

Rosai–Dorfman Disease between Proliferation and Neoplasia

Abstract
Simple Summary
Abstract
1. Introduction
2. Discussion
3. Epidemiology
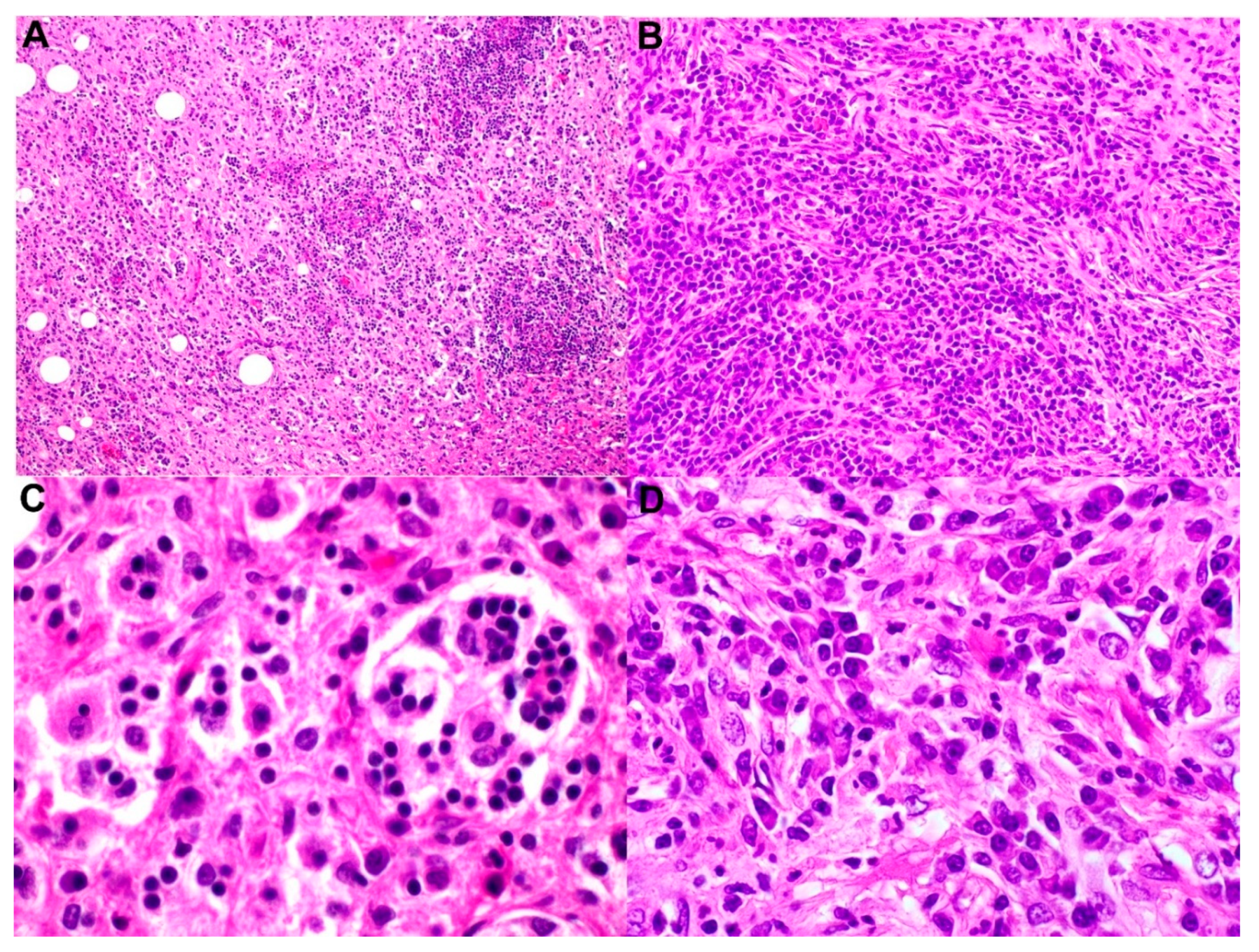
4. Subclassification
5. Clinical Presentation
- Medical history
- Constitutional symptoms;
- Organ affection (head, eyes, ears, nose, and throat; cardiovascular; pulmonary; GI; GU; skin; CNS; and endocrine);
- History of autoimmune disease, LCH, or other histiocytic lesions, as well as hematologic malignancies;
- Family history for children.
- Physical examination
- Lymphadenopathy;
- Organomegaly;
- Cutaneous and extranodal lesions;
- Neurologic changes.
- Radiological evaluation
- All patients should have whole-body positron emission tomography (PET)/computed tomography (CT);
- Selected patients should have CT sinuses with contrast, high-resolution CT chest, magnetic resonance imaging of orbit/brain with contrast and magnetic resonance imaging of spine with contrast;
- Selected patients for organ-specific ultrasound.
- Laboratory evaluation
- Complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate;
- Serum immunoglobulins;
- Coagulation studies, C-reactive protein, uric acid, and lactate dehydrogenase (LDH);
- Hemolysis panel (Coombs, haptoglobin, reticulocytes, and blood smear);
- Panel for autoimmune diseases (ALPS panel, antinuclear antibody, rheumatoid factor, HLA B27);
- NGS targeted gene mutations in RAF-RAS-MEK-ERK pathway;
- If familial form is suspected, then NGS test for SLC29A3 (germline mutations);
- Bone-marrow biopsy (if cytopenia or abnormal peripheral blood smear are present);
- Lumbar puncture for CNS involvement.
- Subspecialty consultation as needed
- Dermatology and ophthalmology evaluation before initiating MEK-inhibitor therapy.
6. RDD with Concurrent Disorders
7. Histopathology and Immunochemistry
7.1. Benign Disorders
- Sinus histiocytosis, which is a benign nonspecific reaction in a reactive lymph node with increased histiocytes. This entity does not show emperipolesis, and few histiocytes stain for S100.
