
MITF Is Regulated by Redox Signals Controlled by the Selenoprotein Thioredoxin Reductase 1


 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Immunochemical Analysis and Western Blot
2.3. 2-D Gel Analysis of Oxidized Protein Complexes
2.4. Thioredoxin Reductase Activity Assay
2.5. Tyrosinase Activity Assay
2.6. Analysis of H2O2-Induced Protein Oxidation
2.7. RNA Extraction and Analysis by qPCR and RNAseq
2.8. Statistics
3. Results
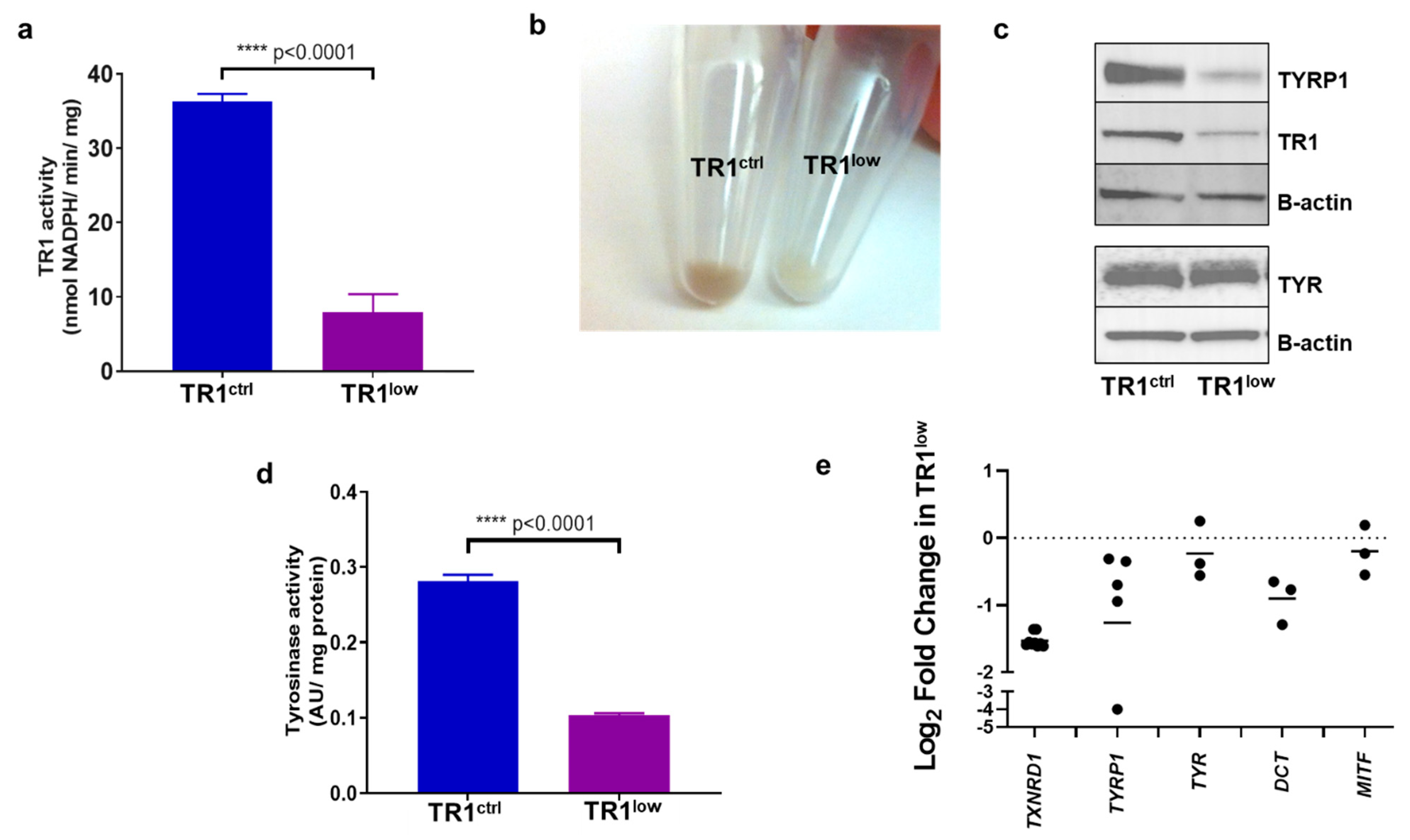
3.1. Pigmentation in Melanocytes Is Decreased by TR1 Knockdown
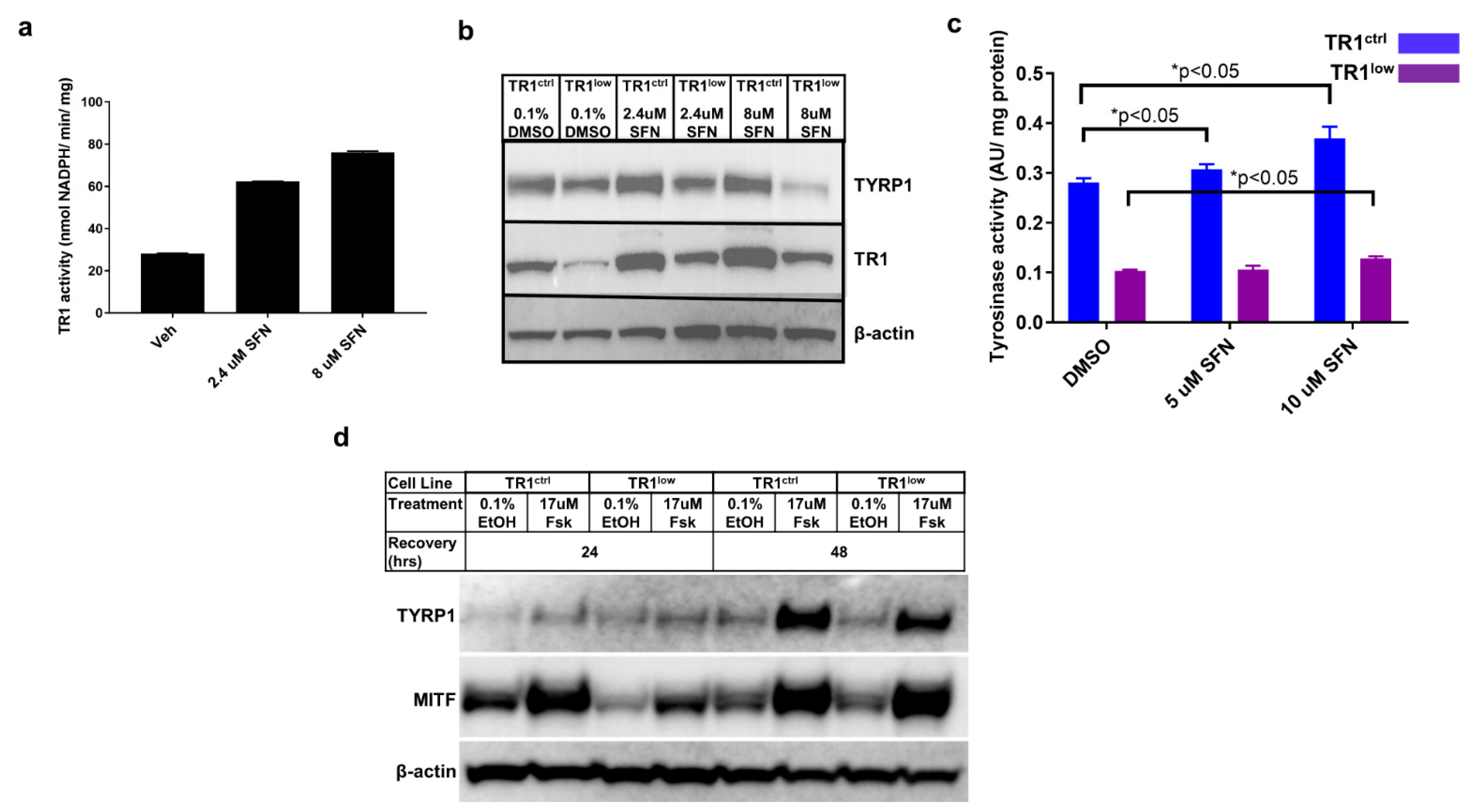
3.2. Alterations in TR1 Activity Affect TYRP1 and MITF Protein Levels as well as Tyrosinase Activity
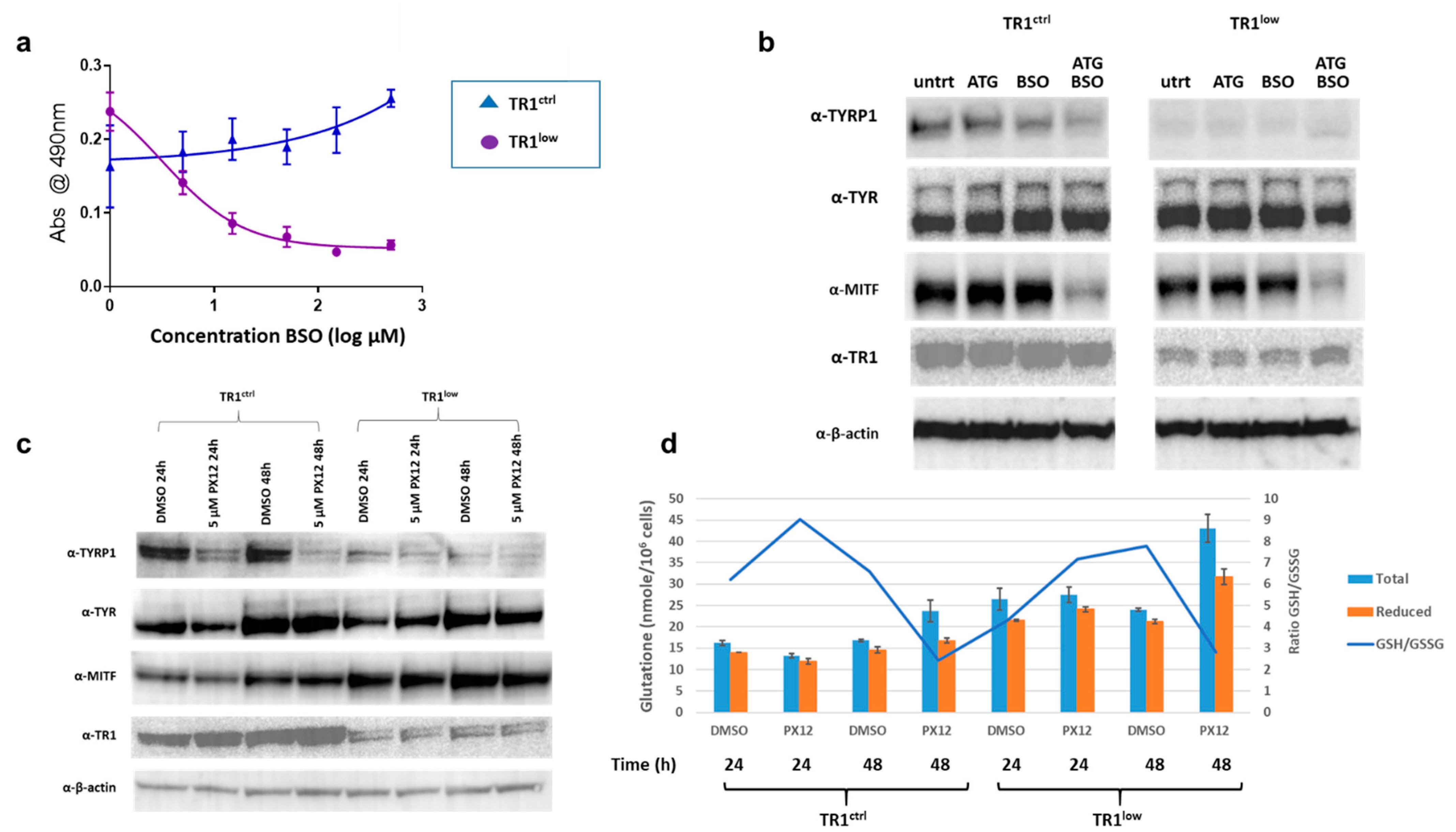
3.3. The TR1/TRX1 Network Cooperates with the GSH Network in the Regulation of TYRP1 and MITF
3.4. MITF and PRX1 Are Reversibly Modified by H2O2 and Loss of TR1 in Melanocytes
3.5. Disruption of TXNRD1 in Melanoma Cells Using CRISPR/Cas9 Results in Loss of Expression of the Melanocyte-Specific Isoform of MITF (MITF-M)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. U. S. Cancer Statistics Public Database. Available online: https://www.cdc.gov/cancer/uscs/public-use/index.htm (accessed on 10 June 2022).
