
Targeting Apoptosis in AML: Where Do We Stand?

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
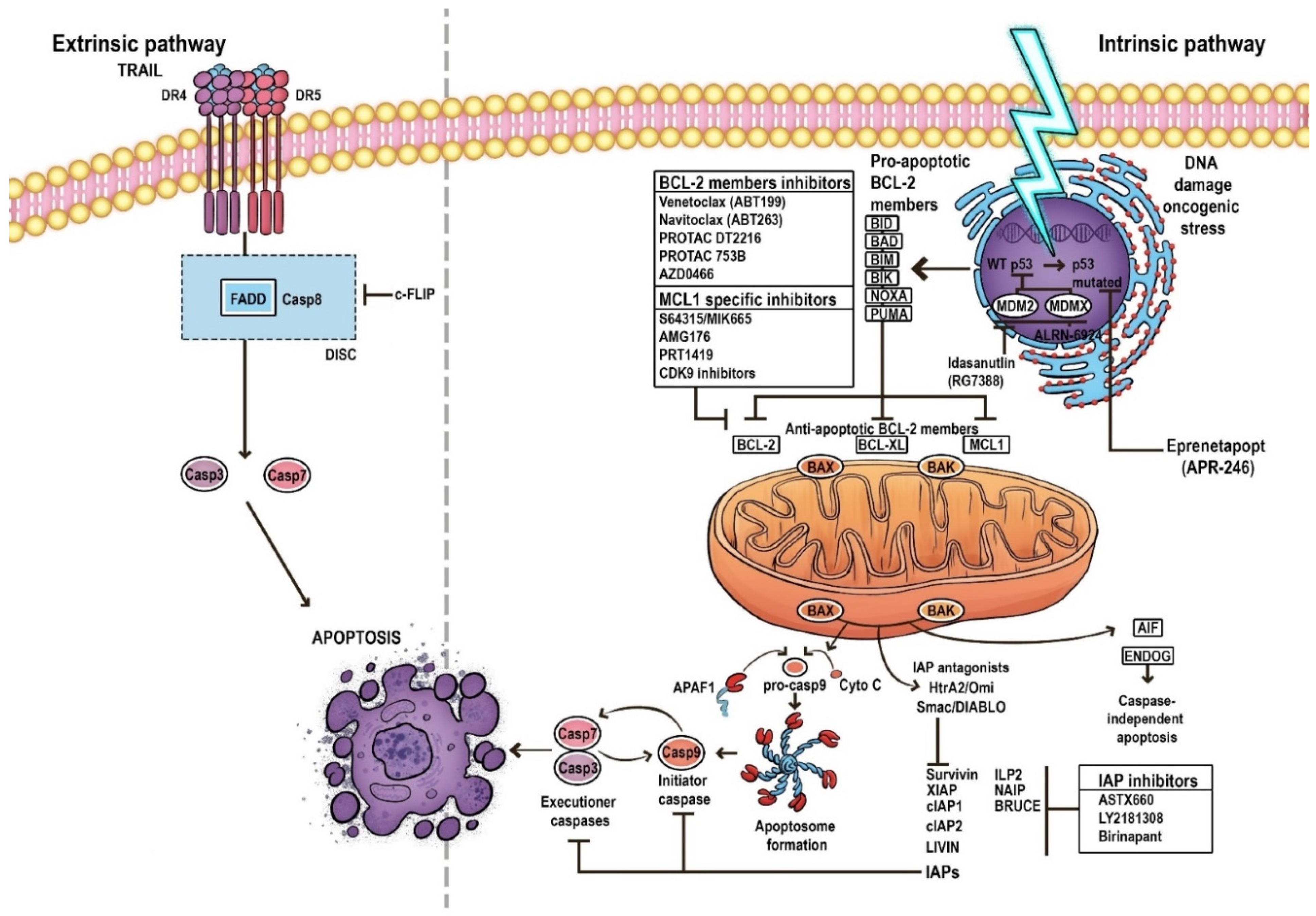
2. Apoptosis
3. Pathways of Apoptosis
4. The Mechanism of the Mitochondrial Apoptosis Pathway
5. BCL-2 Family
6. Evasion of Apoptosis in AML
7. An Overview of the Impact of Dysregulation in the Expression of Apoptosis Genes on AML
8. Targeting BCL-2 in AML
9. In the Grip of Apoptosis—The Desired Future after Venetoclax
9.1. Targeting BCL-XL
9.2. Targeting MCL-1
9.3. Targeting IAPs
9.4. Targeting p53—Dependent Apoptosis Pathways
10. Ferroptosis—A New Type of Regulated Cell Death
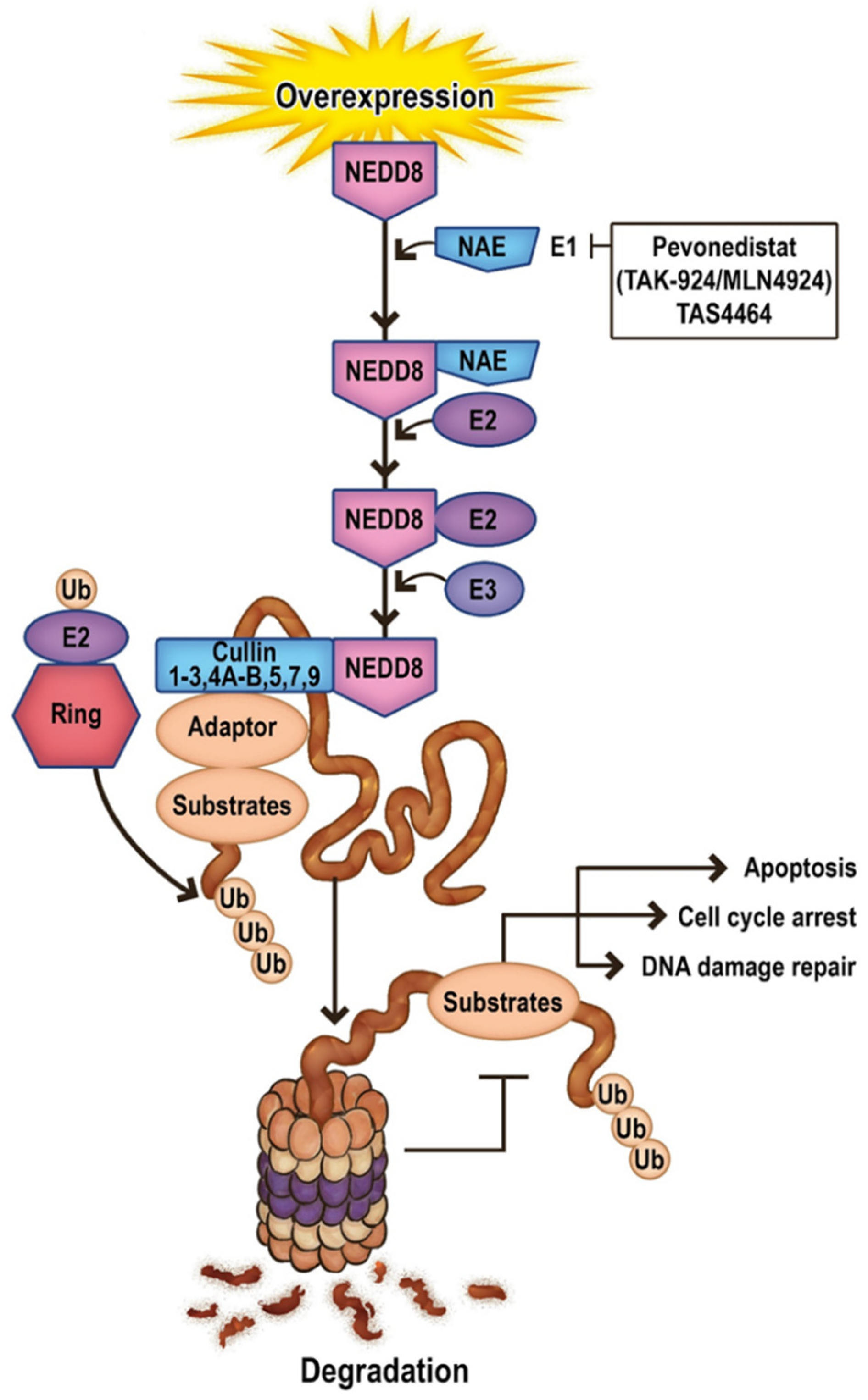
11. Future Directions in Apoptosis-Related Neddylation
12. Targeting Neddylation in AML
12.1. Pevonedistat
12.2. TAS4464
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Halik, A.; Arends, C.M.; Bullinger, L.; Damm, F.; Frick, M. Refining AML Treatment: The Role of Genetics in Response and Resistance Evaluation to New Agents. Cancers 2022, 14, 1689. [Google Scholar] [CrossRef]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. N. Eng. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef]
- Hu, X.-M.; Li, Z.-X.; Lin, R.-H.; Shan, J.-Q.; Yu, Q.-W.; Wang, R.-X.; Liao, L.-S.; Yan, W.-T.; Wang, Z.; Shang, L.; et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Front. Cell Dev. Biol. 2021, 9, 634690. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef]
- Valentin, R.; Grabow, S.; Davids, M.S. The rise of apoptosis: Targeting apoptosis in hematologic malignancies. Blood 2018, 132, 1248–1264. [Google Scholar] [CrossRef]
- Testa, U.; Riccioni, R. Deregulation of apoptosis in acute myeloid leukemia. Haematologica 2007, 92, 81–94. [Google Scholar] [CrossRef]
- Li, B.; Dou, S.-X.; Yuan, J.-W.; Liu, Y.-R.; Li, W.; Ye, F.; Wang, P.-Y.; Li, H. Intracellular transport is accelerated in early apoptotic cells. Proc. Natl. Acad. Sci. USA 2018, 115, 12118–12123. [Google Scholar] [CrossRef] [PubMed]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; D’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Letai, A. Apoptosis and Cancer. Annu. Rev. Cancer Biol. 2017, 1, 275–294. [Google Scholar] [CrossRef]
- Koren, E.; Fuchs, Y. Modes of Regulated Cell Death in Cancer. Cancer Discov. 2021, 11, 245–265. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.H.; Clark, A.C. Death by Caspase Dimerization. In Protein Dimerization and Oligomerization in Biology; Springer: Berlin/Heidelberg, Germany, 2012; Volume 747, pp. 55–73. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta 2020, 1867, 118688. [Google Scholar] [CrossRef] [PubMed]
- Hatok, J.; Racay, P. Bcl-2 family proteins: Master regulators of cell survival. Biomol. Concepts 2016, 7, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Zhang, L.-N.; Li, J.-Y.; Xu, W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther. 2012, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cassier, P.A.; Castets, M.; Belhabri, A.; Vey, N. Targeting apoptosis in acute myeloid leukaemia. Br. J. Cancer 2017, 117, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: P53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef]
- Kitada, S.; Pedersen, I.M.; Schimmer, A.D.; Reed, J.C. Dysregulation of apoptosis genes in hematopoietic malignancies. Oncogene 2002, 21, 3459–3474. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Hunter, A.M.; Sallman, D.A. Current status and new treatment approaches in TP53 mutated AML. Best Pract. Res. Clin. Haematol. 2019, 32, 134–144. [Google Scholar] [CrossRef]
- Wang, C.; Sallman, D.A. What Are the Prospects for Treating TP53 Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia? Cancer J. 2022, 28, 51–61. [Google Scholar] [CrossRef]
- Hou, H.-A.; Chou, W.-C.; Kuo, Y.-Y.; Liu, C.-Y.; Lin, L.-I.; Tseng, M.-H.; Chiang, Y.-C.; Liu, M.-C.; Liu, C.W.; Tang, J.-L.; et al. TP53 mutations in De Novo acute myeloid leukemia patients: Longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosom. Cancer 2019, 58, 875–888. [Google Scholar] [CrossRef]
- Shallis, R.M.; Bewersdorf, J.P.; Stahl, M.F.; Halene, S.; Zeidan, A.M. Are We Moving the Needle for Patients with TP53-Mutated Acute Myeloid Leukemia? Cancers 2022, 14, 2434. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Niparuck, P.; Police, P.; Noikongdee, P.; Siriputtanapong, K.; Limsuwanachot, N.; Rerkamnuaychoke, B.; Chuncharunee, S.; Siriboonpiputtana, T. TP53 mutation in newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Diagn. Pathol. 2021, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. P53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2016, 31, 1296–1305. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Hrdinka, M.; Yabal, M. Inhibitor of apoptosis proteins in human health and disease. Genes Immun. 2019, 20, 641–650. [Google Scholar] [CrossRef]
- Cheung, C.H.A.; Chang, Y.-C.; Lin, T.-Y.; Cheng, S.M.; Leung, E. Anti-apoptotic proteins in the autophagic world: An update on functions of XIAP, Survivin and BRUCE. J. Biomed. Sci. 2020, 27, 31. [Google Scholar] [CrossRef]