- Toxoplasma lymphadenitis usually present with the diagnostic histologic triad of
- Sinusoidal expansion by monocytoid B cells;
- Follicular hyperplasia;
- Epithelioid histiocytes encroaching on reactive germinal centers differ from RDD, since these histiocytes usually occur in small groups with baby granulomatous changes;
- 3.
- Hemophagocytic lymphohistiocytosis (HLH): The patients with HLH present with hemophophagocytosis that cytologically could mimic emperipolesis.
7.2. Neoplastic Disorders
- LCH can be easily differentiated from RDD. Unlike RDD, Langerhans cells in LCH usually show nuclear groves and thin nuclear membranes and are often associated with abundant eosinophils and necrosis. Langerhans cells are positive for CD1a and langerin (CD207), which are negative in RDD, yet both RDD and LCH are positive for S100 [44,45]. An electron microscope shows a specific feature in cytoplasm of the LCH cells, called Birkbeck granules. BRAF V600E is more frequently mutated in LCH than in RDD. Another immunohistochemical stain that can be used to detect underlying BRAF V600E is the BRAF VE1 clone [46]. PD-L1 is more commonly positive in LCH than RDD [46].
- Diagnosing ECD requires a comprehensive study, including clinicopathologic, radiologic, and molecular assessment. Pathognomonic, radiologic features for diagnosis include symmetrical long-bone osteosclerotic lesion of lower limbs and sheathing of the aorta (coating of the aorta by fibrosis). Histologically, it presents with histiocytes with xanthogranulomatous changes and fibrosis against the background of inflammatory cells and Touton giant cells. Similar to RDD, the histiocytes in ECD are also positive for CD63 and CD168, a small subset positive for S-100 that could mimic RDD. However, unlike RDD, the histiocytes in ECD do not show emperiolopoiesis, and are positive for XIIIa and BRAF mutations (>50% of cases) [47];
- Classic Hodgkin lymphoma can rarely be localized in lymph-node sinuses, which can create a diagnostic dilemma in some cases; however, the presence of Reed–Sternberg cells and Hodgkin cells makes the distinction from RDD easier. Hodgkin cells and Reed–Sternberg cells are positive for CD30, CD15, dim PAX5, fascin, and MUM1, and the background histiocytes are positive for CD68 but not S100;
- Anaplastic large-cell lymphoma can present with a sinusoidal pattern, but the hallmark cells with horseshoe nuclei are very distinctive and diffusely positive for CD30 by IHC stains (≥75% of neoplastic cells). For the ALK-positive variant, it harbors ALK translocations and is positive for ALK immunostaining.
- ALK-positive histiocytosis is first described in 3 infants [48]. This entity is divided into three groups: Group 1A (infants with hematopoietic and liver involvement); group 1B (multisystemic diseases); and group 2 (patients with single-organ involvement [49]. Similar to RDD, ALK-positive histiocytosis shows emperipolesis and stains for histiocytic markers (CD168, CD63, CD4, CD14). Many cases also express OCT2 (61%), pERK (46%) and cyclin-D1 (49%). However, this entity harbors many ALK translocations, most commonly KIF5B-ALK, and stains positive for ALK1. S100 is variably expressed, unlike RDD, which is uniformly expressed [49];
- Histiocytic sarcoma shows histiocytic proliferation and usually has marked cytologic atypia, brisk mitotic activity, and is negative for S100. Though emperipolesis could be occasionally identified, unlike RDD, it also has an aggressive clinical course;
- Juvenile xanthogranuloma, most commonly present in children, with a predilection for the head-and-neck region. It is often cutaneous; however, rarely, it can be subcutaneous or intramuscular. Usually, it resolves spontaneously. Histologically, proliferation of numerous mononuclear and multinucleated cells with Touton-like features on a background of inflammatory cells (lymphocytes and eosinophils) are consistently present. There can be variable foamy histiocytes and lipids. Emperipolesis could be seen. Both diseases are positive for CD68 and CD4. However, unlike RDD, the histiocytes are negative for S100 and more frequently positive for Factor XIIIa [50];
- Follicular dendritic cell sarcoma may involve lymph nodes or extranodal sites. Oval-to- spindle cells with eosinophilic cytoplasm forming syncytial sheets. Tumor cells are admixed with small lymphocytes and positive for CD21, CD23, CD35, and clusterin, distinguishing it from RDD. Some cases may express S100 focally.
8. Genetic and Mutational Profile
- Pigmented hypertrichotic dermatosis with insulin-dependent diabetes syndrome: Children present with insulin-dependent diabetes mellitus and pigmented hypertrichosis [67].
