


Epithelial to Mesenchymal Transition as Mechanism of Progression of Pancreatic Cancer: From Mice to Men



{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Complexity of Metastatic Cascade, Embracing Stromal and Mesenchymal Traits, and the Importance of Its Application to Human Pancreatic Cancer
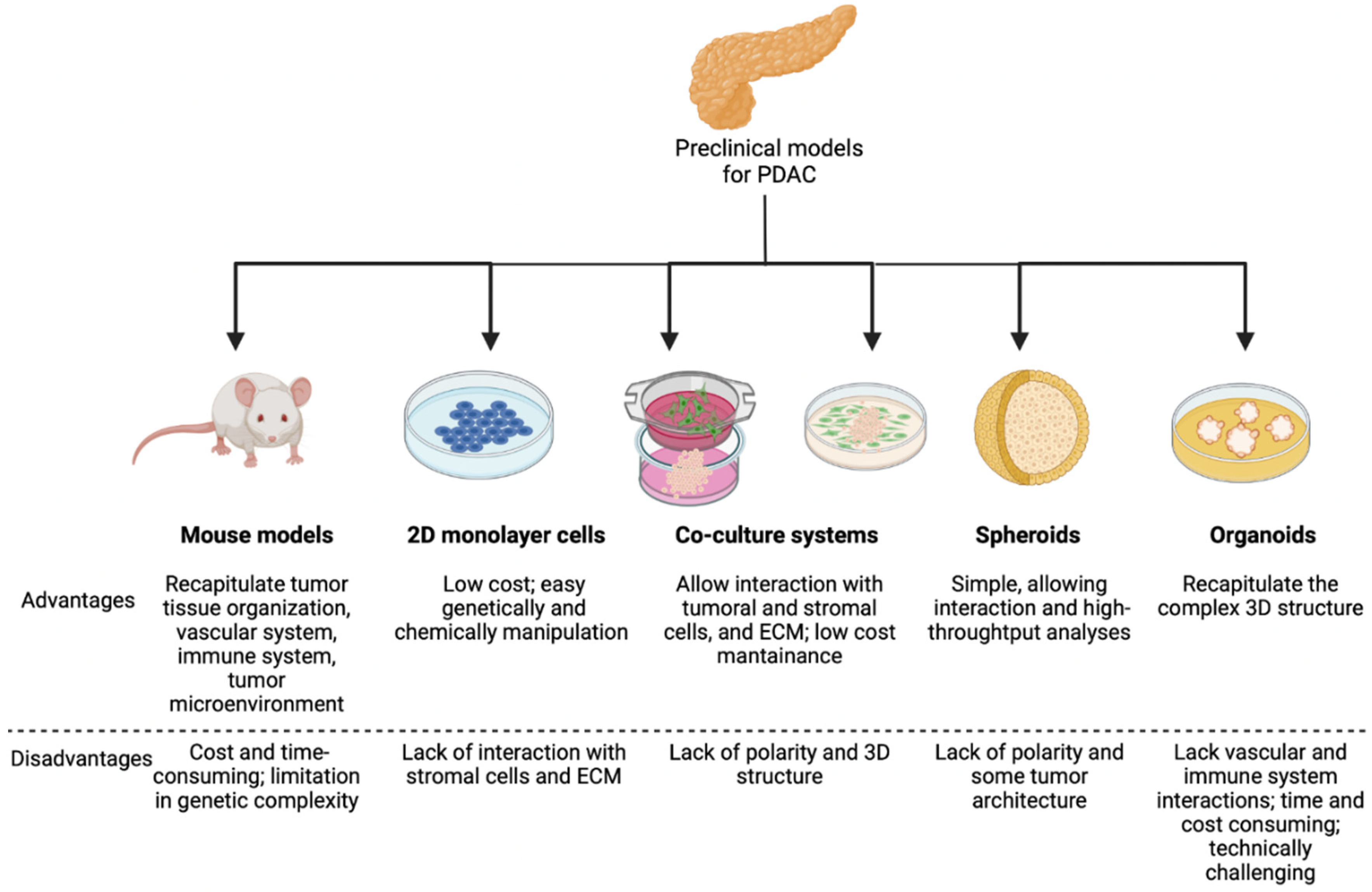
2. Models to Study EMT in Pancreatic Cancer
2.1. Genetically Engineered Mouse Models (GEMMs)
2.2. Heterotypic Human Cell Co-Culture Models
2.3. Spheroids
2.4. Organoids
3. EMT and Human Pancreatic Cancer
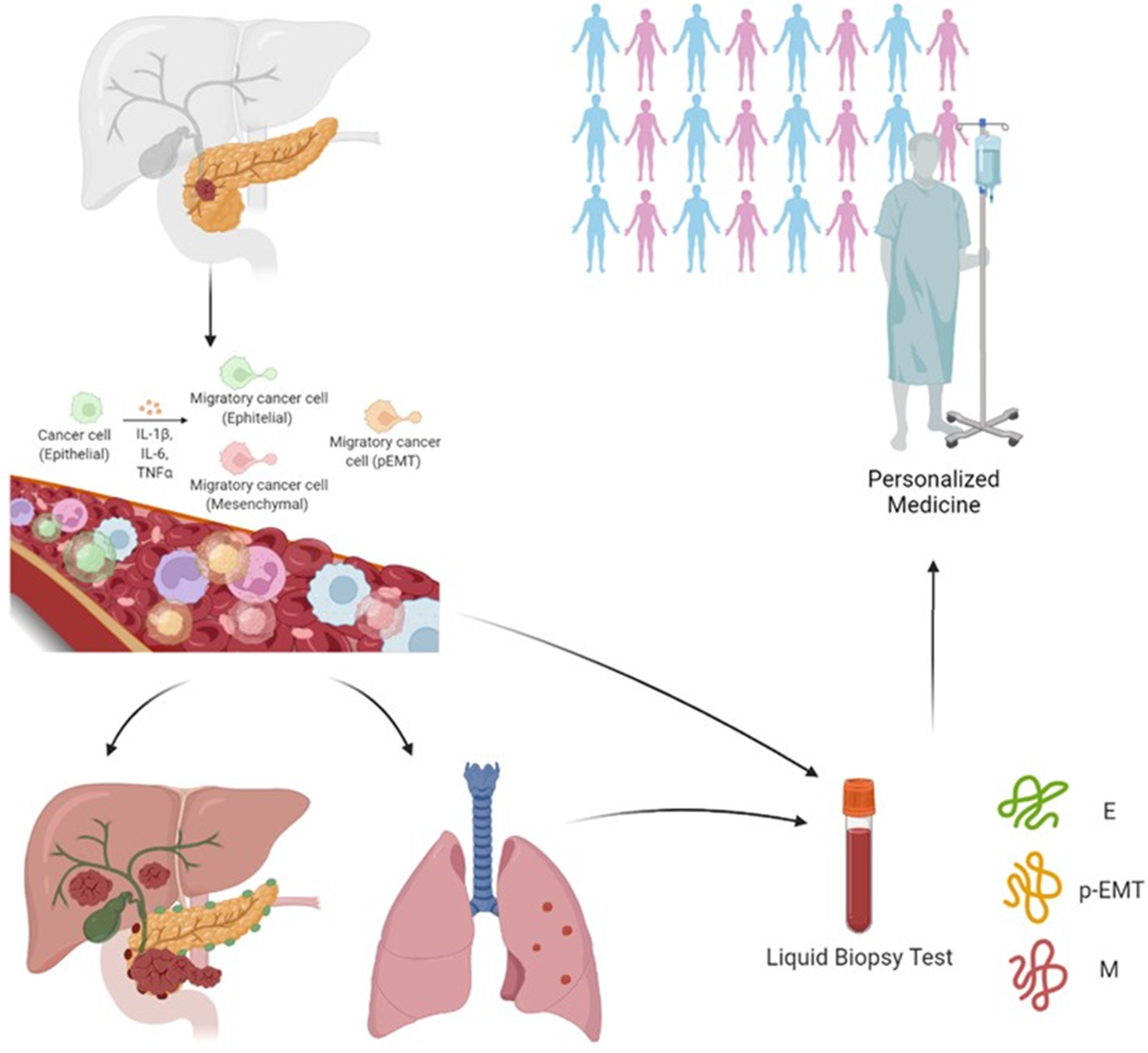
3.1. A Potential Route from Translational Studies to Personalized Medicine?
3.2. EMT, Chemoresistance and Choice of PDAC Treatment
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jones, S.; Chen, W.-D.; Parmigiani, G.; Diehl, F.; Beerenwinkel, N.; Antal, T.; Traulsen, A.; Nowak, M.A.; Siegel, C.; Velculescu, V.E.; et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 4283–4288. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-Cadherin Regulates Metastasis of Pancreatic Cancer In Vivo and Is Suppressed by a SNAIL/HDAC1/HDAC2 Repressor Complex. Gastroenterology 2009, 137, 361–371.e5. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Carstens, J.L.; Yang, S.; de Sampaio, P.C.; Zheng, X.; Barua, S.; McAndrews, K.M.; Rao, A.; Burks, J.K.; Rhim, A.D.; Kalluri, R. Stabilized epithelial phenotype of cancer cells in primary tumors leads to increased colonization of liver metastasis in pancreatic cancer. Cell Rep. 2021, 35, 108990. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- van Roey, R.; Brabletz, T.; Stemmler, M.P.; Armstark, I. Deregulation of Transcription Factor Networks Driving Cell Plasticity and Metastasis in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 753456. [Google Scholar] [CrossRef]
- Lazebnik, Y. What are the hallmarks of cancer? Nat. Rev. Cancer 2010, 10, 232–233. [Google Scholar] [CrossRef]
- Mantovani, A. The Yin-Yang of Tumor-Associated Neutrophils. Cancer Cell 2009, 16, 173–174. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Greco, L.; Rubbino, F.; Morelli, A.; Gaiani, F.; Grizzi, F.; de’Angelis, G.L.; Malesci, A.; Laghi, L. Epithelial to Mesenchy-mal Transition: A Challenging Playground for Translational Research. Current Models and Focus on TWIST1 Relevance and Gastrointestinal Cancers. Int. J. Mol. Sci. 2021, 22, 11469. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Crosstalk of Oncogenic Signaling Pathways during Epithelial–Mesenchymal Transition. Front. Oncol. 2014, 4, 358. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Landström, M.; Moustakas, A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial–mesenchymal transition by TGF-β. Futur. Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef] [PubMed]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta (BBA) Rev. Cancer 2009, 1796, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Deshmukh, A.P.; Hollander, P.D.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2020, 124, 259–269. [Google Scholar] [CrossRef]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e1. [Google Scholar] [CrossRef]
- Laghi, L.; Orbetegli, O.; Bianchi, P.; Zerbi, A.; Di Carlo, V.; Boland, C.R.; Malesci, A. Common occurrence of multiple K-RAS mutations in pancreatic cancers with associated precursor lesions and in biliary cancers. Oncogene 2002, 21, 4301–4306. [Google Scholar] [CrossRef][Green Version]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Laghi, L.; Beghelli, S.; Spinelli, A.; Bianchi, P.; Basso, G.; Di Caro, G.; Brecht, A.; Celesti, G.; Turri, G.; Bersani, S.; et al. Irrelevance of Microsatellite Instability in the Epidemiology of Sporadic Pancreatic Ductal Adenocarcinoma. PLoS ONE 2012, 7, e46002. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Laghi, L.; Malesci, A. Microsatellite Instability and Therapeutic Consequences in Colorectal Cancer. Dig. Dis. 2012, 30, 304–309. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.L.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e1993. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; Guerra, C.; Barbacid, M.; Tuveson, D.A. What We Have Learned About Pancreatic Cancer From Mouse Models. Gastroenterology 2012, 142, 1079–1092. [Google Scholar] [CrossRef]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Anver, M.R.; Biankin, A.V.; Boivin, G.P.; Furth, E.E.; Furukawa, T.; Klein, A.; Klimstra, D.S.; et al. Pathology of Genetically Engineered Mouse Models of Pancreatic Exocrine Cancer: Consensus Report and Recommendations. Cancer Res. 2006, 66, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.-H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16 Ink4a and the p19 Arf -p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef]
- Song, H.Y.; Wang, Y.; Lan, H.; Zhang, Y.X. Expression of Notch receptors and their ligands in pancreatic ductal adenocarcinoma. Exp. Ther. Med. 2018, 16, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, P.; Singh, P.; Wang, Z.; Li, L.; Eragamreddi, S.; Ozkan, S.; Ferrigno, O.; Prunier, C.; Razzaque, M.S.; Xu, K.; et al. TGIF 1 functions as a tumor suppressor in pancreatic ductal adenocarcinoma. EMBO J. 2019, 38, e101067. [Google Scholar] [CrossRef] [PubMed]
