


Exploring the Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
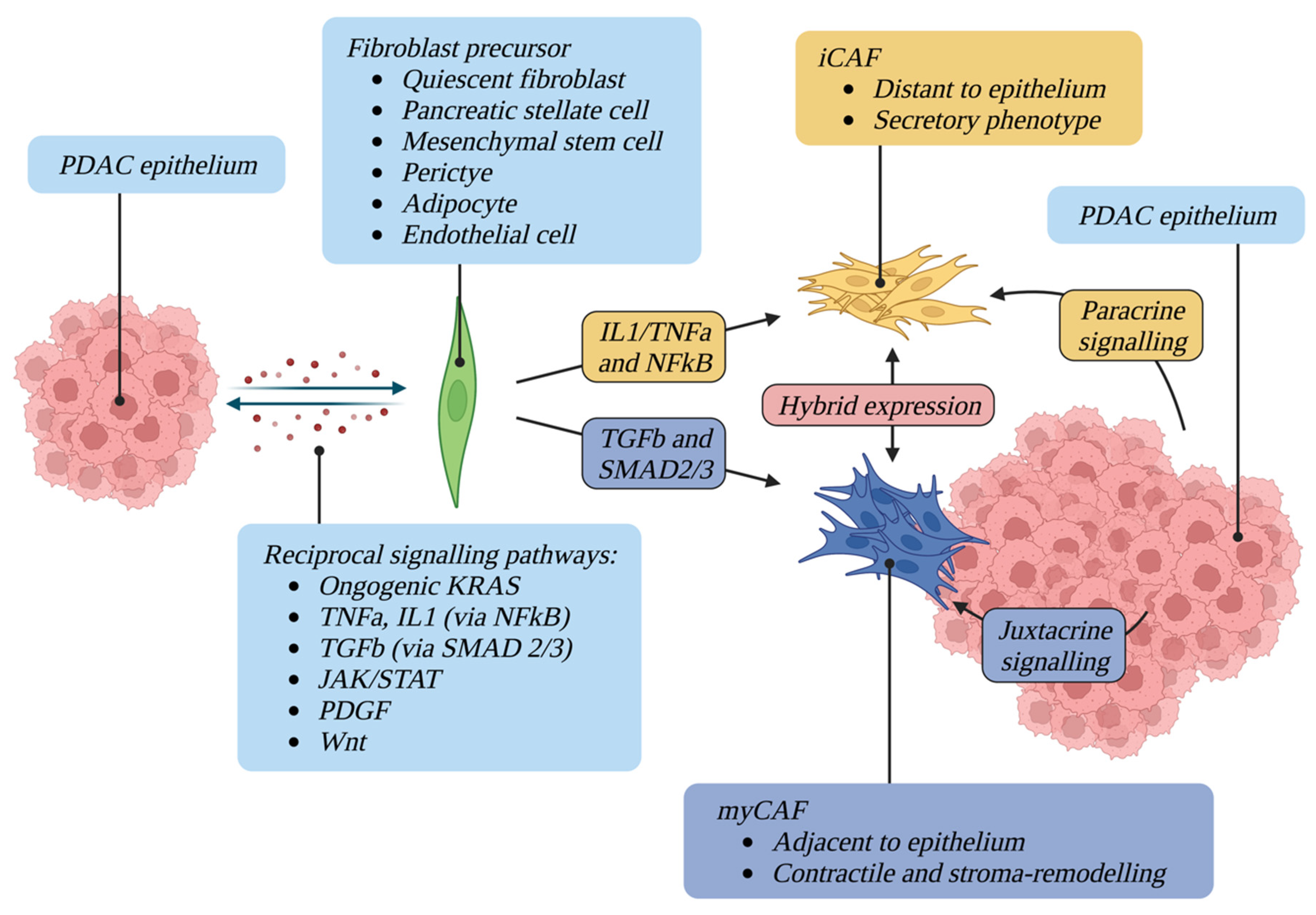
2. CAFs Have Multiple Cells of Origin and Activating Factors
2.1. Origin and Function of Fibroblasts in the Normal Pancreas
2.2. CAF Activating Factors
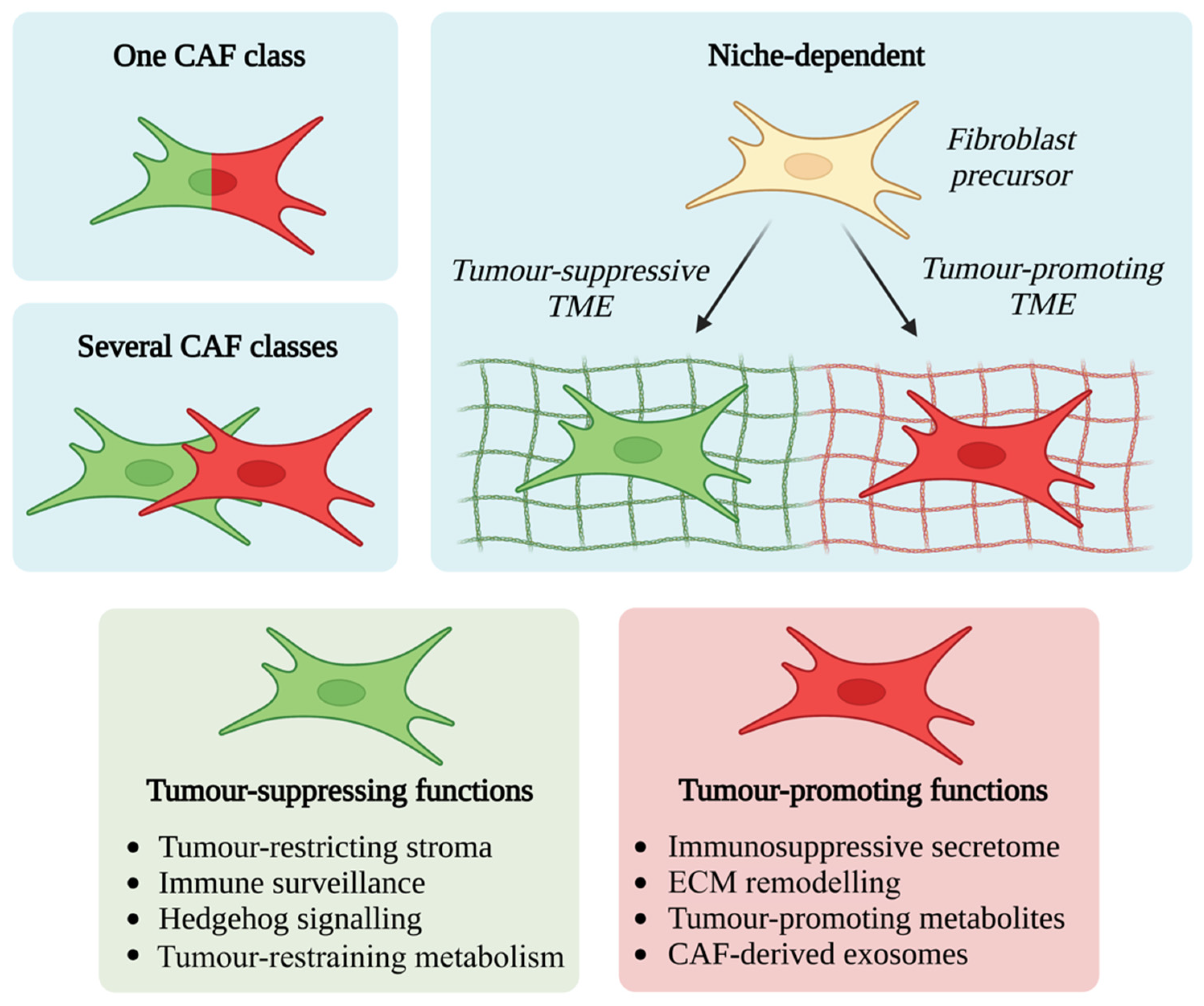
3. CAFs Exhibit Significant Functional and Phenotypic Heterogeneity
3.1. Myofibroblastic CAFs
3.2. Inflammatory CAFs
3.3. Antigen-Presenting CAFs
3.4. Metabolic CAFs
3.5. Complement-Secreting CAFs
3.6. CAF Subtype Markers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | Marker | Name | Location | Ref. |
|---|---|---|---|---|
| panCAF | FAP | Fibroblast activation protein | Cell surface | [56] |
| PDPN | Podoplanin | Cell surface | [15] | |
| Vim | Vimentin | Cytoskeleton | [15] | |
| DCN | Decorin | Extracellular matrix | [15] | |
| myCAF | αSMA (Acta2) | α-Smooth muscle actin | Cytoskeleton | [56] |
| CTGF | Connective tissue growth factor | Extracellular matrix | [56] | |
| COL1A1 | Type 1 collagen (alpha 1) | Extracellular matrix | [56] | |