9. Prognosis
10. Treatment
- No standard therapy has been established for patients with RDD based on results of prospective clinical studies due to the rarity of this disorder. Treatment algorithms are based on retrospective case series, case reports, disease-registry analyses, and expert opinions. Most recently, the NCCN Clinical Practice Guidelines in Oncology, Histiocytic Neoplasms, Version 2.2021, proposed diagnostic and treatment algorithms for patients with RDD [23]. Observation, watch and wait, is considered for asymptomatic or mildly symptomatic patients as 40% of cases with nodal/cutaneous involvement will have spontaneous remission [72];
- Complete surgical resection is used for patients with unifocal areas of involvement. Surgical therapy can also be useful for patients with spinal cord compression or upper-airway obstruction or with large lesions which might cause end-organ damage. Regarding cutaneous RDD, surgery has been found to be the most effective line of treatment for localized disease [73]. Endoscopic resection of sinonasal RDD can help with symptomatic relief [74];
- External-beam radiation therapy can play a role in patients with localized unresectable symptomatic steroid-refractory masses, especially in extranodal locations. The most frequently administered doses ranged from 20 to 30 Gy with 2 Gy per fraction, but higher disease levels have also been reported. Overall response rate (ORR) was approximately 40% in patients treated with doses ranging from 30 to 49 Gy and 27% with doses <30 Gy;
- Corticosteroids have been used in RDD as it was found that steroids decrease the nodal size and symptoms. The optimal dose and duration for this line treatment is ambiguous; however, prednisone (40–70 mg per day) demonstrated variable responses, ranging from failure–no response to complete response in cases with bone, orbit, CNS, and autoimmune-related RDD [75]. According to the consensus recommendations published in 2018, steroids alone do not lead to a stable response among patients with extranodal RDD. It has been found that patients with RDD require a higher dose of prednisone (>0.5 mg/kg per day). Similarly, dexamethasone is effective in RDD with nodal and intracranial lesions [76,77]. A case series of 57 patients reported by Mayo Clinic group showed that corticosteroid treatment was associated with 56% ORR in treatment-naïve patients. Relapses occurred in 53% of patients. When steroids were used as a second-line therapy, the ORR was 67% [15];
- Chemotherapy and immunotherapy have been used to treat RDD with many different chemotherapeutic agents, leading to a mixed treatment responses and adverse events. Some agents were ineffective, such as anthracyclines and alkylating agents; however, vinca alkaloids have shown variable results. Many different regimens have been postulated to have good responses in RDD. A case series on 15 patients with massive lymphadenopathy was reported by the University of Pennsylvania group. The patients were treated with rituximab monotherapy, resulting in 64% progression-free survival at 24 months [80]. Other chemotherapy agents used in refractory RDD with promising activities were nucleoside analogs, such as cladribine (2.1–5 mg/m2 per day for 5 days every 28 days for 6 months) and clofarabine (25 mg/m2 per day for 5 days every 28 days for 6 months) [81,82,83];
- Immunotherapy such as TNF-α inhibitors. thalidomide and lenalidomide were used due to the increased levels of TNF-α and interleukin-6 in patients with RDD. A case report of the patient treated with a low dose (50 mg/day) thalidomide with a dose escalation to 100 mg/day demonstrated promising results [84]. On the one hand, the results of thalidomide therapy have been mixed, and the side effects associated with administration of this agent are not negligible. Lenalidomide, on the other hand, has shown very good responses in patients with refractory nodal and bone RDD and overall better tolerance with fewer side effects, except myelosuppression compared to thalidomide [85]. Rituximab has also been used to treat autoimmune RDD; however, some cases show recurrence or become refractory [86,87,88];
- Targeted therapy with imatinib has been used in a case report with refractory RDD. Imatinib could be an option for systemic involvement by non-LCH disorders [89]. Interestingly, the lesional histiocytes were positive for expression of PDGFRB and KIT/CD117 by immunohistochemical stain, but no underlying mutations in genes coding for these tyrosine kinases were identified. BRAF mutations have rarely been observed in RDD. BRAF inhibitor (dabrafenib) was reported to be used in a patient with concurrent RDD and LCH with clinical and radiological response but increased BRAF-V600E post 13-month therapy; additional MEK inhibitor (trametinib) was added with limited follow-up time [60]. MEK inhibitor (cobimetinib) has shown good results in a patient with KRAS (p.G12R) mutated RDD [53]. Moyon et al. reported that two RDD patients received cobimetinib showed significant pulmonary response with regard to metabolism and tumor size [19].