- Ostapoff, K.T.; Cenik, B.K.; Wang, M.; Ye, R.; Xu, X.; Nugent, D.; Hagopian, M.M.; Topalovski, M.; Rivera, L.B.; Carroll, K.D.; et al. Neutralizing Murine TGFβR2 Promotes a Differentiated Tumor Cell Phenotype and Inhibits Pancreatic Cancer Metastasis. Cancer Res. 2014, 74, 4996–5007. [Google Scholar] [CrossRef]
- Lan, L.; Evan, T.; Li, H.; Hussain, A.; Ruiz, E.J.; Thin, M.Z.; Ferreira, R.M.M.; Ps, H.; Riising, E.M.; Zen, Y.; et al. GREM1 is required to maintain cellular heterogeneity in pancreatic cancer. Nature 2022, 607, 163–168. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, e129212. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.-F.; Chen, C.-Y.; Zhou, J.-Y.; Chen, Y.-S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.-S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.-J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Recouvreux, M.V.; Moldenhauer, M.R.; Galenkamp, K.M.; Jung, M.; James, B.; Zhang, Y.; Lowy, A.; Bagchi, A.; Commisso, C. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Shichi, Y.; Sasaki, N.; Michishita, M.; Hasegawa, F.; Matsuda, Y.; Arai, T.; Gomi, F.; Aida, J.; Takubo, K.; Toyoda, M.; et al. Enhanced morphological and functional differences of pancreatic cancer with epithelial or mesenchymal characteristics in 3D culture. Sci. Rep. 2019, 9, 10871. [Google Scholar] [CrossRef]
- Gagliano, N.; Celesti, G.; Tacchini, L.; Pluchino, S.; Sforza, C.; Rasile, M.; Valerio, V.; Laghi, L.; Conte, V.; Procacci, P. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma: Characterization in a 3D-cell culture model. World J. Gastroenterol. 2016, 22, 4466–4483. [Google Scholar] [CrossRef] [PubMed]
- Bradney, M.J.; Venis, S.M.; Yang, Y.; Konieczny, S.F.; Han, B.; Bradney, M.J.; Venis, S.M.; Yang, Y.; Konieczny, S.F.; Han, B. A Biomimetic Tumor Model of Heterogeneous Invasion in Pancreatic Ductal Adenocarcinoma. Small 2020, 16, 1905500. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Shamir, E.R.; Coutinho, K.; Georgess, D.; Auer, M.; Ewald, A.J. Twist1-positive epithelial cells retain adhesive and proliferative capacity throughout dissemination. Biol. Open 2016, 5, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- Celesti, G.; Di Caro, G.; Bianchi, P.; Grizzi, F.; Basso, G.; Marchesi, F.; Doni, A.; Marra, G.; Roncalli, M.; Mantovani, A.; et al. Presence of Twist1-Positive Neoplastic Cells in the Stroma of Chromosome-Unstable Colorectal Tumors. Gastroenterology 2013, 145, 647–657.e15. [Google Scholar] [CrossRef] [PubMed]
- Sen-Yo, M.; Suehiro, Y.; Kaino, S.; Sakaida, I. TWIST1 hypermethylation is observed in pancreatic cancer. Biomed. Rep. 2012, 1, 31–33. [Google Scholar] [CrossRef]
