibiting the mTOR functional complex mTORC1. |
| Fulvestrant | Selective estrogen receptor downregulator or degrader, competitively binding to estrogen receptors and promoting their degradation. |
| Infigratinib | Pan-inhibitor of human fibroblast growth factor receptors (FGFRs). |
| JQ1 | Small molecule, competitive inhibitor of bromodomain-containing 4 (BRD4), an epigenetic reader acting as transcriptional regulator. |
| Lapatinib | Small molecule, ATP-competitive inhibitor of epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2). |
| Neratinib | Small molecule, irreversible inhibitor of EGFR and HER2. |
| Palbociclib | Cyclin-dependent kinase 4 and 6 inhibitor, preventing retinoblastoma (Rb) protein phosphorylation and leading to cell cycle arrest. |
| Ponatinib | Small molecule, multitargeted tyrosine kinase inhibitor, inhibiting, among others, BCR-ABL, vascular endothelial growth factor receptors (VEGFRs), fibroblast growth factor receptors (FGFRs), TEK Receptor Tyrosine Kinase (TIE2), FMS-like tyrosine kinase 3 (FLT3), KIT, and REarranged during Transfection (RET). |
| Selumetinib | Small molecule, allosteric inhibitor of mitogen-activated protein kinase kinase (MEK or MAPK/ERK kinase) 1 and 2. |
| SHP099 | Small molecule, allosteric inhibitor of Srchomology-2-domain-containing PTP 2 (SHP2). |
| Sorafenib | Small molecule, multitargeted tyrosine kinase inhibitor, inhibiting, among others, RAF, VEGFR 1/2/3, platelet-derived growth factor receptor (PDGFR) beta, KIT, FLT3, FGFR1, and RET. |
| Tamoxifen | Selective estrogen receptor modulator (SERM), competitively inhibiting the binding of estradiol to estrogen receptors. |
| Trametinib | Small molecule, allosteric inhibitor of MEK1 and MEK2. |
| Trastuzumab | Recombinant humanized monoclonal antibody targeting the extracellular domain of HER2. |
| Tucatinib | ATP-competitive, small molecule inhibitor of HER2. |
| Ulixertinib | Small molecule, ATP-competitive inhibitor of extracellular signal-regulated kinase (ERK) 1 and 2. |
| Vemurafenib | ATP-competitive, small-molecule inhibitor of mutant BRAF, including BRAF(V600E). |
References
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1150–1160. [Google Scholar] [CrossRef]
- Johnson, C.; Burkhart, D.L.; Haigis, K.M. Classification of KRAS-Activating Mutations and the Implications for Therapeutic Intervention. Cancer Discov. 2022, 12, 913–923. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 102. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat. Commun. 2021, 12, 1080. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Song, R.X.; McPherson, R.; Kumar, R.; Adam, L.; Jeng, M.H.; Yue, W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 239–256. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Morrison, D.K. A Structure Is Worth a Thousand Words: New Insights for RAS and RAF Regulation. Cancer Discov. 2022, 12, 899–912. [Google Scholar] [CrossRef]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Osaka, N.; Hirota, Y.; Ito, D.; Ikeda, Y.; Kamata, R.; Fujii, Y.; Chirasani, V.R.; Campbell, S.L.; Takeuchi, K.; Senda, T.; et al. Divergent Mechanisms Activating RAS and Small GTPases Through Post-translational Modification. Front. Mol. Biosci. 2021, 8, 642. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Philips, M.R. Post-translational modification of RAS proteins. Curr. Opin. Struct. Biol. 2021, 71, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, T.; Sahmi, M.; Lefrançois, M.; Sicheri, F.; Therrien, M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009, 461, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Stites, E.C.; Yu, H.; Germino, E.A.; Meharena, H.S.; Stork, P.J.S.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 2013, 154, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ognjenović, J.; Banerjee, S.; Karandur, D.; Merk, A.; Kulhanek, K.; Wong, K.; Roose, J.P.; Subramaniam, S.; Kuriyan, J. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019, 366, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Del Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF–MEK1–14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Venkatanarayan, A.; Quinn, J.G.; Phung, W.; Malek, S.; Hymowitz, S.G.; Sudhamsu, J. Dimerization Induced by C-Terminal 14-3-3 Binding Is Sufficient for BRAF Kinase Activation. Biochemistry 2020, 59, 3982–3992. [Google Scholar] [CrossRef]
- Payne, D.M.; Rossomando, A.J.; Martino, P.; Erickson, A.K.; Her, J.H.; Shabanowitz, J.; Hunt, D.F.; Weber, M.J.; Sturgill, T.W. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991, 10, 885–892. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Barbosa, R.; Acevedo, L.A.; Marmorstein, R. The MEK/ERK Network as a Therapeutic Target in Human Cancer. Mol. Cancer Res. 2021, 19, 361–374. [Google Scholar] [CrossRef]
- Sturm, O.E.; Orton, R.; Grindlay, J.; Birtwistle, M.; Vyshemirsky, V.; Gilbert, D.; Calder, M.; Pitt, A.; Kholodenko, B.; Kolch, W. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci. Signal. 2010, 3, ra90. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, B.N.; Hancock, J.F.; Kolch, W. Signalling ballet in space and time. Nat. Rev. Mol. Cell Biol. 2010, 11, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK Activation Downregulates Antiproliferative Genes throughout G1 Phase to Allow Cell-Cycle Progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Sewing, A.; Wiseman, B.; Lloyd, A.C.; Land, H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell. Biol. 1997, 17, 5588–5597. [Google Scholar] [CrossRef]
- Rauch, N.; Rukhlenko, O.S.; Kolch, W.; Kholodenko, B.N. MAPK kinase signalling dynamics regulate cell fate decisions and drug resistance. Curr. Opin. Struct. Biol. 2016, 41, 151–158. [Google Scholar] [CrossRef]