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Destombes, P. Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases). Bull. Soc. Pathol. Exot. Fil. 1965, 58, 1169–1175. [Google Scholar]
- Azoury, F.J.; Reed, R.J. Histiocytosis. Report of an unusual case. N. Engl. J. Med. 1966, 274, 928–930. [Google Scholar] [CrossRef]
- Rosai, J.; Dorfman, R.F. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch. Pathol. 1969, 87, 63–70. [Google Scholar] [PubMed]
- Rosai, J.; Dorfman, R.F. Sinus histiocytosis with massive lymphadenopathy: A pseudolymphomatous benign disorder. Analysis of 34 cases. Cancer 1972, 30, 1174–1188. [Google Scholar] [CrossRef]
- Foucar, E.; Rosai, J.; Dorfman, R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Review of the entity. Semin. Diagn. Pathol. 1990, 7, 19–73. [Google Scholar] [PubMed]
- Abla, O.; Jacobsen, E.; Picarsic, J.; Krenova, Z.; Jaffe, R.; Emile, J.F.; Durham, B.H.; Braier, J.; Charlotte, F.; Donadieu, J.; et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood 2018, 131, 2877–2890. [Google Scholar] [CrossRef]
- Tsang, W.Y.; Yip, T.T.; Chan, J.K. The Rosai-Dorfman disease histiocytes are not infected by Epstein-Barr virus. Histopathology 1994, 25, 88–90. [Google Scholar] [CrossRef]
- Kismet, E.; Koseoglu, V.; Atay, A.A.; Deveci, S.; Demirkaya, E.; Tuncer, K. Sinus histiocytosis with massive lymphadenopathy in three brothers. Pediatr. Int. 2005, 47, 473–476. [Google Scholar] [CrossRef]
- Emile, J.F.; Abla, O.; Fraitag, S.; Horne, A.; Haroche, J.; Donadieu, J.; Requena-Caballero, L.; Jordan, M.B.; Abdel-Wahab, O.; Allen, C.E.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016, 127, 2672–2681. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Frater, J.L.; Maddox, J.S.; Obadiah, J.M.; Hurley, M.Y. Cutaneous Rosai-Dorfman disease: Comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J. Cutan. Med. Surg. 2006, 10, 281–290. [Google Scholar] [CrossRef]
- Ragotte, R.J.; Dhanrajani, A.; Pleydell-Pearce, J.; Del Bel, K.L.; Tarailo-Graovac, M.; van Karnebeek, C.; Terry, J.; Senger, C.; McKinnon, M.L.; Seear, M.; et al. The importance of considering monogenic causes of autoimmunity: A somatic mutation in KRAS causing pediatric Rosai-Dorfman syndrome and systemic lupus erythematosus. Clin. Immunol. 2017, 175, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Vaiselbuh, S.R.; Bryceson, Y.T.; Allen, C.E.; Whitlock, J.A.; Abla, O. Updates on histiocytic disorders. Pediatr. Blood Cancer 2014, 61, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022, 140, 1229–125310. [Google Scholar] [CrossRef] [PubMed]
- Goyal, G.; Ravindran, A.; Young, J.R.; Shah, M.V.; Bennani, N.N.; Patnaik, M.M.; Nowakowski, G.S.; Thanarajasingam, G.; Habermann, T.M.; Vassallo, R.; et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica 2020, 105, 348–357. [Google Scholar] [CrossRef]
- Papo, M.; Cohen-Aubart, F.; Trefond, L.; Bauvois, A.; Amoura, Z.; Emile, J.F.; Haroche, J. Systemic Histiocytosis (Langerhans Cell Histiocytosis, Erdheim-Chester Disease, Destombes-Rosai-Dorfman Disease): From Oncogenic Mutations to Inflammatory Disorders. Curr. Oncol. Rep. 2019, 21, 62. [Google Scholar] [CrossRef]
- Demicco, E.G.; Rosenberg, A.E.; Bjornsson, J.; Rybak, L.D.; Unni, K.K.; Nielsen, G.P. Primary Rosai-Dorfman disease of bone: A clinicopathologic study of 15 cases. Am. J. Surg. Pathol. 2010, 34, 1324–1333. [Google Scholar] [CrossRef]
- Sandoval-Sus, J.D.; Sandoval-Leon, A.C.; Chapman, J.R.; Velazquez-Vega, J.; Borja, M.J.; Rosenberg, S.; Lossos, A.; Lossos, I.S. Rosai-Dorfman disease of the central nervous system: Report of 6 cases and review of the literature. Medicine 2014, 93, 165–175. [Google Scholar] [CrossRef]
- Moyon, Q.; Boussouar, S.; Maksud, P.; Emile, J.F.; Charlotte, F.; Aladjidi, N.; Prevot, G.; Donadieu, J.; Amoura, Z.; Grenier, P.; et al. Lung Involvement in Destombes-Rosai-Dorfman Disease: Clinical and Radiological Features and Response to the MEK Inhibitor Cobimetinib. Chest 2020, 157, 323–333. [Google Scholar] [CrossRef]
- Baker, J.C.; Kyriakos, M.; McDonald, D.J.; Rubin, D.A. Primary Rosai-Dorfman disease of the femur. Skeletal. Radiol. 2017, 46, 129–135. [Google Scholar] [CrossRef]
- Taufiq, M.; Khair, A.; Begum, F.; Akhter, S.; Shamim Farooq, M.; Kamal, M. Isolated Intracranial Rosai-Dorfman Disease. Case Rep. Neurol. Med. 2016, 2016, 1972594. [Google Scholar] [CrossRef][Green Version]
- Cohen Aubart, F.; Idbaih, A.; Emile, J.F.; Amoura, Z.; Abdel-Wahab, O.; Durham, B.H.; Haroche, J.; Diamond, E.L. Histiocytosis and the nervous system: From diagnosis to targeted therapies. Neuro-Oncology 2021, 23, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- Go, R.S.; Jacobsen, E.; Baiocchi, R.; Buhtoiarov, I.; Butler, E.B.; Campbell, P.K.; Coulter, D.W.; Diamond, E.; Flagg, A.; Goodman, A.M.; et al. Histiocytic Neoplasms, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 1277–1303. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.S.; Naden, R.P.; Chari, S.; Choi, H.K.; Della-Torre, E.; Dicaire, J.F.; Hart, P.A.; Inoue, D.; Kawano, M.; Khosroshahi, A.; et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann. Rheum. Dis. 2020, 79, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.T.; Chen, T.C.; Lee, L.Y.; Lu, P.H. IgG4-positive plasma cells in cutaneous Rosai-Dorfman disease: An additional immunohistochemical feature and possible relationship to IgG4-related sclerosing disease. J. Cutan. Pathol. 2009, 36, 1069–1073. [Google Scholar] [CrossRef]