| pSMAD | NA | Cytoplasm/nucleus | [72] | |
| LRRC15 | Leucine-rich repeat-containing 15 | Cell surface | [16] | |
| POSTN | Periostin | Extracellular matrix | [57] | |
| iCAF | IL6 | Interleukin 6 | Secreted cytokine | [56] |
| IL11 | Interleukin 11 | Secreted cytokine | [56] | |
| LIF | Leukaemia inhibitory factor | Secreted cytokine | [56] | |
| PDGFR | Platelet-derived growth factor receptor | Cell surface | [56] | |
| CXCL1 | CXC-motif chemokine ligand 1 | Secreted cytokine | [56] | |
| CXCL2 | CXC-motif chemokine ligand 2 | Secreted cytokine | [56] | |
| Ly6C | Lymphocyte antigen 6c | Cell surface | [15] | |
| pSTAT3 | Signal transducer and activation of transcription | Cytoplasm / nucleus | [72] | |
| ApoD | Apolipoprotein D | Cytoplasm | [57] | |
| CXCL12 | CXC-motif chemokine ligand 12 | Secreted cytokine | [59] | |
| apCAF | HLA-DR/-DP/-DR | Human leucocyte antigen | Cell surface | [15] |
| CD74 | NA | Cell surface | [15] | |
| SLPI | Secretory leucocyte protease inhibitor | Extracellular matrix | [15] | |