- Oberoi-Khanuja, T.K.; Murali, A.; Rajalingam, K. IAPs on the move: Role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis. 2013, 4, e784. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Wierzbowska, A.; Cebula-Obrzut, B.; Pluta, P.; Stępka, K.; Szmigielska-Kapłon, A.; Grzybowska-Izydorczyk, O.; Czemerska, M.; Smolewski, P.; Wrzesien-Kus, A.; et al. Prognostic value of inhibitor of apoptosis protein family expression in patients with acute myeloid leukemia. Leuk. Lymphoma 2015, 56, 2529–2535. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.-M.; Büsselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, F.; Luo, Q.; Wu, X.; Liu, Z.; Chen, H.; Huang, Y. Inhibition of XIAP increases carboplatin sensitivity in ovarian cancer. Onco Targets Ther. 2018, 11, 8751–8759. [Google Scholar] [CrossRef]
- Martinez-Ruiz, G.; Maldonado, V.; Ceballos-Cancino, G.; Grajeda, J.P.R.; Melendez-Zajgla, J. Role of Smac/DIABLO in cancer progression. J. Exp. Clin. Cancer Res. 2008, 27, 48. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, E.; Vachette, P.; Cossu, F.; Malvezzi, F.; Bolognesi, M.; Milani, M. The Activator of Apoptosis Smac-DIABLO Acts as a Tetramer in Solution. Biophys. J. 2015, 108, 714–723. [Google Scholar] [CrossRef][Green Version]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- Karakas, T.; Maurer, U.; Weidmann, E.; Miething, C.C.; Hoelzer, D.; Bergmann, L. High expression of bcl-2 mRNA as a determinant of poor prognosis in acute myeloid leukemia. Ann. Oncol. 1998, 9, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Sahu, G.; Jena, R.K. Clinical Significance of P53 and Bcl-2 in Acute Myeloid Leukemia Patients of Eastern India. Hematol. Rep. 2011, 3, e28. [Google Scholar] [CrossRef] [PubMed]
- Tóthová, E.; Fricova, M.; Stecová, N.; Kafková, A.; Elbertová, A. High expression of Bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Neoplasma 2002, 49, 141–144. [Google Scholar] [PubMed]
- Kornblau, S.M.; Thall, P.F.; Estrov, Z.; Walterscheid, M.; Patel, S.; Theriault, A.; Keating, M.J.; Kantarjian, H.; Estey, E.; Andreeff, M. The prognostic impact of BCL2 protein expression in acute myelogenous leukemia varies with cytogenetics. Clin. Cancer Res. 1999, 5, 1758–1766. [Google Scholar]
- Zhou, J.-D.; Zhang, T.-J.; Xu, Z.-J.; Gu, Y.; Ma, J.-C.; Li, X.-X.; Guo, H.; Wen, X.-M.; Zhang, W.; Yang, L.; et al. BCL2 overexpression: Clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 2019, 14, 68. [Google Scholar] [CrossRef]
- Fuchs, O.; Kostecka, A.; Provazníková, D.; Krásná, B.; Kotlín, R.; Staňková, M.; Kobylka, P.; Dostálová, G.; Zeman, M.; Chochola, M. CCAAT/enhancer-binding protein alpha (CEBPA) polymorphisms and mutations in healthy individuals and in patients with peripheral artery disease, ischaemic heart disease and hyperlipidaemia. Folia Biol. (Praha) 2010, 56, 51–57. [Google Scholar]
- Lange, A.; Almeida, L.; Silva, C.A.; Scheucher, P.; Chahud, F.; Krause, A.; Bohlander, S.; Rego, E. CCAAT/enhancer-binding protein alpha (CEBPA) gene haploinsufficiency does not alter hematopoiesis or induce leukemia in Lck-CALM/AF10 transgenic mice. Braz. J. Med. Biol. Res. 2019, 52, e8424. [Google Scholar] [CrossRef]
- Schaich, M.; Illmer, T.; Seitz, G.; Mohr, B.; Schäkel, U.; Beck, J.F.; Ehninger, G. The prognostic value of Bcl-XL gene expression for remission induction is influenced by cytogenetics in adult acute myeloid leukemia. Haematologica 2001, 86, 470–477. [Google Scholar]
- Bekhet, M.M.; Fouad, N.T.A.; Fathey, H.; El-Sayed, E.; Hendawy, L.M.; Khattab, D. Prognostic value of Bcl2-associated X protein (Bax) expression in adult Egyptian patients with acute myeloid leukemia. Indian J. Med. Res. Pharm. Sci. 2016, 3, 1–7. [Google Scholar]
- Köhler, T.; Schill, C.; Deininger, M.; Krahl, R.; Borchert, S.; Hasenclever, D.; Leiblein, S.; Wagner, O.; Niederwieser, D. High Bad and Bax mRNA expression correlate with negative outcome in acute myeloid leukemia (AML). Leukemia 2002, 16, 22–29. [Google Scholar] [CrossRef]
- Zhu, R.; Li, L.; Nguyen, B.; Seo, J.; Wu, M.; Seale, T.; Levis, M.; Duffield, A.; Hu, Y.; Small, D. FLT3 tyrosine kinase inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute leukemia cells through BIM activation. Signal Transduct. Target. Ther. 2021, 6, 186. [Google Scholar] [CrossRef]
- Lin, V.S.; Lew, T.E.; Handunnetti, S.M.; Blombery, P.; Nguyen, T.; Westerman, D.A.; Kuss, B.J.; Tam, C.S.; Roberts, A.W.; Seymour, J.F.; et al. BTK inhibitor therapy is effective in patients with CLL resistant to venetoclax. Blood 2020, 135, 2266–2270. [Google Scholar] [CrossRef]
- Wei, Y.; Cao, Y.; Sun, R.; Cheng, L.; Xiong, X.; Jin, X.; He, X.; Lu, W.; Zhao, M. Targeting Bcl-2 Proteins in Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 584974. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, L.; Sternberg, D.; Tang, L.; Galinsky, I.; DeAngelo, D.; Stone, R. The FLT3 Internal Tandem Duplication Mutation Prevents Apoptosis in Interleukin-3-Deprived BaF3 Cells Due to Protein Kinase A and Ribosomal S6 Kinase 1–Mediated BAD Phosphorylation at Serine 112. Cancer Res. 2005, 65, 7338–7347. [Google Scholar] [CrossRef]