- Gort, E.H.; Suijkerbuijk, K.P.; Roothaan, S.M.; Raman, V.; Vooijs, M.; van der Wall, E.; van Diest, P.J. Methylation of the TWIST1 Promoter, TWIST1 mRNA Levels, and Immunohistochemical Expression of TWIST1 in Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3325–3330. [Google Scholar] [CrossRef]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.-J.; Hotz, H.G. Epithelial to Mesenchymal Transition: Expression of the Regulators Snail, Slug, and Twist in Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef]
- Lahat, G.; Lubezky, N.; Loewenstein, S.; Nizri, E.; Gan, S.; Pasmanik-Chor, M.; Hayman, L.; Barazowsky, E.; Ben-Haim, M.; Klausner, J.M. Epithelial-to-Mesenchymal Transition (EMT) in Intraductal Papillary Mucinous Neoplasm (IPMN) is Associated with High Tumor Grade and Adverse Outcomes. Ann. Surg. Oncol. 2014, 21, 750–757. [Google Scholar] [CrossRef]
- Reichert, M.; Bakir, B.; Moreira, L.; Pitarresi, J.R.; Feldmann, K.; Simon, L.; Suzuki, K.; Maddipati, R.; Rhim, A.D.; Schlitter, A.M.; et al. Regulation of Epithelial Plasticity Determines Metastatic Organotropism in Pancreatic Cancer. Dev. Cell 2018, 45, 696–711.e8. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Fan, J.; Chen, R.; Ho, Y.-J.; Makohon-Moore, A.P.; LeComte, N.; Zhong, Y.; Hong, J.; Huang, J.; Sakamoto, H.; et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nat. Cancer 2020, 1, 59–74. [Google Scholar] [CrossRef]
- McDonald, O.G.; Maitra, A.; Hruban, R.H. Human Correlates of Provocative Questions in Pancreatic Pathology. Adv. Anat. Pathol. 2012, 19, 351–362. [Google Scholar] [CrossRef]
- Liu, X.; Huang, H.; Remmers, N.; Hollingsworth, M.A. Loss of E-cadherin and epithelial to mesenchymal transition is not required for cell motility in tissues or for metastasis. Tissue Barriers 2014, 2, e969112. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Flores-Figueroa, E.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Hong, J.; Iacobuzio-Donahue, C.A. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef] [PubMed]
- Brunton, H.; Caligiuri, G.; Cunningham, R.; Upstill-Goddard, R.; Bailey, U.-M.; Garner, I.M.; Nourse, C.; Dreyer, S.; Jones, M.; Moran-Jones, K.; et al. HNF4A and GATA6 Loss Reveals Therapeutically Actionable Subtypes in Pancreatic Cancer. Cell Rep. 2020, 31, 107625. [Google Scholar] [CrossRef]
- Sivakumar, S.; de Santiago, I.; Chlon, L.; Markowetz, F. Master Regulators of Oncogenic KRAS Response in Pancreatic Cancer: An Integrative Network Biology Analysis. PLoS Med. 2017, 14, e1002223. [Google Scholar] [CrossRef]
- Available online: https://gco.iarc.fr/today/online-analysis-table?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=39&type=1&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&group_cancer=1&include_nmsc=0&include_nmsc_other=1 (accessed on 23 March 2022).