| SAA3 | Serum amyloid A3 | Secreted apolipoprotein | [15] | |
| S100A4 (FSP1) | Fibroblast-specific protein 1 | Cytoplasm | [68] | |
| meCAF | PLA2G2A | Phospholipase A2 | Cell surface | [57] |
| CRABP2 | Cellular retinoic acid binding protein | Cytoplasm | [57] | |
| Lineage | CD105 | Endoglin | Cell surface | [60] |
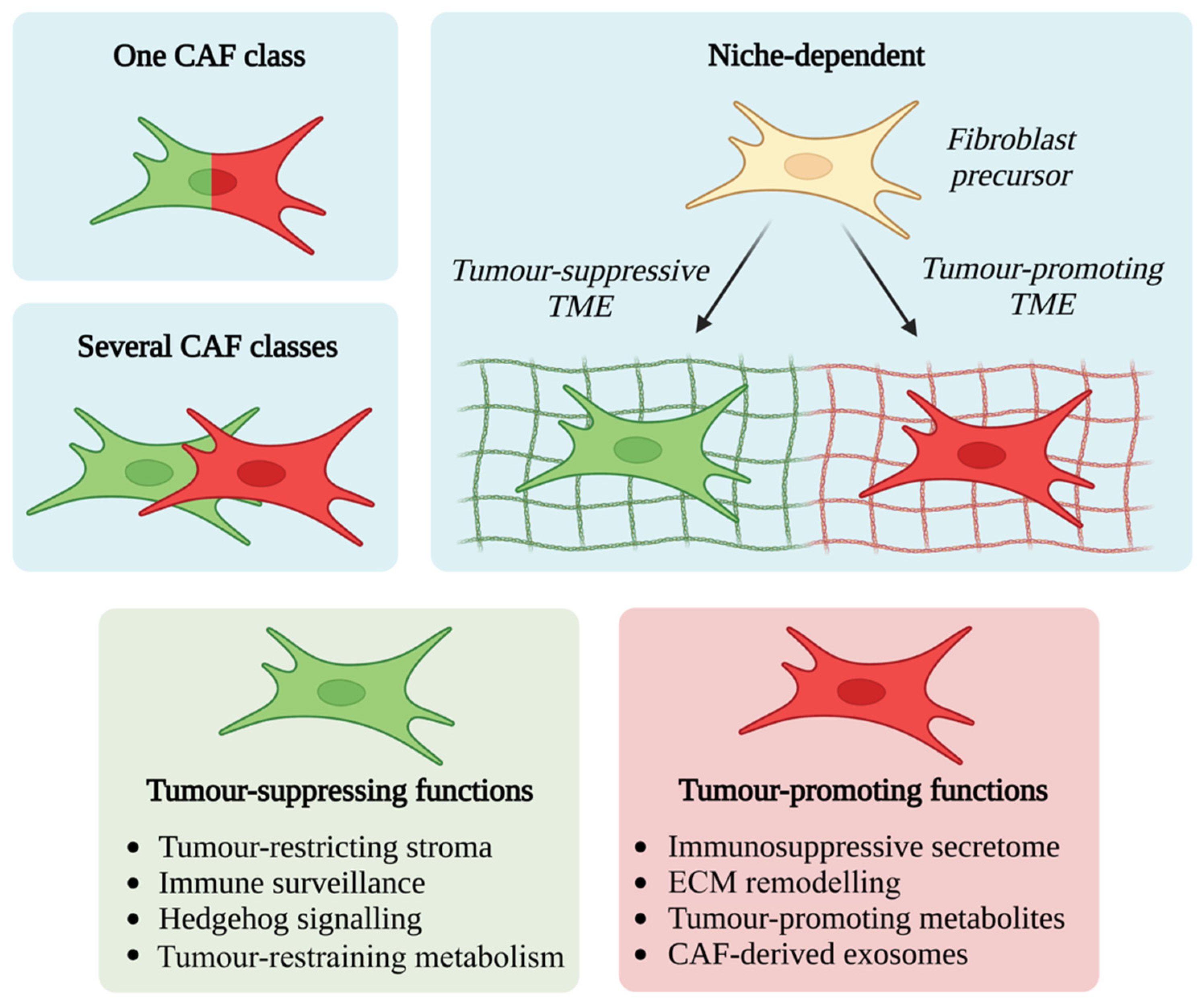
4. CAF Functional Diversity: Friend or Foe?
4.1. Tumour-Promoting CAF Functions
4.2. Tumour-Suppressing CAF Functions