- Kim, K.-T.; Levis, M.; Small, D. Constitutively activated FLT3 phosphorylates BAD partially through Pim-1. Br. J. Haematol. 2006, 134, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD–specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef]
- Li, X.-X.; Zhou, J.-D.; Wen, X.-M.; Zhang, T.-J.; Wu, D.-H.; Deng, Z.-Q.; Zhang, Z.-H.; Lian, X.-Y.; He, P.-F.; Yao, X.-Y.; et al. Increased MCL-1 expression predicts poor prognosis and disease recurrence in acute myeloid leukemia. Onco Targets Ther. 2019, 12, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, C.; Liu, Z.; Hospital, S.; Hu, R. Expression and Relationship of SAMHD1 with Other Apoptotic and Autophagic Genes in Acute Myeloid Leukemia Patients. Acta Haematol. 2019, 143, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Mazzone, C.; Niscola, P.; de Fabritiis, P. TP53 Mutations in Acute Myeloid Leukemia: Still a Daunting Challenge? Front. Oncol. 2021, 10, 610820. [Google Scholar] [CrossRef]
- Ahmadzadeh, A.; Mohammadi, M.H.; Mezginezhad, F.; Nezhad, H.A.; Parkhideh, S.; Khosravi, M.; Khazaei, Z.; Adineh, H.A.; Farsani, M.A. The Expression of the Tp53 Gene in Various Classes of Acute Myeloid Leukemia. World Cancer Res. J. 2018, 5, e1178. [Google Scholar]
- Suliman, G.A.; Mabrouk, M.M.; Rabee, E.S.; Gawaly, A. SMAC/DIABLO gene expression in acute myeloid leukemia patients. Egypt. J. Haematol. 2013, 38, 80–83. [Google Scholar] [CrossRef]
- Pluta, A.; Robak, T.; Cebula, B.; Majchrzak, A.; Pluta, P.; Brzozowski, K.; Stępka, K.; Szmigielska-Kapłon, A.; Grzybowska-Izydorczyk, O.; Czemerska, M.; et al. The role of NF-κB and Smac/DIABLO proteins in the treatment response and survival of acute myeloid leukemia patients. Arch. Med. Sci. 2021, 17, 700–707. [Google Scholar] [CrossRef]
- Saxena, K.; DiNardo, C.; Daver, N.; Konopleva, M. Harnessing Apoptosis in AML. Clin. Lymphoma Myeloma Leuk. 2020, 20, S61–S64. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2018, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.-Z.; Fiedler, W.; Lin, T.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: Results from a phase Ib/II study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Eng. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Wei, A.H.; Panayiotidis, P.; Montesinos, P.; Laribi, K.; Ivanov, V.; Kim, I.; Novak, J.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Author Correction: 6-month follow-up of VIALE-C demonstrates improved and durable efficacy in patients with untreated AML ineligible for intensive chemotherapy. Blood Cancer J. 2021, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Amaya, M.; Strati, P.; Konopleva, M.Y. Venetoclax for AML: Changing the treatment paradigm. Blood Adv. 2019, 3, 4326–4335. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Kadia, T.; Daver, N.; Xiao, L.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax combined with FLAG-IDA induction and consolidation in newly diagnosed acute myeloid leukemia. Am. J. Hematol. 2022, 97, 1035–1043. [Google Scholar] [CrossRef]
- Wang, H.; Mao, L.; Yang, M.; Qian, P.; Lu, H.; Tong, H.; Xie, W.; Zhou, D.; Huang, X.; Wang, Y.; et al. Venetoclax plus 3+7 daunorubicin and cytarabine chemotherapy as first-line treatment for adults with acute myeloid leukaemia: A multicentre, single-arm, phase 2 trial. Lancet Haematol. 2022, 9, e415–e424. [Google Scholar] [CrossRef]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Döhner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.T.; Xiao, Y.; et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef]
- Parry, N.; Wheadon, H.; Copland, M. The application of BH3 mimetics in myeloid leukemias. Cell Death Dis. 2021, 12, 222. [Google Scholar] [CrossRef]
- Anuar, N.N.M.; Hisam, N.S.N.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 564108. [Google Scholar] [CrossRef] [PubMed]
- Arulananda, S.; O’Brien, M.; Evangelista, M.; Harris, T.J.; Steinohrt, N.S.; Jenkins, L.J.; Walkiewicz, M.; O’Donoghue, R.J.J.; Poh, A.R.; Thapa, B.; et al. BCL-XL is an actionable target for treatment of malignant pleural mesothelioma. Cell Death Discov. 2020, 6, 114. [Google Scholar] [CrossRef]
- He, Y.; Khan, S.; Huo, Z.; Lv, D.; Zhang, X.; Liu, X.; Yuan, Y.; Hromas, R.; Xu, M.; Zheng, G.; et al. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol. 2020, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, O.; Bokorova, R. Preclinical Studies of PROTACs in Hematological Malignancies. Cardiovasc. Hematol. Disord. Targets 2021, 21, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S.; et al. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, Q.; Zhang, W.; Andreeff, M.; Jain, N.; Zhang, P.; Zheng, G.; Zhou, D.; Konopleva, M. Targeting BCL-XL and BCL-2 by Protac 753B Effectively Eliminates AML Cells and Enhances Efficacy of Chemotherapy by Targeting Senescent Cells. Blood 2021, 138, 2230. [Google Scholar] [CrossRef]
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhimolea, E.; et al. Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov. 2021, 11, 1542–1561. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Ma, L.; Liu, Y.; Wei, M.; Yao, Y.; Li, C.; Wang, R.; Liu, N.; Dong, Z.; Li, X.; et al. Proteolysis-Targeting Chimera (PROTAC) Modification of Dovitinib Enhances the Antiproliferative Effect against FLT3-ITD-Positive Acute Myeloid Leukemia Cells. J. Med. Chem. 2021, 64, 16497–16511. [Google Scholar] [CrossRef] [PubMed]