- Santarpia, L.; Qi, Y.; Stemke-Hale, K.; Wang, B.; Young, E.J.; Booser, D.J.; Holmes, F.A.; O’Shaughnessy, J.; Hellerstedt, B.; Pippen, J.; et al. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res. Treat. 2012, 134, 333–343. [Google Scholar] [CrossRef]
- Turski, M.L.; Vidwans, S.J.; Janku, F.; Garrido-Laguna, I.; Munoz, J.; Schwab, R.; Subbiah, V.; Rodon, J.; Kurzrock, R. Genomically driven tumors and actionability across histologies: BRAF-mutant cancers as a paradigm. Mol. Cancer Ther. 2016, 15, 533–547. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting alterations in the RAF–MEK pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Gao, Y.; Chang, M.T.; McKay, D.; Na, N.; Zhou, B.; Yaeger, R.; Torres, N.M.; Muniz, K.; Drosten, M.; Barbacid, M.; et al. Allele-specific mechanisms of activation of mek1 mutants determine their properties. Cancer Discov. 2018, 8, 648–661. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.W.; Ambrogio, C.; Bera, A.K.; Li, Q.; Li, X.X.; Li, L.; Son, J.; Gondi, S.; Li, J.; Campbell, E.; et al. KRASQ61H preferentially signals through MAPK in a RAF dimer-dependent manner in non-small cell lung cancer. Cancer Res. 2020, 80, 3719–3731. [Google Scholar] [CrossRef]
- Johnson, C.W.; Seo, H.-S.; Terrell, E.M.; Yang, M.-H.; KleinJan, F.; Gebregiworgis, T.; Gasmi-Seabrook, G.M.C.; Geffken, E.A.; Lakhani, J.; Song, K.; et al. Regulation of GTPase function by autophosphorylation. Mol. Cell 2022, 82, 950–968. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRASG12R mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Haigis, B.K.M.; Cichowski, K. Tissue-specificity in cancer: The rule, not the exception. Science 2019, 363, 1150–1152. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, D.K.; Paulo, J.A.; Sheth, S.; Poulin, E.J.; Popow, O.; Joughin, B.A.; Strasser, S.D.; Starchenko, A.; Gygi, S.P.; Lauffenburger, D.A.; et al. Proteogenomic Network Analysis of Context-Specific KRAS Signaling in Mouse-to-Human Cross-Species Translation. Cell Syst. 2019, 9, 258–270.e6. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-Mediated MEK activation is required for effective mek inhibition in KRAS mutant tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Hazar-Rethinam, M.; Kleyman, M.; Han, G.C.; Liu, D.; Ahronian, L.G.; Shahzade, H.A.; Chen, L.; Parikh, A.R.; Allen, J.N.; Clark, J.W.; et al. Convergent therapeutic strategies to overcome the heterogeneity of acquired resistance in BRAFV600E colorectal cancer. Cancer Discov. 2018, 8, 417–427. [Google Scholar] [CrossRef]
- Whittaker, J.L.; Walker, R.A.; Varley, J.M. Differential expression of cellular oncogenes in benign and malignant human breast tissue. Int. J. Cancer 1986, 38, 651–655. [Google Scholar] [CrossRef]
- Dati, C.; Muraca, R.; Tazartes, O.; Antoniotti, S.; Perroteau, I.; Giai, M.; Cortese, P.; Sismondi, P.; Saglio, G.; De Bortoli, M. c-erbB-2 and ras expression levels in breast cancer are correlated and show a co-operative association with unfavorable clinical outcome. Int. J. Cancer 1991, 47, 833–838. [Google Scholar] [CrossRef]
- Theillet, C.; Lidereau, R.; Escot, C.; Hutzell, P.; Brunet, M.; Gest, J.; Schlom, J.; Callahan, R. Loss of a c-H-ras-1 Allele and Aggressive Human Primary Breast Carcinomas. Cancer Res. 1986, 46, 4776–4781. [Google Scholar]
- Cline, M.J.; Battifora, H.; Yokota, J. Proto-oncogene abnormalities in human breast cancer: Correlations with anatomic features and clinical course of disease. J. Clin. Oncol. 1987, 5, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Biunno, I.; Pozzi, M.R.; Pierotti, M.A.; Pilotti, S.; Cattoretti, G.; Porta, G. Della Structure and expression of oncogenes in surgical specimens of human breast carcinomas. Br. J. Cancer 1988, 57, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Schondorf, T.; Andrack, A.; Niederacher, D.; Scharl, A.; Becker, M.; Engel, H.; Gohring, U.J. H-ras gene amplification or mutation is not common in human primary breast cancer. Oncol. Rep. 1999, 6, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Lidereau, R.; Mathieu-Mahul, D.; Escot, C.; Theillet, C.; Champeme, M.H.; Cole, S.; Mauchauffe, M.; Ali, I.; Amione, J.; Callahan, R.; et al. Genetic variability of proto-oncogenes for breast cancer risk and prognosis. Biochimie 1988, 70, 951–959. [Google Scholar] [CrossRef]
- Garcia, I.; Dietrich, P.Y.; Aapro, M.; Vauthier, G.; Vadas, L.; Engel, E. Genetic Alterations of c-myc, c-erbB-2, and c-Ha-ras Protooncogenes and Clinical Associations in Human Breast Carcinomas. Cancer Res. 1989, 49, 6675–6679. [Google Scholar] [PubMed]
- Göhring, U.J.; Schöndorf, T.; Kiecker, V.R.; Becker, M.; Kurbacher, C.; Scharl, A. Immunohistochemical detection of H-ras protooncoprotein p21 indicates favorable prognosis in node-negative breast cancer patients. Tumor Biol. 1999, 20, 173–183. [Google Scholar] [CrossRef]
- Pereira, C.B.L.; Leal, M.F.; de Souza, C.R.T.; Montenegro, R.C.; Rey, J.A.; Carvalho, A.A.; Assumpção, P.P.; Khayat, A.S.; Pinto, G.R.; Demachki, S.; et al. Prognostic and Predictive Significance of MYC and KRAS Alterations in Breast Cancer from Women Treated with Neoadjuvant Chemotherapy. PLoS ONE 2013, 8, e60576. [Google Scholar] [CrossRef]
- Sivaraman, V.S.; Wang, H.; Nuovo, G.J.; Malbon, C.C. Hyperexpression of mitogen-activated protein kinase in human breast cancer. J. Clin. Investig. 1997, 99, 1478–1483. [Google Scholar] [CrossRef]
- Mueller, H.; Flury, N.; Eppenberger-Castori, S.; Kueng, W.; David, F.; Eppenberger, U. Potential prognostic value of mitogen-activated protein kinase activity for disease-free survival of primary breast cancer patients. Int. J. Cancer 2000, 89, 384–388. [Google Scholar] [CrossRef]