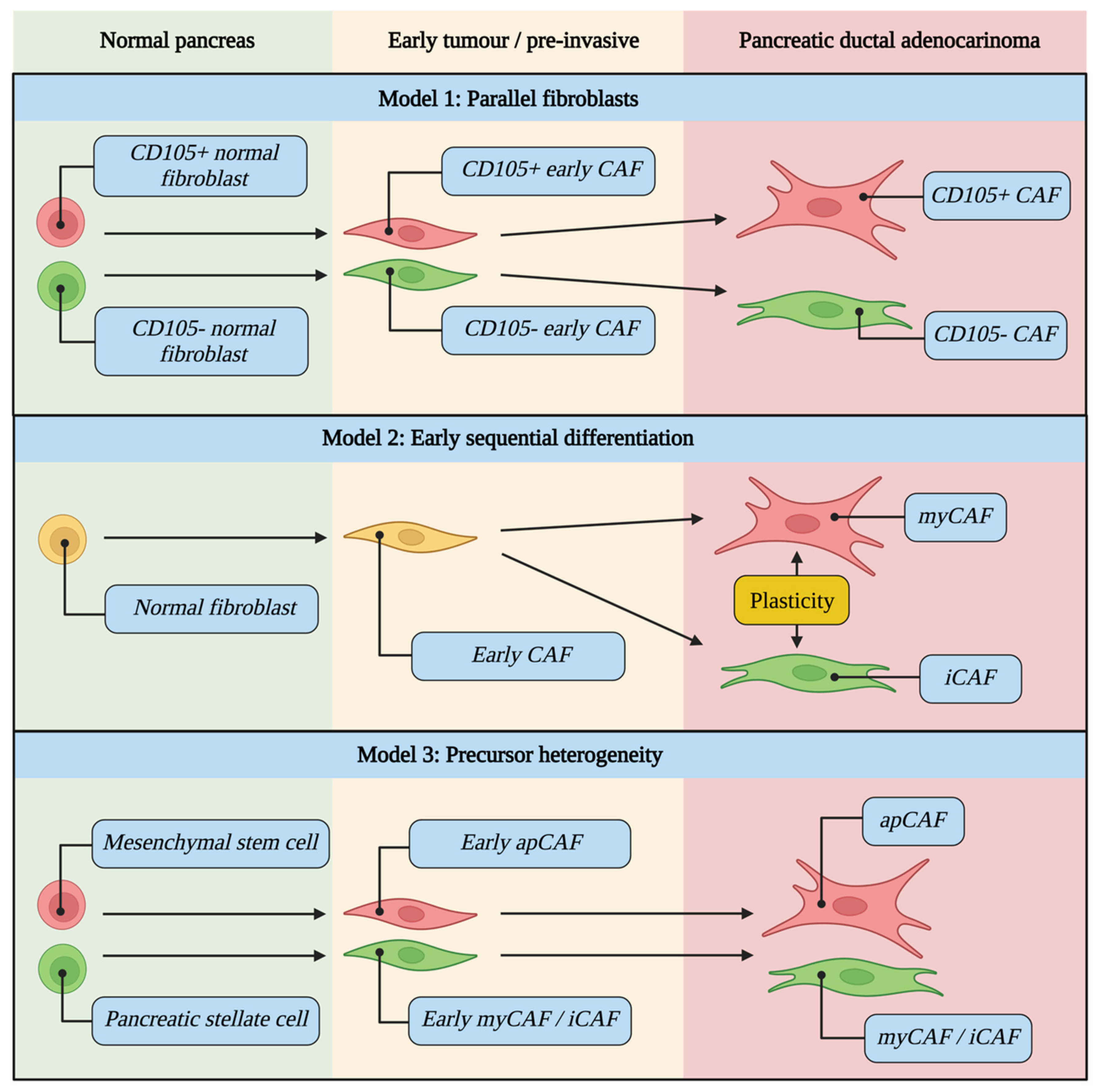
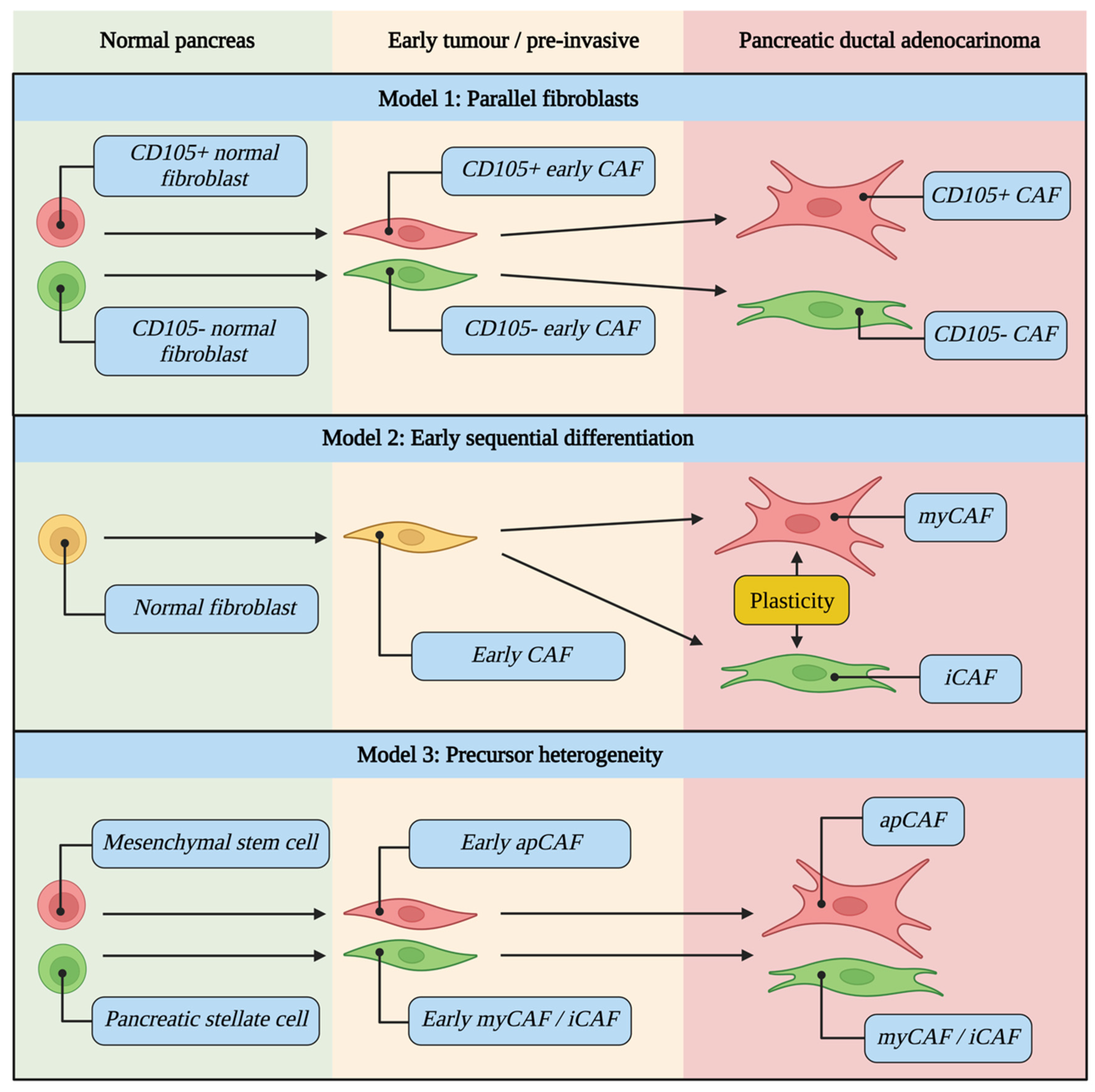
5. CAFs Evolve during Tumour Development and Are Differentially Activated
5.1. CAF Evolutionary Models
5.2. Drivers of CAF Differentiation
6. Tumour-Stroma Crosstalk Influences the TME and Clinical Phenotype
6.1. Crosstalk between CAFs, Immune Cells and Epithelium
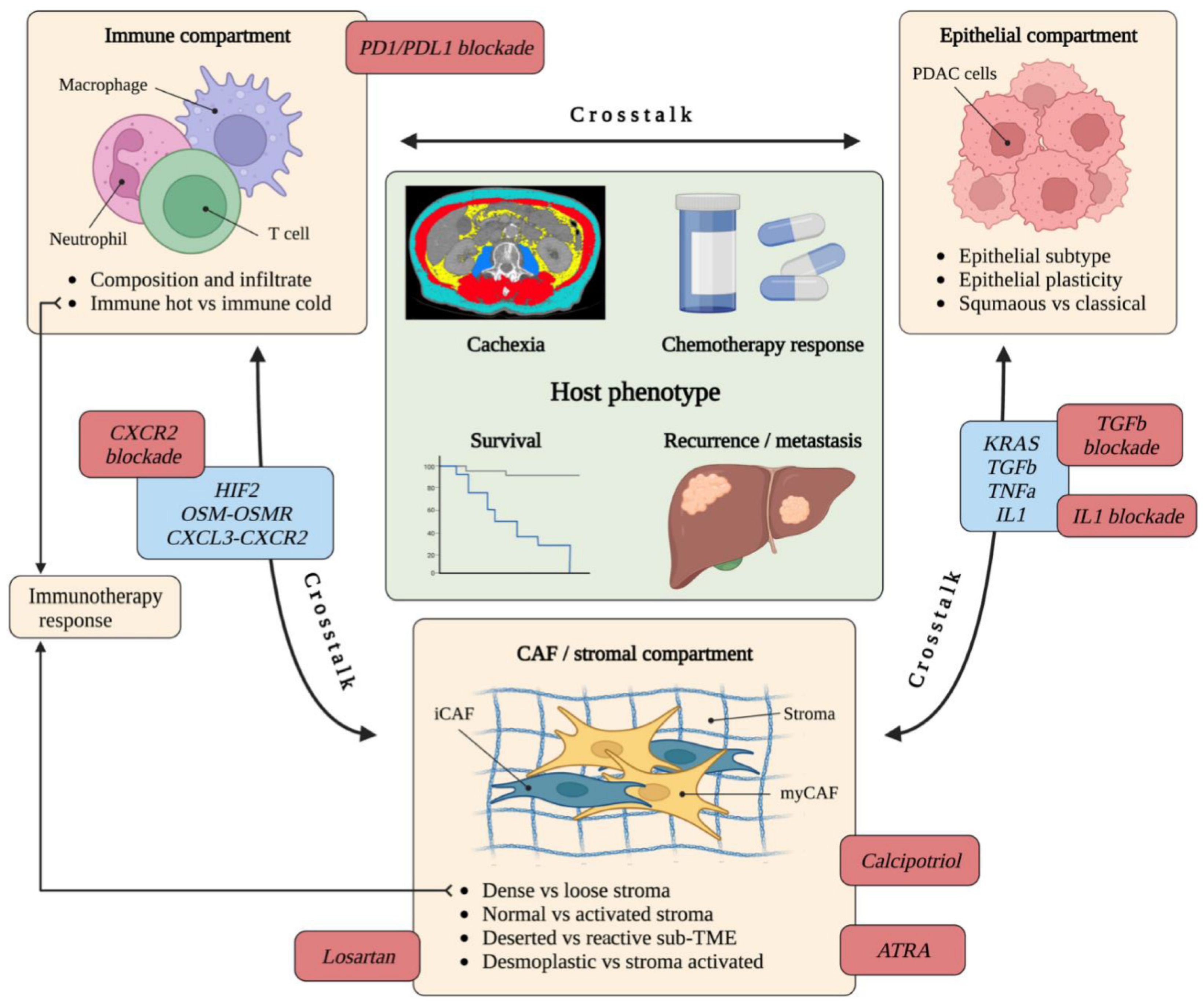
6.2. The Relationship between CAFs, Stromal Subtype and Epithelial Subtype
7. The Role of CAFs in Therapy Response
7.1. Immunotherapy
7.2. Chemotherapy and Radiotherapy
8. Therapeutic Targeting of CAFs Yields Mixed Results
8.1. PSC Reprogramming and Stromal Remodelling
8.2. Targeting Cytokines
8.3. Other Potential Targetable Pathways
9. Discussion and Future Directions
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. International Agency for Research on Cancer—GLOBOCAN 2020. Available online: https://gco.iarc.fr/ (accessed on 30 March 2021).