- Pereira, S.P.; Oldfield, L.; Ney, A.; Hart, P.A.; Keane, M.G.; Pandol, S.J.; Li, D.; Greenhalf, W.; Jeon, C.Y.; Koay, E.J.; et al. Early detection of pancreatic cancer. Lancet Gastroenterol. Hepatol. 2020, 5, 698–710. [Google Scholar] [CrossRef]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef]
- Maitra, A.; Sharma, A.; Brand, R.E.; Van Den Eeden, S.K.; Fisher, W.E.; Hart, P.A.; Hughes, S.J.; Mather, K.J.; Pandol, S.J.; Park, W.G.; et al. A Prospective Study to Establish a New-Onset Diabetes Cohort: From the Consortium for the Study of Chronic Pancreatitis, Diabetes, and Pancreatic Cancer. Pancreas 2018, 47, 1244–1248. [Google Scholar] [CrossRef]
- Sharma, A.; Chari, S.T. Pancreatic Cancer and Diabetes Mellitus. Curr. Treat. Options Gastroenterol. 2018, 16, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Scott, F.I.; Boursi, B.; Reiss, K.A.; Williams, S.; Glick, H.; Yang, Y.-X. Cost-Effectiveness of a Risk-Tailored Pancreatic Cancer Early Detection Strategy Among Patients With New-Onset Diabetes. Clin. Gastroenterol. Hepatol. 2021, 20, 1997–2004.e7. [Google Scholar] [CrossRef] [PubMed]
- Gullo, L.; Tomassetti, P.; Migliori, M.; Casadei, R.; Marrano, D. Do Early Symptoms of Pancreatic Cancer Exist that Can Allow an Earlier Diagnosis? Pancreas 2001, 22, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.G.; Horsfall, L.; Rait, G.; Pereira, S.P. A case–control study comparing the incidence of early symptoms in pancreatic and biliary tract cancer. BMJ Open 2014, 4, e005720. [Google Scholar] [CrossRef] [PubMed]
- Hippisley-Cox, J.; Coupland, C. Identifying patients with suspected pancreatic cancer in primary care: Derivation and validation of an algorithm. Br. J. Gen. Pract. 2012, 62, e38–e45. [Google Scholar] [CrossRef] [PubMed]
- Stapley, S.; Peters, T.; Neal, R.D.; Rose, P.W.; Walter, F.; Hamilton, W. The risk of pancreatic cancer in symptomatic patients in primary care: A large case–control study using electronic records. Br. J. Cancer 2012, 106, 1940–1944. [Google Scholar] [CrossRef] [PubMed]
- Canto, M.I.; Harinck, F.; Hruban, R.H.; Offerhaus, G.J.; Poley, J.-W.; Kamel, I.; Nio, Y.; Schulick, R.S.; Bassi, C.; Kluijt, I.; et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2012, 62, 339–347. [Google Scholar] [CrossRef]
- Kenner, B.J.; Chari, S.T.; Cleeter, D.F.; Go, V.L.W. Early Detection of Sporadic Pancreatic Cancer. Pancreas 2015, 44, 686–692. [Google Scholar] [CrossRef]
- Sheel, A.R.G.; Harrison, S.; Sarantitis, I.; Nicholson, J.A.; Hanna, T.; Grocock, C.; Raraty, M.; Ramesh, J.; Farooq, A.; Costello, E.; et al. Identification of Cystic Lesions by Secondary Screening of Familial Pancreatic Cancer (FPC) Kindreds Is Not Associated with the Stratified Risk of Cancer. Am. J. Gastroenterol. 2019, 114, 155–164. [Google Scholar] [CrossRef]
- Kimura, W.; Moriya, T.; Hanada, K.; Abe, H.; Yanagisawa, A.; Fukushima, N.; Ohike, N.; Shimizu, M.; Hatori, T.; Fujita, N.; et al. Multicenter Study of Serous Cystic Neoplasm of the Japan Pancreas Society. Pancreas 2012, 41, 380–387. [Google Scholar] [CrossRef]
- Del Chiaro, M.; Verbeke, C.; Salvia, R.; Klöppel, G.; Werner, J.; McKay, C.; Friess, H.; Manfredi, R.; Van Cutsem, E.; Löhr, M.; et al. European experts consensus statement on cystic tumours of the pancreas. Dig. Liver Dis. 2013, 45, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Vege, S.S.; Ziring, B.; Jain, R.; Moayyedi, P.; Adams, M.A.; Dorn, S.D.; Dudley-Brown, S.L.; Flamm, S.L.; Gellad, Z.F.; Gruss, C.B.; et al. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015, 148, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, W. The clinical utility of the CA 19-9 tumor-associated antigen. Am. J. Gastroenterol. 1990, 85. [Google Scholar]