- Adeyinka, A.; Nui, Y.; Cherlet, T.; Snell, L.; Watson, P.H.; Murphy, L.C. Activated mitogen-activated protein kinase expression during human breast tumorigenesis and breast cancer progression. Clin. Cancer Res. 2002, 8, 1747–1753. [Google Scholar]
- Von Lintig, F.C.; Dreilinger, A.D.; Varki, N.M.; Wallace, A.M.; Casteel, D.E.; Boss, G.R. Ras activation in human breast cancer. Breast Cancer Res. Treat. 2000, 62, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed]
- Jerjees, D.A.; Alabdullah, M.; Alkaabi, M.; Abduljabbar, R.; Muftah, A.; Nolan, C.; Green, A.R.; Ellis, I.O.; Rakha, E.A. ERK1/2 is related to oestrogen receptor and predicts outcome in hormone-treated breast cancer. Breast Cancer Res. Treat. 2014, 147, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K.; Bamberger, A.M.; Rieck, G.; Grund, D.; Hemminger, G.; Müller, V.; Löning, T. Expression and prognostic relevance of activated extracellular-regulated kinases (ERK 1/2) in breast cancer. Br. J. Cancer 2005, 92, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.T.; Hsu, H.T.; Chang, C.C.; Jiang, M.C.; Yeh, C.M.; Shen, K.H.; Hsu, P.C.; Tai, C.J. High nuclear phosphorylated extracellular signal-regulated kinase expression associated with poor differentiation, larger tumor size, and advanced stage of breast cancer. Pol. J. Pathol. 2013, 64, 163–169. [Google Scholar] [CrossRef]
- Nakopoulou, L.; Mylona, E.; Rafailidis, P.; Alexandrou, P.; Giannopoulou, I.; Keramopoulos, A. Effect of different ERK2 protein localizations on prognosis of patients with invasive breast carcinoma. APMIS 2005, 113, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.L.; Wang, M.C.; Fan, X.L.; Zhong, L.; Ji, S.F.; Sang, G.; Wang, S. Correlation between Raf/MEK/ERKs signaling pathway and clinicopathological features and prognosis for patients with breast cancer having axillary lymph node metastasis. Technol. Cancer Res. Treat. 2018, 17, 1–10. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Kareff, S.; Rodriguez, E.; Dawar, R.; Trabolsi, A.; Gallegos, J.A.O.; Yin, J.; Walker, P.; Kang, I.; Bustos, M.A.; Neman, J.; et al. Molecular characteristics and clinical outcomes of breast cancer with HRAS mutations. J. Clin. Oncol. 2022, 40, 561. [Google Scholar] [CrossRef]
- Kan, Z.; Ding, Y.; Kim, J.; Jung, H.H.; Chung, W.; Lal, S.; Cho, S.; Fernandez-Banet, J.; Lee, S.K.; Kim, S.W.; et al. Multi-omics profiling of younger Asian breast cancers reveals distinctive molecular signatures. Nat. Commun. 2018, 9, 1725. [Google Scholar] [CrossRef] [PubMed]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456.e31. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Lévy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Tiu-Lim, J.W.W.; Yin, J.; Xiu, J.; Korn, W.M.; Lenz, H.-J.; In, G.K.; Torres, E.T.R.; Lu, J.M.; Spicer, D.V.; Xia, B.; et al. Molecular characterization of the Ras-MAPK pathway in metastatic breast cancer. J. Clin. Oncol. 2021, 39, 1034. [Google Scholar] [CrossRef]
- Aftimos, P.; Oliveira, M.; Irrthum, A.; Fumagalli, D.; Sotiriou, C.; Gal-Yam, E.N.; Robson, M.E.; Ndozeng, J.; Di Leo, A.; Ciruelos, E.M.; et al. Genomic and transcriptomic analyses of breast cancer primaries and matched metastases in Aurora, the breast international group (Big) molecular screening initiative. Cancer Discov. 2021, 11, 2796–2811. [Google Scholar] [CrossRef]
- Wallace, M.D.; Pfefferle, A.D.; Shen, L.; McNairn, A.J.; Cerami, E.G.; Fallon, B.L.; Rinaldi, V.D.; Southard, T.L.; Perou, C.M.; Schimenti, J. Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics 2012, 192, 385–396. [Google Scholar] [CrossRef]
- McLaughlin, S.K.; Olsen, S.N.; Dake, B.; De Raedt, T.; Lim, E.; Bronson, R.T.; Beroukhim, R.; Polyak, K.; Brown, M.; Kuperwasser, C.; et al. The RasGAP Gene, RASAL2, Is a Tumor and Metastasis Suppressor. Cancer Cell 2013, 24, 365–378. [Google Scholar] [CrossRef]
- Craig, D.W.; O’Shaughnessy, J.A.; Kiefer, J.A.; Aldrich, J.; Sinari, S.; Moses, T.M.; Wong, S.; Dinh, J.; Christoforides, A.; Blum, J.L.; et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef]
- Dry, J.R.; Pavey, S.; Pratilas, C.A.; Harbron, C.; Runswick, S.; Hodgson, D.; Chresta, C.; McCormack, R.; Byrne, N.; Cockerill, M.; et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res. 2010, 70, 2264–2273. [Google Scholar] [CrossRef]
- Huang, E.; Ishida, S.; Pittman, J.; Dressman, H.; Bild, A.; Kloos, M.; D’Amico, M.; Pestell, R.G.; West, M.; Nevins, J.R. Gene expression phenotypic models that predict the activity of oncogenic pathways. Nat. Genet. 2003, 34, 226–230. [Google Scholar] [CrossRef]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.B.; Harpole, D.; Lancaster, J.M.; Berchuck, A.; et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef]
- Bild, A.H.; Parker, J.S.; Gustafson, A.M.; Acharya, C.R.; Hoadley, K.A.; Anders, C.; Marcom, P.K.; Carey, L.A.; Potti, A.; Nevins, J.R.; et al. An integration of complementary strategies for gene-expression analysis to reveal novel therapeutic opportunities for breast cancer. Breast Cancer Res. 2009, 11, R55. [Google Scholar] [CrossRef]
- Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Kim, J.W.; Wang, Q.; Crawford, M.D.; Datto, M.B.; Kelley, M.; Mathey-Prevot, B.; Potti, A.; et al. A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6994–6999. [Google Scholar] [CrossRef]
- Creighton, C.J.; Hilger, A.M.; Murthy, S.; Rae, J.M.; Chinnaiyan, A.M.; El-Ashry, D. Activation of mitogen-activated protein kinase in estrogen receptor α-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor α-negative human breast tumors. Cancer Res. 2006, 66, 3903–3911. [Google Scholar] [CrossRef]