- Cancer Research, U.K. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer (accessed on 4 April 2022).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-Associated Stromal Fibroblasts Promote Pancreatic Tumor Progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein–α. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Sun, B.-F.; Chen, C.-Y.; Zhou, J.-Y.; Chen, Y.-S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.-S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Die Cellularpathologie in Ihrer Begrundung Auf Physiologische Und Pathologische Gewebelehre; August Hirschwald: Berlin, Germany, 1858. [Google Scholar]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Huang, H.; Brekken, R.A. Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am. J. Physiol. Physiol. 2020, 319, C233–C243. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schneider, E.; Groß, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.; Mirza, T.A.; Feakins, R.M.; Seedhar, A.; Elia, G.; Hart, I.R.; Kocher, H.M. Organotypic Culture Model of Pancreatic Cancer Demonstrates that Stromal Cells Modulate E-Cadherin, β-Catenin, and Ezrin Expression in Tumor Cells. Am. J. Pathol. 2009, 175, 636–648. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.E.; Scales, M.K.; Allen, B.L.; Di Magliano, M.P. Pancreatic Fibroblast Heterogeneity: From Development to Cancer. Cells 2020, 9, 2464. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic Acid–Induced Pancreatic Stellate Cell Quiescence Reduces Paracrine Wnt–β-Catenin Signaling to Slow Tumor Progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.V.; Mielgo, A. Macrophages and Fibroblasts, Key Players in Cancer Chemoresistance. Front. Cell Dev. Biol. 2018, 6, 131. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; Mccaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Geleta, B.; Tout, F.S.; Lim, S.C.; Sahni, S.; Jansson, P.J.; Apte, M.V.; Richardson, D.R.; Kovačević, Ž. Targeting Wnt/tenascin C-mediated cross talk between pancreatic cancer cells and stellate cells via activation of the metastasis suppressor NDRG1. J. Biol. Chem. 2022, 298. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 2010, 17, 523. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Tjomsland, V.; Spångeus, A.; Välilä, J.; Sandström, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-stroma IL1β-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Bassi, D.E.; Klein-Szanto, A.J.; Cukierman, E. Stroma-Derived Three-Dimensional Matrices Are Necessary and Sufficient to Promote Desmoplastic Differentiation of Normal Fibroblasts. Am. J. Pathol. 2005, 167, 475–488. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.; Luong, T.; Cao, X.; Han, B.; Shah, N.; Franco-Barraza, J.; Han, L.; Shenoy, V.; Lelkes, P.; Cukierman, E. Rigidity controls human desmoplastic matrix anisotropy to enable pancreatic cancer cell spread via extracellular signal-regulated kinase 2. Matrix Biol. 2018, 81, 50–69. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.L.; Sholl, L.M.; Ben-Aharon, I.; Beck, A.H.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.M.; Whitesell, L.; et al. The Reprogramming of Tumor Stroma by HSF1 Is a Potent Enabler of Malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.; Tchou, J.; et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ Pancreatic Stellate Cells Enhance the Progression of Pancreatic Cancer. Gastroenterology 2010, 139, 1041–1051.e8. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016, 3, 346–360.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Bernard, V.; Semaan, A.; Huang, J.; Lucas, F.A.S.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244.e20. [Google Scholar]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2018, 248, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Djurec, M.; Graña, O.; Lee, A.; Troulé, K.; Espinet, E.; Cabras, L.; Navas, C.; Blasco, M.T.; Martín-Díaz, L.; Burdiel, M.; et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E1147–E1156. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. eBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef]
- Hirayama, K.; Kono, H.; Nakata, Y.; Akazawa, Y.; Wakana, H.; Fukushima, H.; Fujii, H. Expression of podoplanin in stromal fibroblasts plays a pivotal role in the prognosis of patients with pancreatic cancer. Surg. Today 2018, 48, 110–118. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, G.; Levi-Galibov, O.; David, E.; Bornstein, C.; Giladi, A.; Dadiani, M.; Mayo, A.; Halperin, C.; Pevsner-Fischer, M.; Lavon, H.; et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Cancer 2020, 1, 692–708. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.-J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; LeBleu, V.S.; Carstens, J.; Sugimoto, H.; Zheng, X.; Malasi, S.; Saur, D.; Kalluri, R. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol. Med. 2018, 10, e9085. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Pinese, M.; Jamieson, N.B.; Scarlett, C.J.; Colvin, E.K.; Pajic, M.; Johns, A.L.; Humphris, J.L.; Wu, J.; Cowley, M.J. Precision Oncology in Surgery: Patient Selection for Operable Pancreatic Cancer. Ann. Surg. 2020, 272, 366–376. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef] [Green Version]
- Principe, D.R.; DeCant, B.; Mascariñas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2”, TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 2020, 48, 101417. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing Role of Contact Guidance in Intrastromal T-cell Trapping in Human Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer–Associated Fibroblasts. Cancer Discov. 2021, 12, 484–501. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [Green Version]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid–Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef]
- Parker, S.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef] [PubMed]
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Heiden, M.G.V.; et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res. 2019, 79, 5723–5733. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [Green Version]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot Clinical Trial of Hedgehog Pathway Inhibitor GDC-0449 (Vismodegib) in Combination with Gemcitabine in Patients with Metastatic Pancreatic Adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [Green Version]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2019, 122, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Olive Lab, IPI-926-03. 2012. Available online: https://www.olivelab.org/ipi-926-03.html (accessed on 4 August 2022).