- Gilioli, D.; Fusco, S.; Giannetti, K. Therapy-Induced Senescence as an Anti-Cancer and Immune-Stimulatory Strategy. Blood 2021, 138, 4419. [Google Scholar] [CrossRef]
- Bond, M.J.; Crews, C.M. Proteolysis targeting chimeras (PROTACs) come of age: Entering the third decade of targeted protein degradation. RSC Chem. Biol. 2021, 2, 725–742. [Google Scholar] [CrossRef]
- Konopleva, M.; Jain, N.; Andersen, C.L.; Francisco, N.C.; Elgeioushi, N.; Hobson, R.; Scott, M.; Stone, J.; Sharma, S.; Gutierrez, P.M.; et al. NIMBLE: A Phase I/II Study of AZD0466 Monotherapy or in Combination in Patients with Advanced Hematological Malignancies. Blood 2021, 138, 2353. [Google Scholar] [CrossRef]
- Patterson, C.M.; Balachander, S.B.; Grant, I.; Pop-Damkov, P.; Kelly, B.; McCoull, W.; Parker, J.; Giannis, M.; Hill, K.J.; Gibbons, F.D.; et al. Design and optimisation of dendrimer-conjugated Bcl-2/xL inhibitor, AZD0466, with improved therapeutic index for cancer therapy. Commun. Biol. 2021, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 67. [Google Scholar] [CrossRef]
- Sancho, M.; Leiva, D.; Lucendo, E.; Orzáez, M. Understanding MCL1: From cellular function and regulation to pharmacological inhibition. FEBS J. 2021. [Google Scholar] [CrossRef]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Quinn, B.A.; Dash, R.; Azab, B.; Sarkar, S.; Das, S.K.; Kumar, S.; Oyesanya, R.A.; Dasgupta, S.; Dent, P.; Grant, S.; et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin. Investig. Drugs 2011, 20, 1397–1411. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Foster, M.C.; Blackford, A.L.; Litzow, M.R.; Morris, L.E.; Strickland, S.A.; Lancet, J.E.; Bose, P.; Levy, M.Y.; Tibes, R.; et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7+3) in newly diagnosed acute myeloid leukemia. Haematologica 2015, 100, 1172–1179. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Lee, D.J.; Frattini, M.G.; Fine, G.D.; Costas, J.; Kolibaba, K.; Anthony, S.P.; Bearss, D.J.; Smith, B.D. Phase I Study of Alvocidib Followed by 7+3 (Cytarabine + Daunorubicin) in Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 60–69. [Google Scholar] [CrossRef]
- Cidado, J.; Boiko, S.; Proia, T.; Ferguson, D.; Criscione, S.W.; Martin, M.S.; Pop-Damkov, P.; Su, N.; Franklin, V.N.R.; Chilamakuri, C.S.R.; et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2020, 26, 922–934. [Google Scholar] [CrossRef]
- Cetraro, P.; Plaza-Diaz, J.; MacKenzie, A.; Abadía-Molina, F. A Review of the Current Impact of Inhibitors of Apoptosis Proteins and Their Repression in Cancer. Cancers 2022, 14, 1671. [Google Scholar] [CrossRef]
- Ball, S.; Borthakur, G. Apoptosis targeted therapies in acute myeloid leukemia: An update. Expert Rev. Hematol. 2020, 13, 1373–1386. [Google Scholar] [CrossRef]
- Schimmer, A.D.; Herr, W.; Hänel, M.; Borthakur, G.; Frankel, A.; Horst, H.-A.; Martin, S.; Kassis, J.; Desjardins, P.; Seiter, K.; et al. Addition of AEG35156 XIAP Antisense Oligonucleotide in Reinduction Chemotherapy Does Not Improve Remission Rates in Patients with Primary Refractory Acute Myeloid Leukemia in a Randomized Phase II Study. Clin. Lymphoma Myeloma Leuk. 2011, 11, 433–438. [Google Scholar] [CrossRef]
- Mita, M.M.; LoRusso, P.M.; Papadopoulos, K.P.; Gordon, M.S.; Mita, A.C.; Ferraldeschi, R.; Keer, H.; Oganesian, A.; Su, X.Y.; Jueliger, S.; et al. A Phase I Study of ASTX660, an Antagonist of Inhibitors of Apoptosis Proteins, in Adults with Advanced Cancers or Lymphoma. Clin. Cancer Res. 2020, 26, 2819–2826. [Google Scholar] [CrossRef]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Erba, H.P.; Sayar, H.; Juckett, M.; Lahn, M.; André, V.; Callies, S.; Schmidt, S.; Kadam, S.; Brandt, J.T.; Van Bockstaele, D.; et al. Safety and Pharmacokinetics of the Antisense Oligonucleotide (ASO) LY2181308 as a Single-Agent or in Combination with Idarubicin and Cytarabine in Patients with Refractory or Relapsed Acute Myeloid Leukemia (AML). Investig. N. Drugs 2013, 31, 1023–1034. [Google Scholar] [CrossRef]
- Li, Y.; Lu, W.; Yang, J.; Edwards, M.; Jiang, S. Survivin as a biological biomarker for diagnosis and therapy. Expert Opin. Biol. Ther. 2021, 21, 1429–1441. [Google Scholar] [CrossRef]
- Frey, M.N.V.; Luger, S.; Mangan, J.; Zebrowski, B.A.; Loren, M.A.W.; Minderman, H.; Baird, J.; Porter, D.L.; Hexner, E.O.; Kumar, M.A.J.; et al. A Phase I Study Using Single Agent Birinapant in Patients with Relapsed Myelodysplastic Syndrome and Acute Myelogenous Leukemia. Blood 2014, 124, 3758. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Cheung, C.H.A. An Updated Review of Smac Mimetics, LCL161, Birinapant, and GDC-0152 in Cancer Treatment. Appl. Sci. 2020, 11, 335. [Google Scholar] [CrossRef]