- Mirzoeva, O.K.; Das, D.; Heiser, L.M.; Bhattacharya, S.; Siwak, D.; Gendelman, R.; Bayani, N.; Wang, N.J.; Neve, R.M.; Guan, Y.; et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009, 69, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.L.; Adams, J.R.; Liu, J.C.; Loch, A.J.; Wong, R.G.; Jo, C.E.B.; Beck, L.A.; Santhanam, D.R.; Weiss, L.; Mei, X.; et al. Ras signaling is a key determinant for metastatic dissemination and poor survival of luminal breast cancer patients. Cancer Res. 2015, 75, 4960–4972. [Google Scholar] [CrossRef] [PubMed]
- Wagle, M.C.; Kirouac, D.; Klijn, C.; Liu, B.; Mahajan, S.; Junttila, M.; Moffat, J.; Merchant, M.; Huw, L.; Wongchenko, M.; et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis. Oncol. 2018, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Way, G.P.; Sanchez-Vega, F.; La, K.; Armenia, J.; Chatila, W.K.; Luna, A.; Sander, C.; Cherniack, A.D.; Mina, M.; Ciriello, G.; et al. Machine Learning Detects Pan-cancer Ras Pathway Activation in The Cancer Genome Atlas. Cell Rep. 2018, 23, 172–180.e3. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, S.; Wang, Y.; Zhang, S.; Wong, K.C. Identification of pan-cancer Ras pathway activation with deep learning. Brief. Bioinform. 2021, 22, bbaa258. [Google Scholar] [CrossRef]
- Liu, D.; Zhou, K. BRAF/MEK Pathway is Associated with Breast Cancer in ER-dependent Mode and Improves ER Status-based Cancer Recurrence Prediction. Clin. Breast Cancer 2020, 20, 41–50.e8. [Google Scholar] [CrossRef]
- Zivadinovic, D.; Watson, C.S. Membrane estrogen receptor-alpha levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res. 2005, 7, 130–144. [Google Scholar] [CrossRef]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zhang, X.T.; Shen, P.; Loggie, B.W.; Chang, Y.C.; Deuel, T.F. A variant of estrogen receptor-α, hER-α36: Transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Britton, D.J.; Hutcheson, I.R.; Knowlden, J.M.; Barrow, D.; Giles, M.; McClelland, R.A.; Gee, J.M.W.; Nicholson, R.I. Bidirectional cross talk between ERα and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res. Treat. 2006, 96, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.M.; Miller, T.W.; Balko, J.M.; Kuba, M.G.; Sánchez, V.; Smith, R.A.; Liu, S.; González-Angulo, A.M.; Mills, G.B.; Ye, F.; et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. 2011, 71, 6773–6784. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.T.; Alexi, X.; Opdam, M.; Schuurman, K.; Voorwerk, L.; Sanders, J.; van der Noort, V.; Boven, E.; Zwart, W.; Linn, S.C. IGF-1R pathway activation as putative biomarker for linsitinib therapy to revert tamoxifen resistance in ER-positive breast cancer. Int. J. Cancer 2020, 146, 2348–2359. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Cohen, O.; Kowalski, K.J.; Kusiel, J.G.; Buendia-Buendia, J.E.; Cuoco, M.S.; Exman, P.; Wander, S.A.; Waks, A.G.; Nayar, U.; et al. Acquired FGFR and FGF alterations confer resistance to Estrogen Receptor (ER) targeted therapy in ERþ metastatic breast cancer. Clin. Cancer Res. 2020, 26, 5974–5989. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Truong, T.H.; Ostrander, J.H.; Lange, C.A. 90 Years of Progesterone: Steroid receptors as MAPK signaling sensors in breast cancer: Let the fates decide. J. Mol. Endocrinol. 2020, 65, T35–T48. [Google Scholar] [CrossRef]
- McGlynn, L.M.; Kirkegaard, T.; Edwards, J.; Tovey, S.; Cameron, D.; Twelves, C.; Bartlett, J.M.S.; Cooke, T.G. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin. Cancer Res. 2009, 15, 1487–1495. [Google Scholar] [CrossRef]
- Gee, J.M.W.; Robertson, J.F.R.; Ellis, I.O.; Nicholson, R.I. Phosphorylation of Erk1/2 Mitogen-Activated Protein Kinase Is Associated with Poor Response To Anti-Hormonal Therapy and decreased patient survival in clinical breast cancer. Int. J. Cancer 2001, 95, 247–254. [Google Scholar] [CrossRef]
- Zheng, A.; Kallio, A.; Härkönen, P. Tamoxifen-induced rapid death of MCF-7 breast cancer cells is mediated via extracellularly signal-regulated kinase signaling and can be abrogated by estrogen. Endocrinology 2007, 148, 2764–2777. [Google Scholar] [CrossRef]
- Pearson, A.; Proszek, P.; Pascual, J.; Fribbens, C.; Shamsher, M.K.; Kingston, B.; O’Leary, B.; Herrera-Abreu, M.T.; Cutts, R.J.; Garcia-Murillas, I.; et al. Inactivating NF1 mutations are enriched in advanced breast cancer and contribute to endocrine therapy resistance. Clin. Cancer Res. 2020, 26, 608–622. [Google Scholar] [CrossRef]
- Sokol, E.S.; Feng, Y.X.; Jin, D.X.; Basudan, A.; Lee, A.V.; Atkinson, J.M.; Chen, J.; Stephens, P.J.; Frampton, G.M.; Gupta, P.B.; et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann. Oncol. 2019, 30, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Vafeiadou, V.; Hany, D.; Picard, D. Hyperactivation of MAPK Induces Tamoxifen Resistance in SPRED2-Deficient ERα-Positive Breast Cancer. Cancers 2022, 14, 954. [Google Scholar] [CrossRef] [PubMed]
- Fribbens, C.; Murillas, I.G.; Beaney, M.; Hrebien, S.; O’Leary, B.; Kilburn, L.; Howarth, K.; Epstein, M.; Green, E.; Rosenfeld, N.; et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann. Oncol. 2018, 29, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K.; Winterhalder, R.; Mamot, C.; Hasler-Strub, U.; Rochlitz, C.; Mueller, A.; Berset, C.; Wiliders, H.; Perey, L.; Rudolf, C.B.; et al. Fulvestrant with or without selumetinib, a MEK 1/2 inhibitor, in breast cancer progressing after aromatase inhibitor therapy: A multicentre randomised placebo-controlled double-blind phase II trial, SAKK 21/08. Eur. J. Cancer 2015, 51, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Thottassery, J.V.; Sun, Y.; Westbrook, L.; Rentz, S.S.; Manuvakhova, M.; Qu, Z.; Samuel, S.; Upshaw, R.; Cunningham, A.; Kern, F.G. Prolonged extracellular signal-regulated kinase 1/2 activation during fibroblast growth factor 1- of heregulin β1-induced antiestrogen-resistant growth of breast cancer cells is resistant to mitogen-activated protein/extracellular regulated kinase kinase inhibitors. Cancer Res. 2004, 64, 4637–4647. [Google Scholar] [CrossRef]