- Torphy, R.J.; Wang, Z.; True-Yasaki, A.; Volmar, K.E.; Rashid, N.; Yeh, B.; Johansen, J.S.; Hollingsworth, M.A.; Yeh, J.J.; Collisson, E.A. Stromal Content Is Correlated With Tissue Site, Contrast Retention, and Survival in Pancreatic Adenocarcinoma. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, H.; Van Cutsem, E.; Bendell, J.; Hidalgo, M.; Li, C.-P.; Salvo, M.G.; Macarulla, T.; Sahai, V.; Sama, A.; Greeno, E.; et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Investig. New Drugs 2018, 36, 683–695. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Uppal, N.; Wagner, S.A.; Bendell, J.C.; Beck, J.T.; Wade III, S.M.; Nemunaitis, J.J.; Stella, P.J.; Pipas, J.M.; Wainberg, Z.A.; et al. Randomized, double-blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J. Clin. Oncol. 2015, 33, 4039–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, A.T.; Mitchell, J.T.; Kiemen, A.L.; Fujikura, K.; Fedor, H.; Gambichler, B.; Deshpande, A.; Wu, P.H.; Sidiropoulos, D.N.; Erbe, R.; et al. Spatial transcriptomics of FFPE pancreatic intraepithelial neoplasias reveals cellular and molecular alterations of progression to pancreatic ductal carcinoma. bioRxiv 2022. [Google Scholar] [CrossRef]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalappillil, J.S.; Caligiuri, G.; Nadella, S.; Alagesan, B.; Yordanov, G.N.; Shakiba, M.; Kaminow, B.; Hinds, J.; Perez-Mancera, P.A.; Preall, J.; et al. Abstract 3647: Oncogenic Kras drives cancer-associated fibroblast heterogeneity and substate changes in pancreatic cancer. Cancer Res. 2022, 82, 3647. [Google Scholar] [CrossRef]
- Caligiuri, G.; Thalappillil, J.; Hinds, J.; Courtois, E.T.; Flynn, W.F.; Robson, P.; Dobin, A.; Park, Y.; Tuveson, D.A. Abstract 1563: Spatial transcriptomics reveals heterogeneity and pathway dependencies of cancer associated fibroblasts in pancreatic ductal adenocarcinoma. Cancer Res. 2022, 82, 1563. [Google Scholar] [CrossRef]
- Verginadis, I.I.; Avgousti, H.; Monslow, J.; Skoufos, G.; Chinga, F.; Kim, K.; Leli, N.M.; Karagounis, I.V.; Bell, B.I.; Velalopoulou, A.; et al. A stromal Integrated Stress Response activates perivascular cancer-associated fibroblasts to drive angiogenesis and tumour progression. Nature 2022, 24, 940–953. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef]
- Chijimatsu, R.; Kobayashi, S.; Takeda, Y.; Kitakaze, M.; Tatekawa, S.; Arao, Y.; Nakayama, M.; Tachibana, N.; Saito, T.; Ennishi, D.; et al. Establishment of a reference single-cell RNA sequencing dataset for human pancreatic adenocarcinoma. iScience 2022, 25, 104659. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wu, J.; Wu, J.; Jing, X.; Du, Q.; Hui, X.; et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut 2021, 71, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Hogg, E.K.J.; Below, C.R.; Kononov, A.; Blanco-Gomez, A.; Heider, F.; Xu, J.; Hutton, C.; Zhang, X.; Scheidt, T.; et al. Heterocellular OSM-OSMR signalling reprograms fibroblasts to promote pancreatic cancer growth and metastasis. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.J.G.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology 2022, 162, 2018–2031. [Google Scholar] [CrossRef]
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Jagadeesh, K.A.; Guo, J.A.; Hoffman, H.I.; Yadollahpour, P.; Reeves, J.W.; Mohan, R.; Drokhlyansky, E.; Van Wittenberghe, N.; Ashenberg, O.; et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet. 2022, 54, 1178–1191. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Upstill-Goddard, R.; Legrini, A.; Biankin, A.V.; Jamieson, N.B.; Chang, D.K.; Allison, S.; Beraldi, D.; Cameron, E.; Cooke, S.L.; et al. Genomic and Molecular Analyses Identify Molecular Subtypes of Pancreatic Cancer Recurrence. Gastroenterology 2022, 162, 320–324.e4. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.L.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e1993. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Winter, P.S.; Navia, A.W.; Williams, H.L.; DenAdel, A.; Lowder, K.E.; Galvez-Reyes, J.; Kalekar, R.L.; Mulugeta, N.; Kapner, K.S.; et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021, 184, 6119–6137.e26. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: A phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Bednar, F.; Pasca di Magliano, M. Context-dependent immune responses explain pancreatic cancer immunoresistance. Cancer Cell 2020, 37, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.R.; Jain, R.; et al. Open-label, Phase I Study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.-Q.; Wang, M.-F.; Chen, H.-L.; Shang, D.; Das, J.K.; Song, J. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol. Cancer 2020, 19, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell–Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Candido, J.B.; Morton, J.P.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; de Sampaio, P.C.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022, 12, 1580–1597. [Google Scholar] [CrossRef] [PubMed]