- Menon, M.P.; Evbuomwan, M.O.; Rosai, J.; Jaffe, E.S.; Pittaluga, S. A subset of Rosai-Dorfman disease cases show increased IgG4-positive plasma cells: Another red herring or a true association with IgG4-related disease? Histopathology 2014, 64, 455–459. [Google Scholar] [CrossRef]
- Zhang, X.; Hyjek, E.; Vardiman, J. A subset of Rosai-Dorfman disease exhibits features of IgG4-related disease. Am. J. Clin. Pathol. 2013, 139, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Perry, A.M.; Cao, W.; Smith, L.M.; Hsi, E.D.; Liu, X.; Mo, J.Q.; Dotlic, S.; Mosunjac, M.; Talmon, G.; et al. Relationship between Rosai-Dorfman disease and IgG4-related disease: Study of 32 cases. Am. J. Clin. Pathol. 2013, 140, 395–402. [Google Scholar] [CrossRef]
- O’Malley, D.P.; Duong, A.; Barry, T.S.; Chen, S.; Hibbard, M.K.; Ferry, J.A.; Hasserjian, R.P.; Thompson, M.A.; Richardson, M.S.; Jaffe, R.; et al. Co-occurrence of Langerhans cell histiocytosis and Rosai-Dorfman disease: Possible relationship of two histiocytic disorders in rare cases. Mod. Pathol. 2010, 23, 1616–1623. [Google Scholar] [CrossRef]
- Cohen-Barak, E.; Rozenman, D.; Schafer, J.; Krausz, J.; Dodiuk-Gad, R.; Gabriel, H.; Shani-Adir, A. An unusual co-occurrence of Langerhans cell histiocytosis and Rosai-Dorfman disease: Report of a case and review of the literature. Int. J. Dermatol. 2014, 53, 558–563. [Google Scholar] [CrossRef]
- Hervier, B.; Haroche, J.; Arnaud, L.; Charlotte, F.; Donadieu, J.; Neel, A.; Lifermann, F.; Villabona, C.; Graffin, B.; Hermine, O.; et al. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood 2014, 124, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Razanamahery, J.; Diamond, E.L.; Cohen-Aubart, F.; Plate, K.H.; Lourida, G.; Charlotte, F.; Helias-Rodzewicz, Z.; Goyal, G.; Go, R.S.; Dogan, A.; et al. Erdheim-Chester disease with concomitant Rosai-Dorfman like lesions: A distinct entity mainly driven by MAP2K1. Haematologica 2020, 105, e5–e8. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, F.; Chilosi, M.; Menestrina, F.; Scarpa, A.; Pelicci, P.G.; Amorosi, E.; Fiore-Donati, L.; Knowles, D.M., 2nd. Immunohistological analysis of Rosai-Dorfman histiocytosis. A disease of S-100 + CD1-histiocytes. Virchows Arch. A 1987, 411, 129–135. [Google Scholar] [CrossRef]
- Eisen, R.N.; Buckley, P.J.; Rosai, J. Immunophenotypic characterization of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Semin. Diagn Pathol. 1990, 7, 74–82. [Google Scholar]
- Paulli, M.; Rosso, R.; Kindl, S.; Boveri, E.; Marocolo, D.; Chioda, C.; Agostini, C.; Magrini, U.; Facchetti, F. Immunophenotypic characterization of the cell infiltrate in five cases of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Hum. Pathol. 1992, 23, 647–654. [Google Scholar] [CrossRef]
- Aoyama, K.; Terashima, K.; Imai, Y.; Katsushima, N.; Okuyama, Y.; Niikawa, K.; Mukada, T.; Takahashi, K. Sinus histiocytosis with massive lymphadenopathy. A histogenic analysis of histiocytes found in the fourth Japanese case. Acta Pathol. Jpn. 1984, 34, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Paljakka, P.; Haveri, P.; Saxen, E. Sinus histiocytosis with massive lymphadenopathy. A nodal and extranodal proliferation of S-100 protein positive histiocytes? Am. J. Clin. Pathol. 1987, 88, 270–277. [Google Scholar] [CrossRef]
- Garces, S.; Medeiros, L.J.; Patel, K.P.; Li, S.; Pina-Oviedo, S.; Li, J.; Garces, J.C.; Khoury, J.D.; Yin, C.C. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod. Pathol. 2017, 30, 1367–1377. [Google Scholar] [CrossRef]
- Baraban, E.; Sadigh, S.; Rosenbaum, J.; Van Arnam, J.; Bogusz, A.M.; Mehr, C.; Bagg, A. Cyclin D1 expression and novel mutational findings in Rosai-Dorfman disease. Br. J. Haematol. 2019, 186, 837–844. [Google Scholar] [CrossRef]
- Garces, S.; Medeiros, L.J.; Marques-Piubelli, M.L.; Coelho Siqueira, S.A.; Miranda, R.N.; Cuglievan, B.; Sriganeshan, V.; Medina, A.M.; Garces, J.C.; Saluja, K.; et al. Cyclin D1 expression in Rosai-Dorfman disease: A near-constant finding that is not invariably associated with mitogen-activated protein kinase/extracellular signal-regulated kinase pathway activation. Hum. Pathol. 2022, 121, 36–45. [Google Scholar] [CrossRef]
- Abdulla, Z.; Turley, H.; Gatter, K.; Pezzella, F. Immunohistological recognition of cyclin D1 expression by non-lymphoid cells among lymphoid neoplastic cells. APMIS 2014, 122, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Y.; Sun, L.; Zhai, Q. Cyclin D1 expression by histiocytes may mimic cyclin D1-positive proliferation centres of chronic lymphocytic leukaemia/small lymphocytic lymphoma. Pathol.-Res. Pract. 2018, 214, 72–75. [Google Scholar] [CrossRef]