- Peng, W.-X.; Huang, J.; Yang, L.; Gong, A.-H.; Mo, Y.-Y. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol. Cancer 2017, 16, 161. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Kim, C.Y.; Oh, J.H.; Kim, M.H. Nr4a1 regulates tamoxifen resistance by suppressing erk signaling in er-positive breast cancer. Cells 2021, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhou, J.; Luo, X.; Chen, M.; Chen, Y.; Wang, J.; Xiong, H.; Ying, X.; Hu, W.; Zhao, W.; et al. KLF4 overcomes tamoxifen resistance by suppressing MAPK signaling pathway and predicts good prognosis in breast cancer. Cell. Signal. 2018, 42, 165–175. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Li, M.; Tang, W.; Pratap, U.P.; Luo, Y.; Altwegg, K.A.; Li, X.; Zou, Y.; Zhu, H.; et al. Interaction of transcription factor AP-2 gamma with proto-oncogene PELP1 promotes tumorigenesis by enhancing RET signaling. Mol. Oncol. 2021, 15, 1146–1161. [Google Scholar] [CrossRef]
- Mao, M.; Hu, D.; Yang, J.; Chen, Y.; Zhang, X.; Shen, J.; Teng, R.; Zhou, J.; Wang, L. Regulation of tamoxifen sensitivity by the PLAC8/MAPK pathway axis is antagonized by curcumin-induced protein stability change. J. Mol. Med. 2021, 99, 845–858. [Google Scholar] [CrossRef]
- Menendez, J.A.; Mehmi, I.; Papadimitropoulou, A.; Steen, T.V.; Cuyàs, E.; Verdura, S.; Espinoza, I.; Vellon, L.; Atlas, E.; Lupu, R. Fatty acid synthase is a key enabler for endocrine resistance in heregulin-overexpressing luminal b-like breast cancer. Int. J. Mol. Sci. 2020, 21, 7611. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhu, Q.; Liu, M.; Tu, G.; Li, Q.; Yuan, J.; Wen, S.; Yang, G. GPER promotes tamoxifen-resistance in ER+ breast cancer cells by reduced Bim proteins through MAPK/Erk-TRIM2 signaling axis. Int. J. Oncol. 2017, 51, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Janes, P.W.; Daly, R.J.; DeFazio, A.; Sutherland, R.L. Activation of the Ras signalling pathway in human breast cancer cells overexpressing erbB-2. Oncogene 1994, 9, 3601–3608. [Google Scholar] [PubMed]
- Kirouac, D.C.; Du, J.; Lahdenranta, J.; Onsum, M.D.; Nielsen, U.B.; Schoeberl, B.; McDonagh, C.F. HER2+ Cancer Cell Dependence on PI3K vs. MAPK Signaling Axes Is Determined by Expression of EGFR, ERBB3 and CDKN1B. PLoS Comput. Biol. 2016, 12, e1004827. [Google Scholar] [CrossRef]
- She, Q.B.; Chandarlapaty, S.; Ye, Q.; Lobo, J.; Haskell, K.M.; Leander, K.R.; DeFeo-Jones, D.; Huber, H.E.; Rosen, N. Breast tumor cells with P13K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS ONE 2008, 3, e3065. [Google Scholar] [CrossRef] [PubMed]
- Maadi, H.; Soheilifar, M.H.; Choi, W.; Moshtaghian, A.; Wang, Z. Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers 2021, 13, 3540. [Google Scholar] [CrossRef]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.A.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Smith, A.E.; Ferraro, E.; Safonov, A.; Morales, C.B.; Lahuerta, E.J.A.; Li, Q.; Kulick, A.; Ross, D.; Solit, D.B.; de Stanchina, E.; et al. HER2 + breast cancers evade anti-HER2 therapy via a switch in driver pathway. Nat. Commun. 2021, 12, 6667. [Google Scholar] [CrossRef]
- Alexander, P.B.; Chen, R.; Gong, C.; Yuan, L.; Jasper, J.S.; Ding, Y.; Markowitz, G.J.; Yang, P.; Xu, X.; McDonnell, D.P.; et al. Distinct receptor tyrosine kinase subsets mediate anti-HER2 drug resistance in breast cancer. J. Biol. Chem. 2017, 292, 748–759. [Google Scholar] [CrossRef]
- Wu, D.; Jia, H.-Y.; Wei, N.; Li, S.-J. POU4F1 confers trastuzumab resistance in HER2-positive breast cancer through regulating ERK1/2 signaling pathway. Biochem. Biophys. Res. Commun. 2020, 533, 533–539. [Google Scholar] [CrossRef]
- Zazo, S.; González-Alonso, P.; Martín-Aparicio, E.; Chamizo, C.; Luque, M.; Sanz-Álvarez, M.; Mínguez, P.; Gómez-López, G.; Cristóbal, I.; Caramés, C.; et al. Autocrine CCL5 effect mediates trastuzumab resistance by ERK pathway activation in HER2-positive breast cancer. Mol. Cancer Ther. 2020, 19, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar] [PubMed]
- Sánchez-Muñoz, A.; Gallego, E.; de Luque, V.; Pérez-Rivas, L.G.; Vicioso, L.; Ribelles, N.; Lozano, J.; Alba, E. Lack of evidence for KRAS oncogenic mutations in triple-negative breast cancer. BMC Cancer 2010, 10, 136. [Google Scholar] [CrossRef]
- Tilch, E.; Seidens, T.; Cocciardi, S.; Reid, L.E.; Byrne, D.; Simpson, P.T.; Vargas, A.C.; Cummings, M.C.; Fox, S.B.; Lakhani, S.R.; et al. Mutations in EGFR, BRAF and RAS are rare in triple-negative and basal-like breast cancers from Caucasian women. Breast Cancer Res. Treat. 2014, 143, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Grob, T.J.; Heilenkötter, U.; Geist, S.; Paluchowski, P.; Wilke, C.; Jaenicke, F.; Quaas, A.; Wilczak, W.; Choschzick, M.; Sauter, G.; et al. Rare oncogenic mutations of predictive markers for targeted therapy in triple-negative breast cancer. Breast Cancer Res. Treat. 2012, 134, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Cook, R.S.; Vaught, D.B.; Kuba, M.G.; Miller, T.W.; Bhola, N.E.; Sanders, M.E.; Granja-Ingram, N.M.; Joshua Smith, J.; Meszoely, I.M.; et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat. Med. 2012, 18, 1052–1059. [Google Scholar] [CrossRef]