- Bolick, N.L.; Geller, A.C. Epidemiology of Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 57–72. [Google Scholar] [CrossRef]
- Courtney, P.T.; Yip, A.T.; Cherry, D.R.; Salans, M.A.; Kumar, A.; Murphy, J.D. Cost-effectiveness of Nivolumab-Ipilimumab Combination Therapy for the Treatment of Advanced Non-Small Cell Lung Cancer. JAMA Netw Open 2021, 4, e218787. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, P.B.; Fain, H.D.; Cassidy, J.P., Jr.; Tran, S.M.; Moos, P.J.; Boucher, K.M.; Gerads, R.; Florell, S.R.; Grossman, D.; Leachman, S.A. Selenium for the prevention of cutaneous melanoma. Nutrients 2013, 5, 725–749. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.A.; Thomas, J.; Cassidy, P.; Robinette, K.; Jenkins, N.; Florell, S.R.; Leachman, S.; Samlowski, W.E.; Grossman, D. N-acetylcysteine protects melanocytes against oxidative stress/damage and delays onset of ultraviolet-induced melanoma in mice. Clin. Cancer Res. 2007, 13, 5952–5958. [Google Scholar] [CrossRef]
- Cassidy, P.B.; Honeggar, M.; Poerschke, R.L.; White, K.; Florell, S.R.; Andtbacka, R.H.; Tross, J.; Anderson, M.; Leachman, S.A.; Moos, P.J. The role of thioredoxin reductase 1 in melanoma metabolism and metastasis. Pigment. Cell Melanoma Res. 2015, 28, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, B.; Snider, G.W.; Fredette, N.; Stumpff, J.; Hondal, R.J.; Heintz, N.H. Resolution of oxidative stress by thioredoxin reductase: Cysteine versus selenocysteine. Redox Biol. 2014, 2, 475–484. [Google Scholar] [CrossRef]
- Go, Y.M.; Roede, J.R.; Walker, D.I.; Duong, D.M.; Seyfried, N.T.; Orr, M.; Liang, Y.; Pennell, K.D.; Jones, D.P. Selective targeting of the cysteine proteome by thioredoxin and glutathione redox systems. Mol. Cell Proteom. 2013, 12, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharm. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Turajlic, S.; Furney, S.J.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann. Oncol. 2014, 25, 959–967. [Google Scholar] [CrossRef]
- Ando, K.; Hirao, S.; Kabe, Y.; Ogura, Y.; Sato, I.; Yamaguchi, Y.; Wada, T.; Handa, H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008, 36, 4327–4336. [Google Scholar] [CrossRef] [PubMed]
- Moos, P.J.; Edes, K.; Cassidy, P.; Massuda, E.; Fitzpatrick, F.A. Electrophilic prostaglandins and lipid aldehydes repress redox-sensitive transcription factors p53 and hypoxia-inducible factor by impairing the selenoprotein thioredoxin reductase. J. Biol. Chem. 2003, 278, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M. Oxygen, evolution and redox signalling in the human brain; quantum in the quotidian. J. Physiol. 2019, 597, 15–28. [Google Scholar] [CrossRef]
- Stocker, S.; van Laer, K.; Mijuskovic, A.; Dick, T.P. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.C.; Sobotta, M.C.; Bausewein, D.; Aller, I.; Hell, R.; Dick, T.P.; Meyer, A.J. Redesign of genetically encoded biosensors for monitoring mitochondrial redox status in a broad range of model eukaryotes. J. Biomol. Screen. 2014, 19, 379–386. [Google Scholar] [CrossRef]