- George, B.; Kantarjian, H.; Baran, N.; Krocker, J.D.; Rios, A. TP53 in Acute Myeloid Leukemia: Molecular Aspects and Patterns of Mutation. Int. J. Mol. Sci. 2021, 22, 10782. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Fujihara, K.M.; Zhang, B.Z.; Jackson, T.D.; Ogunkola, M.O.; Nijagal, B.; Milne, J.V.; Sallman, D.A.; Ang, C.S.; Nikolic, I.; Kearney, C.J. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci. Adv. 2022, 8, 9427. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Gallacher, P.; Wennborg, A.; Hickman, D.K.; et al. Phase II Trial of Eprenetapopt (APR-246) in Combination with Azacitidine (AZA) as Maintenance Therapy for TP53 Mutated AML or MDS Following Allogeneic Stem Cell Transplantation (SCT). Blood 2021, 138, 409. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Gallacher, P.; Wennborg, A.; Hickman, D.K.; et al. Phase I and Expansion Study of Eprenetapopt (APR-246) in Combination with Venetoclax (VEN) and Azacitidine (AZA) in TP53-Mutant Acute Myeloid Leukemia (AML). Blood 2021, 138, 3409. [Google Scholar] [CrossRef]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2021, 107, 403–416. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019, 105, 1539–1551. [Google Scholar] [CrossRef]
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; Kooy, M.V.M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef]
- Munisamy, M.; Mukherjee, N.; Thomas, L.; Pham, A.T.; Shakeri, A.; Zhao, Y.; Kolesar, J.; Rao, P.P.N.; Rangnekar, V.M.; Rao, M. Therapeutic opportunities in cancer therapy: Targeting the p53-MDM2/MDMX interactions. Am. J. Cancer Res. 2021, 11, 5762–5781. [Google Scholar]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef]
- Han, X.; Medeiros, L.J.; Zhang, Y.H.; You, M.J.; Andreeff, M.; Konopleva, M.; Bueso-Ramos, C.E. High Expression of Human Homologue of Murine Double Minute 4 and the Short Splicing Variant, HDM4-S, in Bone Marrow in Patients with Acute Myeloid Leukemia or Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2016, 16, S30–S38. [Google Scholar] [CrossRef]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.-M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–89. [Google Scholar]
- García-Cano, J.; Sánchez-Tena, S.; Sala-Gaston, J.; Figueras, A.; Viñals, F.; Bartrons, R.; Ventura, F.; Rosa, J.L.; Sala-Gaston, J. Regulation of the MDM2-p53 pathway by the ubiquitin ligase HERC2. Mol. Oncol. 2019, 14, 69–86. [Google Scholar] [CrossRef]
- Yee, K.; Papayannidis, C.; Vey, N.; Dickinson, M.J.; Kelly, K.R.; Assouline, S.; Kasner, M.; Seiter, K.; Drummond, M.W.; Yoon, S.-S.; et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study. Leuk. Res. 2020, 100, 106489. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef]
- Daver, N.; Garcia, J.; Jonas, B.; Kevin Kelly, R.; Assouline, S.; Brandwein, J.M.; Fenaux, P.; Olin, R.L.; Martinelli, G.; Paolini, S.; et al. Updated Results from the Venetoclax (Ven) in Combination with Idasanutlin (Idasa) Arm of a Phase 1b Trial in Elderly Patients (Pts) with Relapsed or Refractory (R/R) AML Ineligible for Cytotoxic Chemotherapy. Blood 2019, 134, 229. [Google Scholar] [CrossRef]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Ben Neriah, D.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef]
- Sallman, D.A.; Borate, U.; Cull, E.H.; Donnellan, W.B.; Komrokji, R.S.; Steidl, U.G.; Corvez, M.M.; Payton, M.; Annis, D.A.; Pinchasik, D.; et al. Phase 1/1b Study of the Stapled Peptide ALRN-6924, a Dual Inhibitor of MDMX and MDM2, as Monotherapy or in Combination with Cytarabine for the Treatment of Relapsed/Refractory AML and Advanced MDS with TP53 Wild-Type. Blood 2018, 132, 4066. [Google Scholar] [CrossRef]
- Andreozzi, F.; Massaro, F.; Wittnebel, S.; Spilleboudt, C.; Lewalle, P.; Salaroli, A. New Perspectives in Treating Acute Myeloid Leukemia: Driving towards a Patient-Tailored Strategy. Int. J. Mol. Sci. 2022, 23, 3887. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53–MDM2 interaction by small-molecule inhibitors: Learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Z.; Peng, H. Molecular Mechanisms of Ferroptosis and Its Roles in Hematologic Malignancies. Front. Oncol. 2021, 11, 743006. [Google Scholar] [CrossRef]
- Ju, J.; Song, Y.-N.; Wang, K. Mechanism of Ferroptosis: A Potential Target for Cardiovascular Diseases Treatment. Aging Dis. 2021, 12, 261–276. [Google Scholar] [CrossRef]
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 12. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018, 44, 92–102. [Google Scholar] [CrossRef]
- Zhao, Y.; Morgan, M.A.; Sun, Y. Targeting Neddylation Pathways to Inactivate Cullin-RING Ligases for Anticancer Therapy. Antioxid. Redox Signal. 2014, 21, 2383–2400. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, C.-C.; Zhang, H.-P.; Li, G.-Q.; Li, S.-S. MLN4924 suppresses neddylation and induces cell cycle arrest, senescence and apoptosis in human osteosarcoma. Oncotarget 2016, 7, 45263–45274. [Google Scholar] [CrossRef]
- Sumi, H.; Inazuka, M.; Morimoto, M.; Hibino, R.; Hashimoto, K.; Ishikawa, T.; Kuida, K.; Smith, P.G.; Yoshida, S.; Yabuki, M. An inhibitor of apoptosis protein antagonist T-3256336 potentiates the antitumor efficacy of the Nedd8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924). Biochem. Biophys. Res. Commun. 2016, 480, 380–386. [Google Scholar] [CrossRef]