- Huang, J.; Luo, Q.; Xiao, Y.; Li, H.; Kong, L.; Ren, G. The implication from RAS/RAF/ERK signaling pathway increased activation in epirubicin treated triple negative breast cancer. Oncotarget 2017, 8, 108249–108260. [Google Scholar] [CrossRef]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef]
- Nadeau, R.J.; Toher, J.L.; Yang, X.; Kovalenko, D.; Friesel, R. Regulation of Sprouty2 stability by mammalian seven-in-absentia homolog 2. J. Cell. Biochem. 2007, 100, 151–160. [Google Scholar] [CrossRef]
- House, C.M.; Möller, A.; Bowtell, D.D.L. Siah proteins: Novel drug targets in the Ras and hypoxia pathways. Cancer Res. 2009, 69, 8835–8838. [Google Scholar] [CrossRef]
- Van Reesema, L.L.S.; Zheleva, V.; Winston, J.S.; Jansen, R.J.; O’Connor, C.F.; Isbell, A.J.; Bian, M.; Qin, R.; Bassett, P.T.; Hinson, V.J.; et al. SIAH and EGFR, Two RAS Pathway Biomarkers, are Highly Prognostic in Locally Advanced and Metastatic Breast Cancer. eBioMedicine 2016, 11, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Rojo, F.; González-Navarrete, I.; Bragado, R.; Dalmases, A.; Menéndez, S.; Cortes-Sempere, M.; Suárez, C.; Oliva, C.; Servitja, S.; Rodriguez-Fanjul, V.; et al. Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin. Cancer Res. 2009, 15, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qin, T.; Bi, Z.; Hong, H.; Ding, L.; Chen, J.; Wu, W.; Lin, X.; Fu, W.; Zheng, F.; et al. Rac1 activates non-oxidative pentose phosphate pathway to induce chemoresistance of breast cancer. Nat. Commun. 2020, 11, 1456. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E6. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.; Jechlinger, M.; Beug, H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Biol. 2003, 4, 657–665. [Google Scholar] [CrossRef]
- Zhou, C.; Nitschke, A.M.; Xiong, W.; Zhang, Q.; Tang, Y.; Bloch, M.; Elliott, S.; Zhu, Y.; Bazzone, L.; Yu, D.; et al. Proteomic analysis of tumor necrosis factor-α resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008, 10, R105. [Google Scholar] [CrossRef]
- Hoang, V.T.; Matossian, M.D.; La, J.; Hoang, K.; Ucar, D.A.; Elliott, S.; Burks, H.E.; Wright, T.D.; Patel, S.; Bhatt, A.; et al. Dual inhibition of MEK1/2 and MEK5 suppresses the EMT/migration axis in triple-negative breast cancer through FRA-1 regulation. J. Cell. Biochem. 2021, 122, 835–850. [Google Scholar] [CrossRef]
- Hong, D.; Fritz, A.J.; Zaidi, S.K.; van Wijnen, A.J.; Nickerson, J.A.; Imbalzano, A.N.; Lian, J.B.; Stein, J.L.; Stein, G.S. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J. Cell. Physiol. 2018, 233, 9136–9144. [Google Scholar] [CrossRef]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44HI/CD24lO/-stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Schwarz, L.J.; Bhola, N.E.; Kurupi, R.; Owens, P.; Miller, T.W.; Gómez, H.; Cook, R.S.; Arteaga, C.L. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013, 73, 6346–6358. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605. [Google Scholar] [CrossRef]
- Mittal, S.; Sharma, A.; Balaji, S.A.; Gowda, M.C.; Dighe, R.R.; Kumar, R.V.; Rangarajan, A. Coordinate hyperactivation of notch1 and Ras/MAPK pathways correlates with poor patient survival: Novel therapeutic strategy for aggressive breast cancers. Mol. Cancer Ther. 2014, 13, 3198–3209. [Google Scholar] [CrossRef]
- Prat, A.; Perou, C.M. Mammary development meets cancer genomics. Nat. Med. 2009, 15, 842–844. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef]
- Zdanov, S.; Mandapathil, M.; Eid, R.A.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef]
- Dushyanthen, S.; Teo, Z.L.; Caramia, F.; Savas, P.; Mintoff, C.P.; Virassamy, B.; Henderson, M.A.; Luen, S.J.; Mansour, M.; Kershaw, M.H.; et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat. Commun. 2017, 8, 606. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yi, Z.; Li, J.; Wei, Y.; Feng, R.; Liu, J.; Huang, J.; Chen, Y.; Wang, X.; Sun, J.; et al. FGFR blockade boosts T cell infiltration into triple-negative breast cancer by regulating cancer-associated fibroblasts. Theranostics 2022, 12, 4564–4580. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.G.; Nguyen, L.K. Network rewiring, adaptive resistance and combating strategies in breast cancer. Cancer Drug Resist. 2019, 2, 1106–1126. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Finn, R.S.; Hosmer, W.; Dering, J.; Ginther, C.; Adhami, S.; Kamranpour, N.; Pitts, S.; Desai, A.; Elashoff, D.; et al. Identification of common predictive markers of in vitro response to the Mek inhibitor selumetinib (AZD6244; ARRY-142886) in human breast cancer and non-small cell lung cancer cell lines. Mol. Cancer Ther. 2010, 9, 1985–1994. [Google Scholar] [CrossRef]
- Young, A.; Lou, D.; McCormick, F. Oncogenic and wild-type ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013, 3, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 2017, 168, 817–829.e15. [Google Scholar] [CrossRef]
- Sos, M.L.; Fischer, S.; Ullrich, R.; Peifer, M.; Heuckmann, J.M.; Koker, M.; Heynck, S.; Stückrath, I.; Weiss, J.; Fischer, F.; et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 18351–18356. [Google Scholar] [CrossRef]
- Yu, C.F.; Liu, Z.; Cantley, L.G. ERK Negatively Regulates the Epidermal Growth Factor-mediated Interaction of Gab1 and the Phosphatidylinositol 3-Kinase *. J. Biol. Chem. 2002, 277, 19382–19388. [Google Scholar] [CrossRef]
- Li, X.; Huang, Y.; Jiang, J.; Frank, S.J. ERK-dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling. Cell. Signal. 2008, 20, 2145–2155. [Google Scholar] [CrossRef]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.D. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013, 71, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Yart, A.; Chap, H.; Raynal, P. Phosphoinositide 3-kinases in lysophosphatidic acid signaling: Regulation and cross-talk with the Ras/mitogen-activated protein kinase pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2002, 1582, 107–111. [Google Scholar] [CrossRef]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef]