- Keniry, M.; Pires, M.M.; Mense, S.; Lefebvre, C.; Gan, B.; Justiano, K.; Lau, Y.K.; Hopkins, B.; Hodakoski, C.; Koujak, S.; et al. Survival factor NFIL3 restricts FOXO-induced gene expression in cancer. Genes Dev. 2013, 27, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Stocker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Le Poole, I.C.; van den Berg, F.M.; van den Wijngaar, R.M.; Galloway, D.A.; van Amstel, P.J.; Buffing, A.A.; Smits, H.L.; Westerhof, W.; Das, P.K. Generatjion of a human melanocyte cell line by introduction of HPV16E6 and E7 genes. In Vitro Cell Dev. Biol. Anim. 1997, 31, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Poerschke, R.L.; Moos, P.J. Thioredoxin reductase 1 knockdown enhances selenazolidine cytotoxicity in human lung cancer cells via mitochondrial dysfunction. Biochem. Pharm. 2011, 81, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Osborne, S.A.; Tonissen, K.F. Genomic organisation and alternative splicing of mouse and human thioredoxin reductase 1 genes. BMC Genom. 2001, 2, 10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saraswat, R.; McDonagh, B. Diagonal Electrophoresis for the Detection of Proteins Involved in Disulfide Bonds. Methods Mol. Biol. 2019, 1855, 279–286. [Google Scholar] [PubMed]
- Lin, C.B.; Babiarz, L.; Liebel, F.; Price, E.R.; Kizoulis, M.; Gendimenico, G.J.; Fisher, D.E.; Seiberg, M. Modulation of microphthalmia-associated transcription factor gene expression alters skin pigmentation. J. Investig. Derm. 2002, 119, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Kast, J. Biotin Switch Assays for Quantitation of Reversible Cysteine Oxidation. Methods Enzym. 2017, 585, 269–284. [Google Scholar]
- Ito, S.; Nakanishi, Y.; Valenzuela, R.K.; Brilliant, M.H.; Kolbe, L.; Wakamatsu, K. Usefulness of alkaline hydrogen peroxide oxidation to analyze eumelanin and pheomelanin in various tissue samples: Application to chemical analysis of human hair melanins. Pigment. Cell Melanoma Res. 2011, 24, 605–613. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Ito, S. Advanced chemical methods in melanin determination. Pigment. Cell Res. 2002, 15, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Kadekaro, A.L.; Leachman, S.; Kavanagh, R.J.; Swope, V.; Cassidy, P.; Supp, D.; Sartor, M.; Schwemberger, S.; Babcock, G.; Wakamatsu, K.; et al. Melanocortin 1 receptor genotype: An important determinant of the damage response of melanocytes to ultraviolet radiation. FASEB J. 2010, 24, 3850–3860. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, J.A.; Nobuhisa, T.; Cui, R.; Arya, M.; Spry, M.; Wakamatsu, K.; Igras, V.; Kunisada, T.; Granter, S.R.; Nishimura, E.K.; et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 2006, 443, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Dagnell, M.; Schmidt, E.E.; Arner, E.S.J. The A to Z of modulated cell patterning by mammalian thioredoxin reductases. Free Radic. Biol. Med. 2018, 115, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.L.; Wyant, M.B.; Indra, A.; Ito, S.; Wakamatsu, K.; Merrill, G.F.; Moos, P.J.; Cassidy, P.B.; Leachman, S.A.; Ganguli-Indra, G.; et al. Thioredoxin Reductase 1 Modulates Pigmentation and Photobiology of Murine Melanocytes in vivo. J. Investig. Derm. 2022, 142, 1903–1911.e5. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, P.B.; Liu, T.; Florell, S.R.; Honeggar, M.; Leachman, S.A.; Boucher, K.M.; Grossman, D. A Phase II Randomized Placebo-Controlled Trial of Oral N-acetylcysteine for Protection of Melanocytic Nevi against UV-Induced Oxidative Stress In Vivo. Cancer Prev. Res. 2017, 10, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219. [Google Scholar] [CrossRef]