- Ravindran, A.; Goyal, G.; Go, R.S.; Rech, K.L.; Mayo Clinic Histiocytosis Working, G. Rosai-Dorfman Disease Displays a Unique Monocyte-Macrophage Phenotype Characterized by Expression of OCT2. Am. J. Surg. Pathol. 2021, 45, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Favara, B.E.; Feller, A.C.; Pauli, M.; Jaffe, E.S.; Weiss, L.M.; Arico, M.; Bucsky, P.; Egeler, R.M.; Elinder, G.; Gadner, H.; et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med. Pediatr. Oncol. 1997, 29, 157–166. [Google Scholar] [CrossRef]
- Orkin, S.H.; Nathan, D.G.; Ginsburg, D.; Look, A.T.; Fisher, D.E.; Lux, S. Lymphohistiocytic disorders. In Nathan and Oski’s Hematology of Infancy and Childhood; WB Saunders: Philadelphia, PA, USA, 2003. [Google Scholar]
- Ballester, L.Y.; Cantu, M.D.; Lim, K.P.H.; Sarabia, S.F.; Ferguson, L.S.; Renee Webb, C.; Allen, C.E.; McClain, K.L.; Mohila, C.A.; Punia, J.N.; et al. The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol. Oncol. 2018, 36, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Dagna, L.; Hyman, D.M.; Cavalli, G.; Janku, F.; Estrada-Veras, J.; Ferrarini, M.; Abdel-Wahab, O.; Heaney, M.L.; Scheel, P.J.; et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood 2014, 124, 483–492. [Google Scholar] [CrossRef]
- Chan, J.K.; Lamant, L.; Algar, E.; Delsol, G.; Tsang, W.Y.; Lee, K.C.; Tiedemann, K.; Chow, C.W. ALK+ histiocytosis: A novel type of systemic histiocytic proliferative disorder of early infancy. Blood 2008, 112, 2965–2968. [Google Scholar] [CrossRef]
- Kemps, P.G.; Picarsic, J.; Durham, B.H.; Helias-Rodzewicz, Z.; Hiemcke-Jiwa, L.; van den Bos, C.; van de Wetering, M.D.; van Noesel, C.J.M.; van Laar, J.A.M.; Verdijk, R.M.; et al. ALK-positive histiocytosis: A new clinicopathologic spectrum highlighting neurologic involvement and responses to ALK inhibition. Blood 2022, 139, 256–280. [Google Scholar] [CrossRef]
- McClain, K.L. Histiocytic Diseases of Neonates: Langerhans Cell Histiocytosis, Rosai-Dorfman Disease, and Juvenile Xanthogranuloma. Clin. Perinatol. 2021, 48, 167–179. [Google Scholar] [CrossRef]
- Diamond, E.L.; Durham, B.H.; Haroche, J.; Yao, Z.; Ma, J.; Parikh, S.A.; Wang, Z.; Choi, J.; Kim, E.; Cohen-Aubart, F.; et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016, 6, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Matter, M.S.; Bihl, M.; Juskevicius, D.; Tzankov, A. Is Rosai-Dorfman disease a reactve process? Detection of a MAP2K1 L115V mutation in a case of Rosai-Dorfman disease. Virchows Arch. 2017, 471, 545–547. [Google Scholar] [CrossRef]
- Jacobsen, E.; Shanmugam, V.; Jagannathan, J. Rosai-Dorfman Disease with Activating KRAS Mutation—Response to Cobimetinib. N. Engl. J. Med. 2017, 377, 2398–2399. [Google Scholar] [CrossRef]
- Durham, B.H.; Lopez Rodrigo, E.; Picarsic, J.; Abramson, D.; Rotemberg, V.; De Munck, S.; Pannecoucke, E.; Lu, S.X.; Pastore, A.; Yoshimi, A.; et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat. Med. 2019, 25, 1839–1842. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.H.; Gasilina, A.; Roychoudhury, J.; Clark, J.; McCormack, F.X.; Pressey, J.; Grimley, M.S.; Lorsbach, R.; Ali, S.; Bailey, M.; et al. Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI Insight 2017, 2, e89473. [Google Scholar] [CrossRef]
- Wu, K.J.; Li, S.H.; Liao, J.B.; Chiou, C.C.; Wu, C.S.; Chen, C.C. NRAS Mutations May Be Involved in the Pathogenesis of Cutaneous Rosai Dorfman Disease: A Pilot Study. Biology 2021, 10, 396. [Google Scholar] [CrossRef]
- Fatobene, G.; Haroche, J.; Helias-Rodzwicz, Z.; Charlotte, F.; Taly, V.; Ferreira, A.M.; Abdo, A.N.R.; Rocha, V.; Emile, J.F. BRAF V600E mutation detected in a case of Rosai-Dorfman disease. Haematologica 2018, 103, e377–e379. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.E.; Wachsmann, M.; Oliver, D.; Abedin, Z.; Ye, D.; Burns, D.K.; Raisanen, J.M.; Greenberg, B.M.; Hatanpaa, K.J. BRAF mutation leading to central nervous system rosai-dorfman disease. Ann. Neurol. 2018, 84, 147–152. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, A.L.; Duan, M.H.; Cai, H.; Gao, X.M.; Liu, T.; Sun, J.; Liang, Z.Y.; Zhou, D.B.; Cao, X.X.; et al. Diverse kinase alterations and myeloid-associated mutations in adult histiocytosis. Leukemia 2022, 36, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Mastropolo, R.; Close, A.; Allen, S.W.; McClain, K.L.; Maurer, S.; Picarsic, J. BRAF-V600E-mutated Rosai-Dorfman-Destombes disease and Langerhans cell histiocytosis with response to BRAF inhibitor. Blood Adv. 2019, 3, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, V.; Margolskee, E.; Kluk, M.; Giorgadze, T.; Orazi, A. Rosai-Dorfman Disease Harboring an Activating KRAS K117N Missense Mutation. Head Neck Pathol. 2016, 10, 394–399. [Google Scholar] [CrossRef]
- Gatalica, Z.; Bilalovic, N.; Palazzo, J.P.; Bender, R.P.; Swensen, J.; Millis, S.Z.; Vranic, S.; Von Hoff, D.; Arceci, R.J. Disseminated histiocytoses biomarkers beyond BRAFV600E: Frequent expression of PD-L1. Oncotarget 2015, 6, 19819–19825. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Brand, C.; Schneider, J.W.; Schubert, P. Rosai-Dorfman disease: An overview. J. Clin. Pathol. 2020, 73, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, S.; Artusi, T.; Selleri, L.; Temperani, P.; Zucchini, P.; Vecchi, A.; Emilia, G.; Torelli, U. Sinus histiocytosis with massive lymphadenopathy: Immunological, cytogenetic and molecular studies. Blut 1990, 60, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.V.; Morris, M.R.; Cangul, H.; Gleeson, D.; Straatman-Iwanowska, A.; Davies, N.; Keenan, S.; Pasha, S.; Rahman, F.; Gentle, D.; et al. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010, 6, e1000833. [Google Scholar] [CrossRef]