- Saini, K.S.; Loi, S.; De Azambuja, E.; Metzger-filho, O.; Lamba, M.; Ignatiadis, M.; Dancey, J.E.; Piccart-gebhart, M.J. Targeting the PI3K / AKT / mTOR and Raf / MEK / ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef]
- Balmanno, K.; Chell, S.D.; Gillings, A.S.; Hayat, S.; Cook, S.J. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int. J. Cancer 2009, 125, 2332–2341. [Google Scholar] [CrossRef]
- Meng, J.; Peng, H.; Dai, B.; Guo, W.; Wang, L.; Ji, L.; Minna, J.D.; Chresta, C.M.; Smith, P.D.; Fang, B.; et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886). Cancer Biol. Ther. 2009, 8, 2073–2080. [Google Scholar] [CrossRef]
- Tran, L.M.; Chang, C.J.; Plaisier, S.; Wu, S.; Dang, J.; Mischel, P.S.; Liao, J.C.; Graeber, T.G.; Wu, H. Determining PTEN functional status by network component deduced transcription factor activities. PLoS ONE 2012, 7, e31053. [Google Scholar] [CrossRef]
- Saal, L.H.; Gruvberger-Saal, S.K.; Persson, C.; Lövgren, K.; Jumppanen, M.; Staaf, J.; Jönsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 2008, 40, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Marty, B.; Maire, V.; Gravier, E.; Rigaill, G.; Vincent-Salomon, A.; Kappler, M.; Lebigot, I.; Djelti, F.; Tourdès, A.; Gestraud, P.; et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R101. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Jagani, Z.; Kay, X.X.; Loo, A.; Dorsch, M.; Yao, Y.M.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef]
- Janku, F.; Lee, J.J.; Tsimberidou, A.M.; Hong, D.S.; Naing, A.; Falchook, G.S.; Fu, S.; Luthra, R.; Garrido-Laguna, I.; Kurzrock, R. PIK3CA mutations frequently coexist with ras and braf mutations in patients with advanced cancers. PLoS ONE 2011, 6, e22769. [Google Scholar] [CrossRef]
- She, Q.B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 Is a Key Effector of the Oncogenic Activation of the AKT and ERK Signaling Pathways that Integrates Their Function in Tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Gounder, M.; Rodon, J.; Janku, F.; Lolkema, M.P.; Stephenson, J.J.; Bedard, P.L.; Schuler, M.; Sessa, C.; LoRusso, P.; et al. Phase Ib Study of Combination Therapy with MEK Inhibitor Binimetinib and Phosphatidylinositol 3-Kinase Inhibitor Buparlisib in Patients with Advanced Solid Tumors with RAS/RAF Alterations. Oncologist 2020, 25, e160–e169. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; Von Hoff, D.D.; Eskens, F.; Blumenschein, G.; Richards, D.; Genvresse, I.; Reschke, S.; Granvil, C.; Skubala, A.; Peña, C.; et al. Phase Ib Trial of the PI3K Inhibitor Copanlisib Combined with the Allosteric MEK Inhibitor Refametinib in Patients with Advanced Cancer. Target. Oncol. 2020, 15, 163–174. [Google Scholar] [CrossRef]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef]
- Zawistowski, J.S.; Bevill, S.M.; Goulet, D.R.; Stuhlmiller, T.J.; Beltran, A.S.; Olivares-Quintero, J.F.; Singh, D.; Sciaky, N.; Parker, J.S.; Rashid, N.U.; et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov. 2017, 7, 302–321. [Google Scholar] [CrossRef]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 2016, 6, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.S.; Zhou, J.; Bin Liu, J.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; De Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. Shp2 inhibition prevents adaptive resistance to mek inhibitors in multiple cancer models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Adamopoulos, C.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019, 26, 65–78.e5. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Research article combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600E-mutant colorectal cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef]
- Fey, D.; Croucher, D.R.; Kolch, W.; Kholodenko, B.N. Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front. Physiol. 2012, 3, 355. [Google Scholar] [CrossRef]
- Xue, Z.; Vis, D.J.; Bruna, A.; Sustic, T.; Van Wageningen, S.; Batra, A.S.; Rueda, O.M.; Bosdriesz, E.; Caldas, C.; Wessels, L.F.A.; et al. MAP3K1 and MAP2K4 mutations are associated with sensitivity to MEK inhibitors in multiple cancer models. Cell Res. 2018, 28, 719–729. [Google Scholar] [CrossRef]
- Galiè, M. RAS as Supporting Actor in Breast Cancer. Front. Oncol. 2019, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.; Bergin, O.; Bianchi, A.; McNally, S.; Martin, F. Key signalling nodes in mammary gland development and cancer. Mitogen-activated protein kinase signalling in experimental models of breast cancer progression and in mammary gland development. Breast Cancer Res. 2009, 11, 209. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation Class | Common Sites | Functional Effect * |
|---|---|---|---|
| RAS [3,29] | 1 | G12 | Reduced GTP hydrolysis (reduced GAPs function and RAS intrinsic GTPase activity) |
| 2 | G13, K117, A146 | Increased nucleotide exchange activity (increased GEFs activity and RAS intrinsic nucleotide exchange activity) | |
| 3 | A59, Q61 | Hybrid (reduced GTP hydrolysis and increased nucleotide exchange) | |
| 4 | Multiple | To be determined | |
| BRAF [28] | I | V600 | Constitutively active BRAF functioning as a monomer (“activators”) |
| II | K601E, L597Q, G469A, fusions and in-frame deletions | Constitutively active mutant BRAF dimers (“activators”) | |