- Fahrmann, J.F.; Schmidt, C.M.; Mao, X.; Irajizad, E.; Loftus, M.; Zhang, J.; Patel, N.; Vykoukal, J.; Dennison, J.B.; Long, J.P.; et al. Lead-Time Trajectory of CA19-9 as an Anchor Marker for Pancreatic Cancer Early Detection. Gastroenterology 2021, 160, 1373–1383.e6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bamlet, W.R.; Oberg, A.L.; Chaffee, K.G.; Donahue, G.; Cao, X.J.; Chari, S.; Garcia, B.A.; Petersen, G.M.; Zaret, K.S. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Sci. Transl. Med. 2017, 9, eaah5583. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Ciprani, D.; O’Shea, A.; Liss, A.S.; Yang, R.; Fletcher-Mercaldo, S.; Mino-Kenudson, M.; Castillo, C.F.-D.; Weissleder, R. Extracellular Vesicle Analysis Allows for Identification of Invasive IPMN. Gastroenterology 2021, 160, 1345–1358.e11. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-X.; Yin, S.; Ma, L.; Wheeler, A.; Chen, Y.; Zhang, Y.; Liu, B.; Xiong, J.; Zhang, W.; Hu, J.; et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017, 27, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Mellby, L.D.; Nyberg, A.P.; Johansen, J.S.; Wingren, C.; Nordestgaard, B.G.; Bojesen, S.E.; Mitchell, B.L.; Sheppard, B.C.; Sears, R.C.; Borrebaeck, C.A. Serum Biomarker Signature-Based Liquid Biopsy for Diagnosis of Early-Stage Pancreatic Cancer. J. Clin. Oncol. 2018, 36, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Thege, F.; Santana, S.M.; Lannin, T.B.; Saha, T.N.; Tsai, S.; Maggs, L.; Kochman, M.L.; Ginsberg, G.G.; Lieb, J.G.; et al. Detection of Circulating Pancreas Epithelial Cells in Patients With Pancreatic Cystic Lesions. Gastroenterology 2014, 146, 647–651. [Google Scholar] [CrossRef]
- Pan, Y.; Li, D.; Yang, J.; Wang, N.; Xiao, E.; Tao, L.; Ding, X.; Sun, P.; Li, D. Portal Venous Circulating Tumor Cells Undergoing Epithelial-Mesenchymal Transition Exhibit Distinct Clinical Significance in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 757307. [Google Scholar] [CrossRef]
- Yamada, S.; Fuchs, B.C.; Fujii, T.; Shimoyama, Y.; Sugimoto, H.; Nomoto, S.; Takeda, S.; Tanabe, K.K.; Kodera, Y.; Nakao, A. Epithelial-to-mesenchymal transition predicts prognosis of pancreatic cancer. Surgery 2013, 154, 946–954. [Google Scholar] [CrossRef]
- Khoja, L.; Backen, A.; Sloane, R.; Menasce, L.; Ryder, D.; Krebs, M.; Board, R.; Clack, G.; Hughes, A.; Blackhall, F.; et al. A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br. J. Cancer 2011, 106, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Zoli, W.; Rigaud, M. Circulating tumor cells and epithelial, mesenchymal and stemness markers: Characterization of cell subpopulations. Ann. Transl. Med. 2014, 2, 109. [Google Scholar] [CrossRef]
- Kulemann, B.; Liss, A.S.; Warshaw, A.L.; Seifert, S.; Bronsert, P.; Glatz, T.; Pitman, M.B.; Hoeppner, J. KRAS mutations in pancreatic circulating tumor cells: A pilot study. Tumor Biol. 2015, 37, 7547–7554. [Google Scholar] [CrossRef]
- Dotan, E.; Alpaugh, R.K.; Ruth, K.; Negin, B.P.; Denlinger, C.S.; Hall, M.J.; Astsaturov, I.; McAleer, C.; Fittipaldi, P.; Thrash-Bingham, C.; et al. Prognostic Significance of MUC-1 in Circulating Tumor Cells in Patients With Metastatic Pancreatic Adenocarcinoma. Pancreas 2016, 45, 1131–1135. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Wang, Z.-R.; Chen, C.-L.; Di, L.; Bi, Z.-F.; Li, Z.-H.; Liu, Y.-M. Molecular detection of epithelial-mesenchymal transition markers in circulating tumor cells from pancreatic cancer patients: Potential role in clinical practice. World J. Gastroenterol. 2019, 25, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Semaan, A.; Bernard, V.; Kim, D.U.; Lee, J.J.; Huang, J.; Kamyabi, N.; Stephens, B.M.; Qiao, W.; Varadhachary, G.R.; Katz, M.H.; et al. Characterisation of circulating tumour cell phenotypes identifies a partial-EMT sub-population for clinical stratification of pancreatic cancer. Br. J. Cancer 2021, 124, 1970–1977. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.clinicaltrials.gov/ct2/show/NCT04323917 (accessed on 7 October 2022).