- Moynihan, L.M.; Bundey, S.E.; Heath, D.; Jones, E.L.; McHale, D.P.; Mueller, R.F.; Markham, A.F.; Lench, N.J. Autozygosity mapping, to chromosome 11q25, of a rare autosomal recessive syndrome causing histiocytosis, joint contractures, and sensorineural deafness. Am. J. Hum. Genet. 1998, 62, 1123–1128. [Google Scholar] [CrossRef]
- Cliffe, S.T.; Kramer, J.M.; Hussain, K.; Robben, J.H.; de Jong, E.K.; de Brouwer, A.P.; Nibbeling, E.; Kamsteeg, E.J.; Wong, M.; Prendiville, J.; et al. SLC29A3 gene is mutated in pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum. Mol. Genet. 2009, 18, 2257–2265. [Google Scholar] [CrossRef]
- Bolze, A.; Abhyankar, A.; Grant, A.V.; Patel, B.; Yadav, R.; Byun, M.; Caillez, D.; Emile, J.F.; Pastor-Anglada, M.; Abel, L.; et al. A mild form of SLC29A3 disorder: A frameshift deletion leads to the paradoxical translation of an otherwise noncoding mRNA splice variant. PLoS ONE 2012, 7, e29708. [Google Scholar] [CrossRef]
- Chouk, H.; Ben Rejeb, M.; Boussofara, L.; Elmabrouk, H.; Ghariani, N.; Sriha, B.; Saad, A.; H’Mida, D.; Denguezli, M. Phenotypic intrafamilial variability including H syndrome and Rosai-Dorfman disease associated with the same c.1088G > A mutation in the SLC29A3 gene. Hum. Genom. 2021, 15, 63. [Google Scholar] [CrossRef]
- Maric, I.; Pittaluga, S.; Dale, J.K.; Niemela, J.E.; Delsol, G.; Diment, J.; Rosai, J.; Raffeld, M.; Puck, J.M.; Straus, S.E.; et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am. J. Surg. Pathol. 2005, 29, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Paryani, N.N.; Daugherty, L.C.; O’Connor, M.I.; Jiang, L. Extranodal rosai-dorfman disease of the bone treated with surgery and radiotherapy. Rare Tumors 2014, 6, 5531. [Google Scholar] [CrossRef]
- Pulsoni, A.; Anghel, G.; Falcucci, P.; Matera, R.; Pescarmona, E.; Ribersani, M.; Villiva, N.; Mandelli, F. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Report of a case and literature review. Am. J. Hematol. 2002, 69, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Al-Khateeb, T.H. Cutaneous Rosai-Dorfman Disease of the Face: A Comprehensive Literature Review and Case Report. J. Oral Maxillofac. Surg. 2016, 74, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Zhou, S.H.; Wang, S.Q.; Teng, X.D.; Fan, J. Factors associated with recurrence and therapeutic strategies for sinonasal Rosai-Dorfman disease. Head Neck 2012, 34, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Shulman, S.; Katzenstein, H.; Abramowsky, C.; Broecker, J.; Wulkan, M.; Shehata, B. Unusual presentation of Rosai-Dorfman disease (RDD) in the bone in adolescents. Fetal Pediatr. Pathol. 2011, 30, 442–447. [Google Scholar] [CrossRef]
- McPherson, C.M.; Brown, J.; Kim, A.W.; DeMonte, F. Regression of intracranial rosai-dorfman disease following corticosteroid therapy. Case report. J. Neurosurg. 2006, 104, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Adeleye, A.O.; Amir, G.; Fraifeld, S.; Shoshan, Y.; Umansky, F.; Spektor, S. Diagnosis and management of Rosai-Dorfman disease involving the central nervous system. Neurol. Res. 2010, 32, 572–578. [Google Scholar] [CrossRef]
- Teachey, D.T.; Greiner, R.; Seif, A.; Attiyeh, E.; Bleesing, J.; Choi, J.; Manno, C.; Rappaport, E.; Schwabe, D.; Sheen, C.; et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br. J. Haematol. 2009, 145, 101–106. [Google Scholar] [CrossRef]
- Cooper, S.L.; Arceci, R.J.; Gamper, C.J.; Teachey, D.T.; Schafer, E.S. Successful Treatment of Recurrent Autoimmune Cytopenias in the Context of Sinus Histiocytosis With Massive Lymphadenopathy Using Sirolimus. Pediatr. Blood Cancer 2016, 63, 358–360. [Google Scholar] [CrossRef]
- Namoglu, E.C.; Hughes, M.E.; Plastaras, J.P.; Landsburg, D.J.; Maity, A.; Nasta, S.D. Management and outcomes of sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman Disease). Leuk. Lymphoma 2020, 61, 905–911. [Google Scholar] [CrossRef]
- Simko, S.J.; Tran, H.D.; Jones, J.; Bilgi, M.; Beaupin, L.K.; Coulter, D.; Garrington, T.; McCavit, T.L.; Moore, C.; Rivera-Ortegon, F.; et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr. Blood Cancer 2014, 61, 479–487. [Google Scholar] [CrossRef]
- Aouba, A.; Terrier, B.; Vasiliu, V.; Candon, S.; Brousse, N.; Varet, B.; Hermine, O. Dramatic clinical efficacy of cladribine in Rosai-Dorfman disease and evolution of the cytokine profile: Towards a new therapeutic approach. Haematologica 2006, 91, ECR52. [Google Scholar] [PubMed]