- Cojocari, D.; Smith, B.N.; Purkal, J.J.; Arrate, M.P.; Huska, J.D.; Xiao, Y.; Gorska, A.; Hogdal, L.J.; Ramsey, H.E.; Boghaert, E.R.; et al. Pevonedistat and azacitidine upregulate NOXA (PMAIP1) to increase sensitivity to venetoclax in preclinical models of acute myeloid leukemia. Haematologica 2021, 107, 825–835. [Google Scholar] [CrossRef]
- Snow, A.; Zeidner, J.F. The development of pevonedistat in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML): Hope or hype? Ther. Adv. Hematol. 2022, 13, 20406207221112899. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zheng, Y.; Sun, Y. Neddylation regulation of mitochondrial structure and functions. Cell Biosci. 2021, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Adès, L.; Girshova, L.; Doronin, V.A.; Díez-Campelo, M.; Valcárcel, D.; Kambhampati, S.; Viniou, N.-A.; Woszczyk, D.; Arias, R.D.P.; Symeonidis, A.; et al. Pevonedistat plus azacitidine v.s. azacitidine alone in higher-risk MDS/chronic myelomonocytic leukemia or low-blast-percentage AML. Blood Adv. 2022, 6, 5132–5145. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Ongoing Trial | Intervention/Treatment | Clinical Trial Phase | Serial NCT Number | Current Status |
|---|---|---|---|---|
| CC-90011 given concurrently with Venetoclax and Azacitidine in r/r AML and treatment-naive subjects with AML who are not eligible for intensive induction chemotherapy. | CC-90011, Venetoclax, Azacitidine | Phase I/II | NCT04748848 | Completed |
| Milademetan tosylate with cytarabine with or without ventoclax in treating participants with r/r AML. | Cytarabine, Milademetan Tosylate, Venetoclax | Phase I/II | NCT03634228 | Completed |
| Low-dose cytarabine or azacitidine plus venetoclax and quizartinib in newly diagnosed AML patients aged equal or more than 60 years old. | Azacitidine, Venetoclax, Quizartinib, Cytarabine | Phase I/II | NCT04687761 | Recruiting |
| CA-4948 monotherapy and in combination with azacitidine or venetoclax in AML patients. | Emavusertib, Azacitidine, Venetoclax | Phase I/IIa | NCT04278768 | Active, not recruiting |
| Anti-OX40 antibody PF-04518600 (OX40) with or without venetoclax, avelumab, glasdegib, gemtuzumab ozogamicin, and azacitidine in treating r/r AML. | PF-04518600, Avelumab, Azacitidine, Gemtuzumab Ozogamicin, Glasdegib, Glasdegib Maleate, Venetoclax | Phase Ib/II | NCT03390296 | Active, not recruiting |
| VEN-OM assessing venetoclax with escalating doses of omacetaxine in patients with r/r AML. | Omacetaxine, Venetoclax | Phase Ib/II | NCT04926285 | Recruiting |
| ENAVEN-AML assessing enasidenib in combination with venetoclax in patients with IDH2-mutated r/r AML. | Enasidenib, Venetoclax | Phase Ib/II | NCT04092179 | Recruiting |
| Omacetaxine with venetoclax in treating patients with r/r AML and have a genetic change RUNX1. | Omacetaxine Mepesuccinate, Venetoclax | Phase Ib/II | NCT04874194 | Recruiting |
| IMGN632 with azacitidine and/or venetoclax in patients with relapsed and frontline CD123-positive AML, and antileukemia activity of IMGN632 when administered as monotherapy in patients with MRD+ AML after frontline treatment. | Azacitidine, IMGN632, Venetoclax | Phase Ib/II | NCT04086264 | Recruiting |
| Decitabine/cedazuridine (ASTX727) and venetoclax in combination with ivosidenib or enasidenib in r/r AML. | Decitabine, Cedazuridine, Enasidenib, Ivosidenib, Venetoclax | Phase Ib/II | NCT04774393 | Recruiting |
| Venetoclax in combination with quizartinib in r/r FLT3 (+) AML. | Quizartinib, Venetoclax | Phase Ib/II | NCT03735875 | Active, not recruiting |
| Venetoclax with ivosidenib with or without azacitidine, in treating patients with IDH1-mutated AML. | Azacitidine, Ivosidenib, Venetoclax | Phase Ib/II | NCT03471260 | Recruiting |
| Quizartinib, decitabine, and venetoclax in treating participants with acute myeloid leukemia that is untreated or has come back (relapsed). | Decitabine, Quizartinib, Venetoclax | Phase I/II | NCT03661307 | Recruiting |
| DS-1594b with or without azacitidine, venetoclax, or mini-HCVD in treating patients with r/r AML. | DS-1594b, Azacitidine, Venetoclax, Mini-HCVD | Phase I/II | NCT04752163 | Active, not recruiting |
| Gilteritinib with azacitidine and venetoclax in treating patients with FLT3-mutation positive r/r acute myeloid leukemia. | Azacitidine, Gilteritinib, Venetoclax | Phase I/II | NCT04140487 | Recruiting |
| Venetoclax with azacitidine and pevonedistat in treating patients with newly diagnosed AML. | Azacitidine, Pevonedistat, Venetoclax | Phase I/II | NCT03862157 | Active, not recruiting |
| Venetoclax and lintuzumab-ac225 in patients with with CD33 positive r/r AML. | Lintuzumab-Ac225, Venetoclax, Spironolactone | Phase I/II | NCT03867682 | Recruiting |
| RELAX assessing venetoclax in combination with increasing cytarabine doses plus mitoxantrone in r/r AML. | Venetoclax, Cytarabine, Mitoxantron | Phase I/II | NCT04330820 | Recruiting |
| Gilteritinib given together with ASTX727 and venetoclax in treating patients with FLT3-mutated AML that is newly diagnosed or r/r. | Decitabine, Cedazuridine, Gilteritinib, Venetoclax | Phase I/II | NCT05010122 | Recruiting |