- Kirkpatrick, D.L.; Kuperus, M.; Dowdeswell, M.; Potier, N.; Donald, L.J.; Kunkel, M.; Berggren, M.; Angulo, M.; Powis, G. Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides. Biochem. Pharm. 1998, 55, 987–994. [Google Scholar] [CrossRef]
- Tuncer, S.; Gurbanov, R.; Sheraj, I.; Solel, E.; Esenturk, O.; Banerjee, S. Low dose dimethyl sulfoxide driven gross molecular changes have the potential to interfere with various cellular processes. Sci. Rep. 2018, 8, 14828. [Google Scholar] [CrossRef]
- Shin, H.R.; You, B.R.; Park, W.H. PX-12-induced HeLa cell death is associated with oxidative stress and GSH depletion. Oncol. Lett. 2013, 6, 1804–1810. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Kirkpatrick, D.L.; Belani, C.P.; Friedland, D.; Green, S.B.; Chow, H.H.; Cordova, C.A.; Stratton, S.P.; Sharlow, E.R.; Baker, A.; et al. A Phase I pharmacokinetic and pharmacodynamic study of PX-12, a novel inhibitor of thioredoxin-1, in patients with advanced solid tumors. Clin. Cancer Res. 2007, 13, 2109–2114. [Google Scholar] [CrossRef]
- Li, Z.; Ferguson, L.; Deol, K.K.; Roberts, M.A.; Magtanong, L.; Hendricks, J.M.; Mousa, G.A.; Kilinc, S.; Schaefer, K.; Wells, J.A.; et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat. Chem. Biol. 2022, 18, 751–761. [Google Scholar] [CrossRef]
- Stolwijk, J.M.; Falls-Hubert, K.C.; Searby, C.C.; Wagner, B.A.; Buettner, G.R. Simultaneous detection of the enzyme activities of GPx1 and GPx4 guide optimization of selenium in cell biological experiments. Redox Biol. 2020, 32, 101518. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.E.; Peskin, A.V.; Han, M.H.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidized peroxiredoxin 2 interacts with the protein disulfide- isomerase ERp46. Biochem. J. 2013, 453, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.L.; Nadler, M.; Skoko, J.J.; Bertomeu, T.; Pelosi, A.; Shafaei, P.M.; Levine, K.; Schempf, A.; Pennarun, B.; Yang, B.; et al. A Peroxidase Peroxiredoxin 1-Specific Redox Regulation of the Novel FOXO3 microRNA Target let-7. Antioxid. Redox Signal. 2018, 28, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Stottmeier, B.; Dick, T.P. Redox sensitivity of the MyD88 immune signaling adapter. Free Radic Biol. Med. 2016, 101, 93–101. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef]
- Hemesath, T.J.; Price, E.R.; Takemoto, C.; Badalian, T.; Fisher, D.E. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 1998, 391, 298–301. [Google Scholar] [CrossRef]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992, 20, 3821–3830. [Google Scholar] [CrossRef]
- Yasumoto, K.; Yokoyama, K.; Takahashi, K.; Tomita, Y.; Shibahara, S. Functional analysis of microphthalmia-associated transcription factor in pigment cell-specific transcription of the human tyrosinase family genes. J. Biol. Chem. 1997, 272, 503–509. [Google Scholar] [CrossRef]
- Dolinska, M.B.; Wingfield, P.T.; Young, K.L., 2nd; Sergeev, Y.V. The TYRP1-mediated protection of human tyrosinase activity does not involve stable interactions of tyrosinase domains. Pigment. Cell Melanoma Res. 2019, 32, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed]
- Bondurand, N.; Pingault, V.; Goerich, D.E.; Lemort, N.; Sock, E.; le Caignec, C.; Wegner, M.; Goossens, M. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum. Mol. Genet. 2000, 9, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, B.; Ludwig, A.; Klenovsek, K.; Wegner, M. Cooperative binding of Sox10 to DNA: Requirements and consequences. Nucleic Acids Res. 2002, 30, 5509–5516. [Google Scholar] [CrossRef] [PubMed]