- Sasaki, K.; Pemmaraju, N.; Westin, J.R.; Wang, W.L.; Khoury, J.D.; Podoloff, D.A.; Moon, B.; Daver, N.; Borthakur, G. A single case of rosai-dorfman disease marked by pathologic fractures, kidney failure, and liver cirrhosis treated with single-agent cladribine. Front. Oncol. 2014, 4, 297. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Pavlidakey, P.; Sami, N. Rosai-Dorfman disease successfully treated with thalidomide. JAAD Case Rep. 2016, 2, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Assal, A.; Scherba, M.; Elman, J.; White, R.; Verma, A.; Strakhan, M.; Mohammadi, F.; Janakiram, M. Lenalidomide in the treatment of Rosai Dorfman disease--a first in use report. Am. J. Hematol. 2016, 91, E1. [Google Scholar] [CrossRef] [PubMed]
- Petschner, F.; Walker, U.A.; Schmitt-Graff, A.; Uhl, M.; Peter, H.H. [“Catastrophic systemic lupus erythematosus” with Rosai-Dorfman sinus histiocytosis. Successful treatment with anti-CD20/rutuximab]. Dtsch. Med. Wochenschr. 2001, 126, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Maklad, A.M.; Bayoumi, Y.; Tunio, M.; Alshakweer, W.; Dahar, M.A.; Akbar, S.A. Steroid-resistant extranodal rosai-dorfman disease of cheek mass and ptosis treated with radiation therapy. Case Rep. Hematol. 2013, 2013, 428297. [Google Scholar] [CrossRef]
- Nadal, M.; Kervarrec, T.; Machet, M.C.; Petrella, T.; Machet, L. Cutaneous Rosai-Dorfman Disease Located on the Breast: Rapid Effectiveness of Methotrexate After Failure of Topical Corticosteroids, Acitretin and Thalidomide. Acta Derm. Venereol. 2015, 95, 758–759. [Google Scholar] [CrossRef]
- Utikal, J.; Ugurel, S.; Kurzen, H.; Erben, P.; Reiter, A.; Hochhaus, A.; Nebe, T.; Hildenbrand, R.; Haberkorn, U.; Goerdt, S.; et al. Imatinib as a treatment option for systemic non-Langerhans cell histiocytoses. Arch. Dermatol. 2007, 143, 736–740. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Site | Incidence | Symptoms/Signs | Radiologic Findings |
|---|---|---|---|
| Nodal | |||
| Lymph node | 57% of cases | Bilateral cervical lymphadenopathy or other LN sites, manifested with palpable masses | Enlarged LNs |
| Extranodal | |||
| Skin | 10% of cases | Painless, macular, slow-growing papules, subcutaneous nodules. Any skin site can be affected. | Nodule(s) or mass(es) |
| CNS | <5% of cases (75% intracranial and 25% spinal lesions), more than 300 cases have been reported. Cervical and thoracic regions are the most common areas affected in spinal-dural or epidural lesion | Headaches, seizures, gait difficulty. In familial cases, there is an association with damage to the auditory nerve pathway and deafness. | Dural lesion, extra-axial, homogeneously enhancing, mimicking nodular meningioma; or parenchymal (infratentorial) involvement |
| Orbit | 11% of cases | Presents as a mass in different part of the orbit, e.g., conjunctiva, lacrimal glands, and cornea. It can also present as uveitis. | Orbital mass |
| Head and neck | 11% of cases involving nasal cavity, more common among Asians | Nasal obstruction, epistaxis, and nasal dorsum deformity | Nodules, swelling, mass(es) |
| Intrathoracic | 2% of patients, with pulmonary disease concurrent lymphadenopathy or systemic disease. Cardiac involvement is extremely rare ~0.1–0.2% of cases. | Chronic dry cough, progressive dyspnea, or acute respiratory failure. RDD affecting lower respiratory tract have a high mortality rate, reaching 45%. | Pulmonary nodular consolidation in all lobes of the lungs; pleural effusion with fibrosis or nodules |
| Retroperitoneal /genitourinary tract | Kidneys are affected in approximately 4% of cases. | Abdominal or flank pain, fullness, hematuria, renal failure, hypercalcemia, and/or nephrotic syndrome | Mass(es), hydronephrosis, urethral obstruction |
| Gastrointestinal tract | <1% of cases, commonly in middle-aged women with concurrent nodal or extranodal affection | Abdominal pain, constipation, hematochezia, and intestinal obstruction | Mass(es) |
| Bone | 5% to 10% of cases, usually with concurrent nodal affection | Bone pain and, rarely, pathologic fractures | Cortex-based osteolytic lesion, commonly long bones, vertebrae, and sacrum |
| Gene | Molecular Alteration | References |
|---|---|---|
| ARAF | N217K | [51] |
| MAP2K1 | F53V, L115V, P124R, G128D, V50M, D65M | [38,51,52] |
| KRAS | A146T, A146V, K117N, G12D, G12R, G13S, Q22K, | [38,51,53,55,56,61] |
| NRAS | G13D | [51] |
| CBL | C384Y, GNAQ Q209H | [55] |
| KDM5A | amplification | [55] |
| FBXW7 | E113D | [55] |
| BRAF | V600E, deletion (p. 486–491), Y472C and R188G | [57,58,59] |
| SMAD4 | T521I (variant of unknown significance, 1 case) | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbaz Younes, I.; Sokol, L.; Zhang, L. Rosai–Dorfman Disease between Proliferation and Neoplasia. Cancers 2022, 14, 5271. https://doi.org/10.3390/cancers14215271
Elbaz Younes I, Sokol L, Zhang L. Rosai–Dorfman Disease between Proliferation and Neoplasia. Cancers. 2022; 14(21):5271. https://doi.org/10.3390/cancers14215271
Chicago/Turabian StyleElbaz Younes, Ismail, Lubomir Sokol, and Ling Zhang. 2022. "Rosai–Dorfman Disease between Proliferation and Neoplasia" Cancers 14, no. 21: 5271. https://doi.org/10.3390/cancers14215271
APA StyleElbaz Younes, I., Sokol, L., & Zhang, L. (2022). Rosai–Dorfman Disease between Proliferation and Neoplasia. Cancers, 14(21), 5271. https://doi.org/10.3390/cancers14215271