| III | D594, G466 | Impaired kinase activity; amplify signals from wild-type RAS, forming mutant/wild-type RAF heterodimers (“amplifiers”) | |
| MEK [28,30] | I | Rare in-frame deletions | RAF-independent: activate ERK independent of upstream signaling (“activators”) |
| II | Not concentrated in hotspots | RAF-regulated: mixed properties of “activators” and “amplifiers” | |
| III | Not concentrated in hotspots | RAF-dependent: increase ERK activation only in presence of active RAF (“amplifiers”) |
| Mutations (All Subtypes) | Frequency | References |
|---|---|---|
| KRAS | 0.6–1.0% | [69,70] |
| HRAS | 0.2–0.5% | [69,71] |
| NRAS | 0.1% | [69] |
| BRAF | 0.6% | [69] |
| NF1 | 3.0–3.8% | [5,72] |
| CNAs in TNBC | Frequency | References |
| KRAS | 32% | [5] |
| BRAF | 30% | [5] |
| Gene | Total n. Pts Analyzed * | Pts with Mutation | % Pts with Mutation | N. Mutations | Missense | Truncating ^ | Splice-Site |
|---|---|---|---|---|---|---|---|
| NF1 | 3694 | 74 | 2.0 | 78 | 0 | 57 | 21 |
| KRAS | 3694 | 18 | 0.5 | 18 | 18 | 0 | 0 |
| RASA1 | 1188 | 3 | 0.3 | 3 | 0 | 2 | 1 |
| HRAS | 3694 | 8 | 0.2 | 8 | 8 | 0 | 0 |
| BRAF | 3694 | 2 | 0.1 | 2 | 2 | 0 | 0 |
| ARHGAP35 | 1188 | 2 | 0.2 | 2 | 1 | 1 | 0 |
| JUN | 1188 | 2 | 0.2 | 2 | 2 | 0 | 0 |
| SPRED1 | 1188 | 1 | 0.1 | 1 | 0 | 1 | 0 |
| MAP2K1 | 1188 | 1 | 0.1 | 1 | 1 | 0 | 0 |
| MAPK1 | 1188 | 1 | 0.1 | 1 | 1 | 0 | 0 |
| DUSP4 | 1188 | 1 | 0.1 | 1 | 0 | 0 | 1 |
| NRAS | 3694 | 1 | 0.0 | 1 | 1 | 0 | 0 |
| Gene | Total n. Pts Analyzed * | Pts with CNA | % Pts with CNA | Predominant CNA Type |
|---|---|---|---|---|
| KRAS | 3363 | 76 | 2.3% | Gain/amplification |
| BRAF | 3363 | 59 | 1.8% | Gain/amplification |
| JUN | 3363 | 46 | 1.4% | Gain/amplification |
| RAF1 | 3363 | 39 | 1.2% | Gain/amplification |
| MAPK1 | 3363 | 28 | 0.8% | Gain/amplification |
| HRAS | 3363 | 25 | 0.7% | Gain/amplification |
| NF1 | 3363 | 18 | 0.5% | Deep deletion |
| SPRED1 | 3363 | 16 | 0.5% | Deep deletion |
| RASA1 | 3363 | 9 | 0.3% | Deep deletion |
| PTPN11 | 3363 | 3 | 0.1% | Deep deletion |
| ERF | 3363 | 1 | 0.0% | Deep deletion |
| Gene | Total n. Pts Analyzed | Pts with Mutation | % Pts with Mutation | N. Mutations | Missense | Truncating | Splice-Site |
|---|---|---|---|---|---|---|---|
| NF1 | 1121 | 40 | 4% | 45 | 0 | 35 | 10 |
| KRAS | 1121 | 9 | 0.8% | 9 | 9 | 0 | 0 |
| RASA1 | 1121 | 5 | 0.4% | 5 | 0 | 4 | 1 |
| BRAF | 1121 | 4 | 0.4% | 4 | 4 | 0 | 0 |
| MAP2K1 | 1121 | 3 | 0.3% | 3 | 3 | 0 | 0 |
| PTPN11 | 1121 | 3 | 0.3% | 3 | 3 | 0 | 0 |
| HRAS | 1121 | 2 | 0.2% | 2 | 2 | 0 | 0 |
| ARAF | 1121 | 1 | 0.1% | 1 | 1 | 0 | 0 |
| RAF1 | 1121 | 1 | 0.1% | 1 | 1 | 0 | 0 |
| JUN | 1121 | 1 | 0.1% | 1 | 1 | 0 | 0 |
| MAPK1 | 1121 | 1 | 0.1% | 1 | 1 | 0 | 0 |
| Gene | Total n. Pts Analyzed | Pts with CNA | % Pts with CNA | Predominant CNA Type |
|---|---|---|---|---|
| KRAS | 1121 | 20 | 1.8% | Amplification |
| RAF1 | 1121 | 9 | 0.8% | Amplification |
| JUN | 1121 | 8 | 0.7% | Amplification |
| MAPK1 | 1121 | 7 | 0.6% | Amplification |
| HRAS | 1121 | 7 | 0.6% | Amplification |
| BRAF | 1121 | 2 | 0.2% | Amplification |
| SPRED1 | 331 | 5 | 1.5% | Deep deletion |
| NF1 | 1121 | 10 | 0.9% | Deep deletion |
| ERF | 331 | 3 | 0.9% | Deep deletion |
| RASA1 | 1121 | 5 | 0.4% | Deep deletion |
| PTPN11 | 1121 | 1 | 0.1% | Deep deletion |
| Variable | Levels | Coefficient (=log(HR)) | SE Coefficient | p |
|---|---|---|---|---|
| Tumor classification | ||||
| T1 | 0 | |||
| T2 | 0.338 | 0.093 | 0.0003 | |
| T3-4 | 0.600 | 0.170 | 0.0004 | |
| Nodal classification | ||||
| N0 | 0 | |||
| N1 | 0.418 | 0.099 | 2.48 × 10−5 | |
| N2 | 0.983 | 0.125 | 3.79 × 10−15 | |
| N3 | 1.471 | 0.147 | <2 × 10−16 | |
| Grading | ||||
| G1 | 0 | |||
| G2 | −0.150 | 0.573 | 0.793 | |
| G3 | 0.559 | 0.566 | 0.324 | |
| tt (grading) | ||||
| G1 | 0 | |||
| G2 | 0.340 | 0.302 | 0.261 | |
| G3 | 0.009 | 0.302 | 0.975 | |
| PAM50 subtype | ||||
| Luminal A | 0 | |||
| Luminal B | 0.923 | 0.331 | 0.0053 | |
| HER2 | 2.038 | 0.348 | 4.66 × 10−9 | |
| Basal | 2.686 | 0.357 | 5.15 × 10−14 | |
| Claudin-low | 1.666 | 0.393 | 2.24 × 10−5 | |
| Normal | 1.054 | 0.465 | 0.0235 | |
| tt (PAM50 subtype) | ||||
| Luminal A | 0 | |||
| Luminal B | −0.163 | 0.170 | 0.3364 | |
| HER2 | −0.719 | 0.192 | 0.0002 | |
| Basal | −1.469 | 0.218 | 1.54 × 10−11 | |
| Claudin-low | −0.856 | 0.224 | 0.0001 | |
| Normal | −0.248 | 0.241 | 0.3030 | |
| NF1 gene alteration | ||||
| Negative | 0 | |||
| Positive | 1.237 | 0.365 | 0.0007 | |
| tt (NF1 alteration) | ||||
| Negative | 0 | |||
| Positive | −0.671 | 0.277 | 0.0153 | |
| RAF1 gene alteration | ||||
| Negative | 0 | |||
| Positive | 0.726 | 0.308 | 0.0184 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocca, A.; Braga, L.; Volpe, M.C.; Maiocchi, S.; Generali, D. The Predictive and Prognostic Role of RAS–RAF–MEK–ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets. Cancers 2022, 14, 5306. https://doi.org/10.3390/cancers14215306
Rocca A, Braga L, Volpe MC, Maiocchi S, Generali D. The Predictive and Prognostic Role of RAS–RAF–MEK–ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets. Cancers. 2022; 14(21):5306. https://doi.org/10.3390/cancers14215306
Chicago/Turabian StyleRocca, Andrea, Luca Braga, Maria Concetta Volpe, Serena Maiocchi, and Daniele Generali. 2022. "The Predictive and Prognostic Role of RAS–RAF–MEK–ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets" Cancers 14, no. 21: 5306. https://doi.org/10.3390/cancers14215306
APA StyleRocca, A., Braga, L., Volpe, M. C., Maiocchi, S., & Generali, D. (2022). The Predictive and Prognostic Role of RAS–RAF–MEK–ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets. Cancers, 14(21), 5306. https://doi.org/10.3390/cancers14215306