| Liposome-encapsulated daunorubicin-cytarabine (CPX-351) and venetoclax in r/r or untreated AML patients. | CPX-351, Venetoclax | Phase II | NCT03629171 | Recruiting |
| Venetoclax, cladribine, low dose cytarabine, and azacitidine in treating patients with AML that has previously not been treated. | Azacitidine, Cladribine, Cytarabine, Venetoclax | Phase II | NCT03586609 | Recruiting |
| AML secondary to myeloproliferative neoplasms unfit for intensive chemotherapy investigating a treatment combination including decitabine and venetoclax. | Venetoclax, Decitabine | Phase II | NCT04763928 | Recruiting |
| Azacitidine, venetoclax, and trametinib in r/r AML. | Azacitidine, Trametinib, Venetoclax | Phase II | NCT04487106 | Recruiting |
| Venetoclax together with busulfan, cladribine, and fludarabine in treating patients with high-risk acute myeloid leukemia who are undergoing stem cell transplant. | Busulfan, Cladribine, Fludarabine Phosphate, HSCT, Thiotepa, Venetoclax | Phase II | NCT04708054 | Recruiting |
| STIMULUS-AML1 assessing the efficacy of MBG453 in combination with the HMA azacitidine and venetoclax. | MBG453, Venetoclax, Azacitidine | Phase II | NCT04150029 | Active, not recruiting |
| (V-FIRST) assessing the efficacy of venetoclax in combination with fludarabine, cyratabine, and idarubicine in induction for AML patients with poor prognosis. | Venetoclax, Fludarabine, Cyratabine, Idarubicine | Phase II | NCT03455504 | Recruiting |
| Decitabine, venetoclax, and ponatinib for the treatment of Philadelphia chromosome-positive AML. | Decitabine, Ponatinib, Venetoclax | Phase II | NCT04188405 | Recruiting |
| Venetoclax plus azacitidine in newly diagnosed AML in patients who cannot receive intensive chemotherapy. | Low-dose Venetoclax, Azacitidine | Phase II | NCT05048615 | Recruiting |
| Venetoclax and ASTX727 in relapsed/refractory (r/r) AML or unfit patients and the same combination for higher-risk AML patients without FLT3 (NCT04817241). | Decitabine, Cedazuridine, Venetoclax | Phase II | NCT04746235 | Recruiting |
| Azacytidine + venetoclax versus conventional cytotoxic chemotherapy in induction-eligible AML patients. | Cytarabine, Idarubicin, Daunorubicin, Venetoclax, Azacitidine | Phase II | NCT04801797 | Recruiting |
| Azacitidine and venetoclax with or without pembrolizumab in treating older patients with newly diagnosed acute myeloid leukemia who are ineligible or who refuse intensive chemotherapy. | Azacitidine, Pembrolizumab, Venetoclax | Phase II | NCT04284787 | Recruiting |
| Venetoclax and sequential busulfan, cladribine, and fludarabine phosphate before donor stem cell transplant in treating patients with AML. | Busulfan, Cladribine, Fludarabine, Venetoclax | Phase II | NCT02250937 | Active, not recruiting |
| Venetoclax and azacitidine for non-elderly adult patients with acute myeloid leukemia. | Venetoclax, Azacitidine | Phase II | NCT03573024 | Recruiting |
| BP1001 (a liposomal Grb2 antisense oligonucleotide) in combination with venetoclax plus decitabine in patients with AML who are ineligible for intensive induction therapy. | BP1001, Ventoclax, Decitabine | Phase IIa | NCT02781883 | Recruiting |
| Therapeutic efficacy of (venetoclax and decitabine) versus conventional “7 + 3” chemotherapy in induction young patients with AML. | Venetoclax, Decitabine, Cytarabine, Idarubicin | Phase III | NCT05177731 | Recruiting |
| ENHANCE-3 study comparing the efficacy of magrolimab + venetoclax + azacitidine versus placebo + venetoclax + azacitidine in unfit de novo AML. | Magrolimab, Venetoclax, Azacitidine | Phase III | NCT05079230 | Recruiting |
| ENHANCE-2 comparing the efficacy of magrolimab + azacitidine versus venetoclax + azacitidine in adults with previously untreated TP53 mutant acute myeloid leukemia (AML) who are appropriate for non-intensive therapy as measured by overall survival (OS). | Magrolimab, Venetoclax, Azacitidine, Cytarabine, Daunorubicin, Idarubicin | Phase III | NCT04778397 | Recruiting |
| VIALE-T assessing the efficacy of venetoclax in combination with azacitidine to improve Relapse Free Survival (RFS) in AML participants compared to Best Supportive Care (BSC) when given as maintenance therapy following allo-HSCT. | Venetoclax, Azacitidine | Phase III | NCT04161885 | Recruiting |
| Author; Year | Target Disease | Intervention Arm | Control Arm | Number of Patients | Important Findings |
|---|---|---|---|---|---|
| BCL2 inhibitor (Venetoclax) | |||||
| Konopleva et al. 2016 | High-risk r/r AML or patients unfit for intensive chemotherapy | Ven | - | 32 |
|
| |||||
| |||||
| DiNardo et al. 2019 | Naïve AML or patients unfit for intensive chemotherapy | Ven + HMA (AZA/DEC) | - | 145 |
|
| |||||
| |||||
| Wei et al. 2019 | Naïve AML or patients unfit for intensive chemotherapy | Ven + LDAC | - | 82 |
|
| |||||
| DiNardo et al. 2020 VIALE-A | Naïve AML, ineligible for intensive chemotherapy | Ven + AZA | Placebo + AZA | 431 |
|
| |||||
| |||||