- Codutti, L.; van Ingen, H.; Vascotto, C.; Fogolari, F.; Corazza, A.; Tell, G.; Quadrifoglio, F.; Viglino, P.; Boelens, R.; Esposito, G. The solution structure of DNA-free Pax-8 paired box domain accounts for redox regulation of transcriptional activity in the pax protein family. J. Biol. Chem. 2008, 283, 33321–33328. [Google Scholar] [CrossRef] [PubMed]
- Kambe, F.; Nomura, Y.; Okamoto, T.; Seo, H. Redox regulation of thyroid-transcription factors, Pax-8 and TTF-1, is involved in their increased DNA-binding activities by thyrotropin in rat thyroid FRTL-5 cells. Mol. Endocrinol. 1996, 10, 801–812. [Google Scholar] [PubMed]
- Eichhoff, O.M.; Weeraratna, A.; Zipser, M.C.; Denat, L.; Widmer, D.S.; Xu, M.; Kriegl, L.; Kirchner, T.; Larue, L.; Dummer, R.; et al. Differential LEF1 and TCF4 expression is involved in melanoma cell phenotype switching. Pigment. Cell Melanoma Res. 2011, 24, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Yasumoto, K.; Takeda, K.; Takahashi, K.; Fukuzaki, A.; Orikasa, S.; Shibahara, S. Melanocyte-specific microphthalmia-associated transcription factor isoform activates its own gene promoter through physical interaction with lymphoid-enhancing factor 1. J. Biol. Chem. 2002, 277, 28787–28794. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Li, K.; Jiang, L.L.; He, M.F.; Pu, C.H.; Kang, D.; Xie, J. Developmental toxicity of auranofin in zebrafish embryos. J. Appl. Toxicol. 2017, 37, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Marzioni, D. Natural and synthetic compounds in Ovarian Cancer: A focus on NRF2/KEAP1 pathway. Pharm. Res. 2022, 183, 106365. [Google Scholar] [CrossRef] [PubMed]
- Marrot, L.; Jones, C.; Perez, P.; Meunier, J.R. The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment. Cell Melanoma Res. 2008, 21, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.Y.; Sohn, K.C.; Kim, J.H.; Kim, S.M.; Kim, C.H.; Hwang, J.S.; Lee, J.H.; Kim, C.D.; Yoon, T.J. Impact of NAD(P)H:quinone oxidoreductase-1 on pigmentation. J. Investig. Derm. 2010, 130, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Glineur, C.; Davioud-Charvet, E.; Vandenbunder, B. The conserved redox-sensitive cysteine residue of the DNA-binding region in the c-Rel protein is involved in the regulation of the phosphorylation of the protein. Biochem. J. 2000, 352 Pt 2, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Doka, E.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, A.; Cheng, Q.; et al. Control of protein function through oxidation and reduction of persulfidated states. Sci Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef]
- Rodriguez, K.M.; Buch-Larsen, S.C.; Kirby, I.T.; Siordia, I.R.; Hutin, D.; Rasmussen, M.; Grant, D.M.; David, L.L.; Matthews, J.; Nielsen, M.L.; et al. Chemical genetics and proteome-wide site mapping reveal cysteine MARylation by PARP-7 on immune-relevant protein targets. Elife 2021, 10, e60480. [Google Scholar] [CrossRef]
- Putker, M.; Vos, H.R.; van Dorenmalen, K.; de Ruiter, H.; Duran, A.G.; Snel, B.; Burgering, B.M.; Vermeulen, M.; Dansen, T.B. Evolutionary acquisition of cysteines determines FOXO paralog-specific redox signaling. Antioxid. Redox Signal. 2015, 22, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Bismuth, K.; Maric, D.; Arnheiter, H. MITF and cell proliferation: The role of alternative splice forms. Pigment. Cell Res. 2005, 18, 349–359. [Google Scholar] [CrossRef]