- Versteijne, E.; van Dam, J.L.; Suker, M.; Janssen, Q.P.; Groothuis, K.; Akkermans-Vogelaar, J.M.; Besselink, M.G.; Bonsing, B.A.; Buijsen, J.; Busch, O.R.; et al. Neoadjuvant Chemoradiotherapy Versus Upfront Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Long-Term Results of the Dutch Randomized PREOPANC Trial. J. Clin. Oncol. 2022, 40, 1220–1230. [Google Scholar] [CrossRef]
- Froeling, F.E.; Kocher, H. Homeostatic Restoration of Desmoplastic Stroma Rather Than Its Ablation Slows Pancreatic Cancer Progression. Gastroenterology 2015, 148, 849–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; Desouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Ene–Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin–Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carapuça, E.F.; Gemenetzidis, E.; Feig, C.; Bapiro, E.T.; Williams, M.D.; Wilson, A.S.; Delvecchio, F.R.; Arumugam, P.; Grose, R.P.; Lemoine, N.R.; et al. Anti-stromal treatment together with chemotherapy targets multiple signalling pathways in pancreatic adenocarcinoma. J. Pathol. 2016, 239, 286–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, H.M.; Basu, B.; Froeling, F.E.; Sarker, D.; Slater, S.; Carlin, D.; Desouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. STARPAC clinical trial. Pancreatology 2020, 20, e17. [Google Scholar] [CrossRef]
- Precision-Panc. 2022. Available online: www.precisionpanc.org (accessed on 3 September 2022).
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Moro, C.F.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 17444. [Google Scholar] [CrossRef]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Mace, A.T.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2016, 67, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Z.; Ju, H.Q.; Aguilar, M.; Gocho, T.; Li, H.; Iida, T.; Lee, H.; Fan, X.; Zhou, H.; Ling, J.; et al. IL1 receptor antagonist inhibits pancreatic cancer growth by abrogating NF-κB activation. Clin. Cancer Res. 2016, 22, 1432–1444. [Google Scholar] [CrossRef] [Green Version]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, S.; et al. NFκB in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via up-regulation of CXCL12. Gastroenterology 2018, 155, 880–891.e8. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [Green Version]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFβ receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Ikeda, M.; Takahashi, H.; Kondo, S.; Lahn, M.M.F.; Ogasawara, K.; Benhadji, K.A.; Fujii, H.; Ueno, H. Phase 1b study of galunisertib in combination with gemcitabine in Japanese patients with metastatic or locally advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2017, 79, 1169–1177. [Google Scholar] [CrossRef]
- Melisi, D.; Oh, D.-Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef]
- Liu, D.; Gong, J.; Liu, T.; Li, K.; Yin, X.; Liu, Y.; Wang, Y.; Wang, L.; Wang, W.; Zhang, Y.; et al. Phase 1 study of SHR-1701, a bifunctional fusion protein targeting PD-L1 and TGF-β, in patients with advanced solid tumors. J. Clin. Oncol. 2021, 39, 2503. [Google Scholar] [CrossRef]
- Xie, J.; Meng, Z.; Chen, L.; Hua, Y.; Li, H. Phase II study of SHR-1701 combined with famitinib in the treatment of advanced pancreatic cancer or biliary tract cancer. J. Clin. Oncol. 2022, 40, 568. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizutani, Y.; Iida, T.; Ohno, E.; Ishikawa, T.; Kinoshita, F.; Kuwatsuka, Y.; Imai, M.; Shimizu, S.; Tsuruta, T.; Enomoto, A.; et al. Safety and efficacy of MIKE-1 in patients with advanced pancreatic cancer: A study protocol for an open-label phase I/II investigator-initiated clinical trial based on a drug repositioning approach that reprograms the tumour stroma. BMC Cancer 2022, 22, 1–11. [Google Scholar] [CrossRef]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGF βR2-mutant tumors exposes vulnerabilities to stromal TGF β blockade in pancreatic cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Carstens, J.L.; de Sampaio, P.C.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [Green Version]
- Ando, R.; Sakai, A.; Iida, T.; Kataoka, K.; Mizutani, Y.; Enomoto, A. Good and Bad Stroma in Pancreatic Cancer: Relevance of Functional States of Cancer-Associated Fibroblasts. Cancers 2022, 14, 3315. [Google Scholar] [CrossRef]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, J.R.; Lundberg, E.; Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet. 2022, 1–19. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryce, A.S.; Dreyer, S.B.; Froeling, F.E.M.; Chang, D.K. Exploring the Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2022, 14, 5302. https://doi.org/10.3390/cancers14215302
Bryce AS, Dreyer SB, Froeling FEM, Chang DK. Exploring the Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers. 2022; 14(21):5302. https://doi.org/10.3390/cancers14215302
Chicago/Turabian StyleBryce, Adam S., Stephan B. Dreyer, Fieke E. M. Froeling, and David K. Chang. 2022. "Exploring the Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer" Cancers 14, no. 21: 5302. https://doi.org/10.3390/cancers14215302
APA StyleBryce, A. S., Dreyer, S. B., Froeling, F. E. M., & Chang, D. K. (2022). Exploring the Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers, 14(21), 5302. https://doi.org/10.3390/cancers14215302