- Available online: https://worldwide.espacenet.com/patent/search?q=EP13197367 (accessed on 7 October 2022).
- Court, C.; Ankeny, J.S.; Sho, S.; Winograd, P.; Hou, S.; Song, M.; Wainberg, Z.A.; Girgis, M.D.; Graeber, T.G.; Agopian, V.G.; et al. Circulating Tumor Cells Predict Occult Metastatic Disease and Prognosis in Pancreatic Cancer. Ann. Surg. Oncol. 2018, 25, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Kulemann, B.; Rösch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Pancreatic cancer: Circulating Tumor Cells and Primary Tumors show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 4510. [Google Scholar] [CrossRef]
- Brugge, W.R. Endoscopic approach to the diagnosis and treatment of pancreatic disease. Curr. Opin. Gastroenterol. 2013, 29, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Kerner, C.; Wilfert, W.; Mueller, M.; Thiery, J.; Hauss, J.; Witzigmann, H. Detection of disseminated pancreatic cells by amplification of cytokeratin-19 with quantitative RT-PCR in blood, bone marrow and peritoneal lavage of pancreatic carcinoma patients. World J. Gastroenterol. 2007, 13, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, G.; Roskams, T.; van Pelt, J.; Houtmeyers, F.; Aerts, R.; Topal, B. Perioperative cancer cell dissemination detected with a real-time RT-PCR assay for EpCAM is not associated with worse prognosis in pancreatic ductal adenocarcinoma. BMC Cancer 2011, 11, 47. [Google Scholar] [CrossRef]
- Soeth, E.; Grigoleit, U.; Moellmann, B.; Röder, C.; Schniewind, B.; Kremer, B.; Kalthoff, H.; Vogel, I. Detection of tumor cell dissemination in pancreatic ductal carcinoma patients by CK 20 RT-PCR indicates poor survival. J. Cancer Res. Clin. Oncol. 2005, 131, 669–676. [Google Scholar] [CrossRef]
- Okubo, K.; Uenosono, Y.; Arigami, T.; Mataki, Y.; Matsushita, D.; Yanagita, S.; Kurahara, H.; Sakoda, M.; Kijima, Y.; Maemura, K.; et al. Clinical impact of circulating tumor cells and therapy response in pancreatic cancer. Eur. J. Surg. Oncol. (EJSO) 2017, 43, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhu, Y.; Zhang, Z.; Zhang, C.; Huang, X.; Yuan, Z. Clinical significance of pancreatic circulating tumor cells using combined negative enrichment and immunostaining-fluorescence in situ hybridization. J. Exp. Clin. Cancer Res. 2016, 35, 66. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Valero, V.; Saunders, T.; Blackford, A.L.; Griffin, J.F.; Poling, J.; Hruban, R.H.; Anders, R.A.; Herman, J.; Zheng, L.; et al. Circulating Tumor Cell Phenotype Predicts Recurrence and Survival in Pancreatic Adenocarcinoma. Ann. Surg. 2016, 264, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Blackford, A.L.; Weiss, M.J.; Cameron, J.L.; He, J.; Goggins, M.; Rasheed, Z.A.; Wolfgang, C.L.; Wood, L.D. Circulating Tumor Cells Expressing Markers of Tumor-Initiating Cells Predict Poor Survival and Cancer Recurrence in Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 2681–2690. [Google Scholar] [CrossRef]
- Kurihara, T.; Itoi, T.; Sofuni, A.; Itokawa, F.; Tsuchiya, T.; Tsuji, S.; Ishii, K.; Ikeuchi, N.; Tsuchida, A.; Kasuya, K.; et al. Detection of circulating tumor cells in patients with pancreatic cancer: A preliminary result. J. Hepato-Biliary-Pancreatic Surg. 2008, 15, 189–195. [Google Scholar] [CrossRef] [PubMed]
- de Albuquerque, A.; Kubisch, I.; Ernst, D.; Breier, G.; Stamminger, G.; Fersis, N.; Stölzel, U.; Boese-Landgraf, J.; Eichler, A.; Kaul, S. Development of a molecular multimarker assay for the analysis of circulating tumor cells in adenocarci-noma patients. Clin. Lab. 2012, 58, 373–384. [Google Scholar] [PubMed]