- Murakami, M.; Iwata, Y.; Funaba, M. Expression and transcriptional activity of alternative splice variants of Mitf exon 6. Mol. Cell Biochem. 2007, 303, 251–257. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Pogenberg, V.; Ogmundsdottir, M.H.; Bergsteinsdottir, K.; Schepsky, A.; Phung, B.; Deineko, V.; Milewski, M.; Steingrimsson, E.; Wilmanns, M. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012, 26, 2647–2658. [Google Scholar] [CrossRef]
- Moller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Nili, M.; Mukherjee, A.; Shinde, U.; David, L.; Rotwein, P. Defining the disulfide bonds of insulin-like growth factor-binding protein-5 by tandem mass spectrometry with electron transfer dissociation and collision-induced dissociation. J. Biol. Chem. 2012, 287, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Koludrovic, D.; Davidson, I. MITF, the Janus transcription factor of melanoma. Future Oncol. 2013, 9, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Hamm, M.; Sohier, P.; Petit, V.; Raymond, J.H.; Delmas, V.; le Coz, M.; Gesbert, F.; Kenny, C.; Aktary, Z.; Pouteaux, M.; et al. BRN2 is a non-canonical melanoma tumor-suppressor. Nat. Commun. 2021, 12, 3707. [Google Scholar] [CrossRef] [PubMed]
- Yardman-Frank, M.J.; Fisher, D.E. Skin pigmentation and its control: From ultraviolet radiation to stem cells. Exp. Derm. 2021, 30, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, M.; Sartini, D.; Molinelli, E.; Campagna, R.; Pozzi, V.; Salvolini, E.; Simonetti, O.; Campanati, A.; Offidani, A. The Double-Edged Sword of Oxidative Stress in Skin Damage and Melanoma: From Physiopathology to Therapeutical Approaches. Antioxidants 2022, 11, 612. [Google Scholar] [CrossRef]
- Poerschke, R.L.; Fritz, K.S.; Franklin, C.C. Methods to detect protein glutathionylation. Curr. Protoc. Toxicol. 2013, 57, 6 17 1–6 17 18. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kline, C.D.; Anderson, M.; Bassett, J.W.; Kent, G.; Berryman, R.; Honeggar, M.; Ito, S.; Wakamatsu, K.; Indra, A.K.; Moos, P.J.; et al. MITF Is Regulated by Redox Signals Controlled by the Selenoprotein Thioredoxin Reductase 1. Cancers 2022, 14, 5011. https://doi.org/10.3390/cancers14205011
Kline CD, Anderson M, Bassett JW, Kent G, Berryman R, Honeggar M, Ito S, Wakamatsu K, Indra AK, Moos PJ, et al. MITF Is Regulated by Redox Signals Controlled by the Selenoprotein Thioredoxin Reductase 1. Cancers. 2022; 14(20):5011. https://doi.org/10.3390/cancers14205011
Chicago/Turabian StyleKline, Chelsey D., Madeleine Anderson, John W. Bassett, Gail Kent, Rachel Berryman, Matthew Honeggar, Shosuke Ito, Kazumasa Wakamatsu, Arup K. Indra, Philip J. Moos, and et al. 2022. "MITF Is Regulated by Redox Signals Controlled by the Selenoprotein Thioredoxin Reductase 1" Cancers 14, no. 20: 5011. https://doi.org/10.3390/cancers14205011
APA StyleKline, C. D., Anderson, M., Bassett, J. W., Kent, G., Berryman, R., Honeggar, M., Ito, S., Wakamatsu, K., Indra, A. K., Moos, P. J., Leachman, S. A., & Cassidy, P. B. (2022). MITF Is Regulated by Redox Signals Controlled by the Selenoprotein Thioredoxin Reductase 1. Cancers, 14(20), 5011. https://doi.org/10.3390/cancers14205011