- Bissolati, M.; Sandri, M.T.; Burtulo, G.; Zorzino, L.; Balzano, G.; Braga, M. Portal vein-circulating tumor cells predict liver metastases in patients with resectable pancreatic cancer. Tumor Biol. 2014, 36, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, F.; Ning, N.; Chen, Q.; Yang, Z.; Guo, Y.; Xu, D.; Zhang, D.; Zhan, T.; Cui, W. Patterns of circulating tumor cells identified by CEP8, CK and CD45 in pancreatic cancer. Int. J. Cancer 2014, 136, 1228–1233. [Google Scholar] [CrossRef]
- Earl, J.; Garcia-Nieto, S.; Martinez-Avila, J.C.; Montans, J.; Sanjuanbenito, A.; Rodríguez-Garrote, M.; Lisa, E.; Mendía, E.; Lobo, E.; Malats, N.; et al. Circulating tumor cells (CTC) and KRAS mutant circulating free DNA (cfDNA) detection in peripheral blood as biomarkers in patients diagnosed with exocrine pancreatic cancer. BMC Cancer 2015, 15, 797. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzis, G.; Groot, V.P.; Yu, J.; Ding, D.; Teinor, J.A.; Javed, A.A.; Wood, L.D.; Burkhart, R.A.; Cameron, J.L.; Makary, M.A.; et al. Circulating Tumor Cells Dynamics in Pancreatic Adenocarcinoma Correlate With Disease Status. Ann. Surg. 2018, 268, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Schuth, S.; Le Blanc, S.; Krieger, T.G.; Jabs, J.; Schenk, M.; Giese, N.A.; Büchler, M.W.; Eils, R.; Conrad, C.; Strobel, O. Patient-specific modeling of stroma-mediated chemoresistance of pancreatic cancer using a three-dimensional organoid-fibroblast co-culture system. J. Exp. Clin. Cancer Res. 2022, 41, 312. [Google Scholar] [CrossRef]
- Hwang, H.J.; Oh, M.-S.; Lee, D.W.; Kuh, H.-J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wang, Z.; Li, Y.-Y.; Yu, B.-H.; Zhang, F.; Li, H.-Y. miR-33a suppresses the nuclear translocation of β-catenin to enhance gemcitabine sensitivity in human pancreatic cancer cells. Tumor Biol. 2015, 36, 9395–9403. [Google Scholar] [CrossRef]
- Martinelli, P.; Cañamero, M.; del Pozo, N.; Madriles, F.; Zapata, A.; Real, F.X. Gata6 is required for complete acinar differentiation and maintenance of the exocrine pancreas in adult mice. Gut 2012, 62, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.-H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef]
- Palamaris, K.; Felekouras, E.; Sakellariou, S. Epithelial to Mesenchymal Transition: Key Regulator of Pancreatic Ductal Adenocarcinoma Progression and Chemoresistance. Cancers 2021, 13, 5532. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.-L.; Bochnakian, A.; Toussaint, P.; Gilson, P.; Harlé, A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals 2021, 14, 740. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, L.; Rubbino, F.; Laghi, L. Epithelial to Mesenchymal Transition as Mechanism of Progression of Pancreatic Cancer: From Mice to Men. Cancers 2022, 14, 5797. https://doi.org/10.3390/cancers14235797
Greco L, Rubbino F, Laghi L. Epithelial to Mesenchymal Transition as Mechanism of Progression of Pancreatic Cancer: From Mice to Men. Cancers. 2022; 14(23):5797. https://doi.org/10.3390/cancers14235797
Chicago/Turabian StyleGreco, Luana, Federica Rubbino, and Luigi Laghi. 2022. "Epithelial to Mesenchymal Transition as Mechanism of Progression of Pancreatic Cancer: From Mice to Men" Cancers 14, no. 23: 5797. https://doi.org/10.3390/cancers14235797
APA StyleGreco, L., Rubbino, F., & Laghi, L. (2022). Epithelial to Mesenchymal Transition as Mechanism of Progression of Pancreatic Cancer: From Mice to Men. Cancers, 14(23), 5797. https://doi.org/10.3390/cancers14235797