| Wei et al. 2021 VIALE-C | Naïve AML, ineligible for intensive chemotherapy | Ven + LDAC | Placebo + LDAC | 211 |
|
| |||||
| |||||
| IAP Antagonists | |||||
| Schimmer et al. 2011 | r/r AML | AEG35156 + high-dose cytarabine and idarubicin | High-dose Ara-C + Idarubicin | 40 |
|
| |||||
| Erba et al. 2013 | r/r AML | LY2181308 alone or with Idarubicin + Ara-C | - | 24 |
|
| |||||
| |||||
| |||||
| Frey et al. 2014 | r/r AML or high risk MDS refractory to HMA | Birinapant | - | 20 |
|
| CDK9 inhibitors—Indirect MCL1 Inhibitors | |||||
| Zeidner et al. 2021 | Naïve AML ≤ 65 years | Alvocidib | - | 32 |
|
| Cytarabine |
| ||||
| Daunorubicin |
| ||||
| Zeidner et al. 2015 | Naïve AML with intermediate/adverse-risk cytogenetics | Alvocidib, Cytarabine, Mitoxantrone | Cytarabine | 165 |
|
| (FLAM) | +Daunorubicin |
| |||
| Targeting p53 | |||||
| 1. The restoration of wild-type TP53 | |||||
| Sallman et al. 2021 | TP53-mutant AML with 20–30% marrow blasts or MDS | Eprenetapopt (APR-246) + AZA | - | 55 (n = 11 AML) |
|
| |||||
| NCT03072043 | |||||
| Cluzeau et al. | TP53-mutant AML or MDS | Eprenetapopt (APR-246) + AZA | - | 53 (n = 18 AML) |
|
| 2021 |
| ||||
| |||||
| NCT03588078 | |||||
| 2. MDM2 Inhibitors | |||||
| Yee et al. 2021 | r/r AML, s-AML, t-AML, or naïve AML, ineligible for intensive chemotherapy | Idasanutlin (RG-7388) + Ara-C | Idasanutlin | 122 |
|
| |||||
| Daver et al. 2019 | r/r AML, ineligible for intensive chemotherapy | Idasanutlin + VEN | - | 49 |
|
| |||||
| Agents and Ongoing Trials | Target Disease | Intervention Arm | Control Arm | Estimated Enrollment | Design; Clinical Trial Phase | Serial NCT Number | Current Status |
|---|---|---|---|---|---|---|---|
| BCL-2, BCL-XL, BCL-W inhibitors | |||||||
| Navitoclax; BCL-2/BCL-XL/BCL-W inhibitor; | r/r AML Previously Treated with Venetoclax | Navitoclax, Venetoclax, Decitabine | - | 24 | Phase Ib, Open-Label study | NCT05222984 | Recruiting |
| AZD0466 BCL-2/BCL-XL inhibitor NIMBLE | r/r AML | AZD0466 | - | 141 | Phase I/II, Open-Label study | NCT04865419 | Recruiting |
| Direct MCL1 Inhibitors | |||||||
| S64315/MIK665 | r/r AML, ineligible for intensive chemotherapy | S64315 + AZA | - | 180 | Phase I/II, Open-Label study | NCT04629443 | Recruiting |
| S64315/MIK665 | r/r AML, ineligible for intensive chemotherapy | S64315 + VEN | - | 40 | Phase Ib, Open-Label study | NCT03672695 | Recruiting |
| S64315/MIK665 | r/r AML, ineligible for intensive chemotherapy | S64315+ S65487 (VOB560) | - | 170 | Phase Ib, Open-Label study | NCT04702425 | Recruiting |
| AMG176 | r/r AML | AMG176 ± AZA | - | 175 | Phase I, Open-Label study | NCT02675452 | Recruiting |
| PRT1419 | r/r AML | PRT1419 | - | 36 | Phase I, Open-Label study | NCT04543305 | Active, not recruiting |
| PRT1419 | r/r AML | PRT1419 | - | 30 | Phase I, Open-Label study | NCT05107856 | Recruiting |
| AZD5991 | r/r AML, ineligible for intensive chemotherapy | AZD5991 ± VEN | - | 144 | Phase I/II, Open-Label study | NCT03218683 | Terminated |
| CDK9 inhibitors—Indirect MCL1 Inhibitors | |||||||
| Alvocidib | r/r AML | Alvocidib + VEN | - | 36 | Phase Ib, Open-Label study | NCT03441555 | Completed |
| Dinaciclib/MK7965 | r/r AML | Dinaciclib (MK7965) + VEN | - | 48 | Phase Ib, Open-Label study | NCT03484520 | Active, not recruiting |
| CYC065 | r/r AML | CYC065 + VEN | - | 25 | Phase I, Open-Label study | NCT04017546 | Active, not recruiting |
| AZD4573 | r/r AML | AZD4573 | - | 44 | Phase I, Open-Label study | NCT03263637 | Completed |
| IAP Antagonists | |||||||
| ASTX660 | r/r AML | ASTX660 ± ASTX727 | - | 68 | Phase I, Open-Label study | NCT04155580 | Terminated |
| Targeting p53 | |||||||
| 1. The restoration of wild-type TP53 | |||||||
| Eprenetapopt/APR-246 | TP53-mutant AML | Eprenetapopt (APR-246) + AZA + VEN | - | 51 | Phase I, Open-Label study | NCT04214860 | Completed |
| Eprenetapopt/APR-246 | TP53-mutant AML after allo-HSCT | Eprenetapopt (APR-246) + AZA | - | 33 | Phase II, Open-Label study | NCT03931291 | Completed |
| 2. MDM2 Inhibitors | |||||||
| Idasanutlin | Newly diagnosed AML | Idasanutlin+ Cytarabine + Daunorubicin | - | 24 | Phase Ib/II, Open-Label Study | NCT03850535 | Terminated |
| Idasanutlin | r/r AML, ineligible for intensive chemotherapy | Idasanutlin + Cobimetinib + Venetoclax | - | 88 | Phase Ib, Open-Label Study | NCT02670044 | Completed |
| Idasanutlin | r/r AML | Idasanutlin + Ara-C | Placebo + Ara-C | 447 | Phase III, Double-Blind, Randomized Study | NCT02545283 | Terminated |
| 3. MDM2/MDMX inhibitors | |||||||
| ALRN-6924 | r/r AML | ALRN-6924 ± Ara-C | - | 55 | Phase I/Ib Open-Label Study | NCT02909972 | Completed |
| Targeting Neddylation (NAE Inhibitors) | |||||||
|---|---|---|---|---|---|---|---|
| Agents and Ongoing Trials | Target Disease | Intervention Arm | Control Arm | Estimated Enrollment | Design; Clinical Trial Phase | Serial NCT Number | Current Status |
| Pevonedistat/TAK-924/MLN4924 | AML, ineligible for intensive chemotherapy | Pevonedistat + VEN + AZA | VEN + AZA | 164 | Phase II, Open-Label study | NCT04266795 | Active, not recruiting |
| Pevonedistat/TAK-924/MLN4924 | r/r AML | Pevonedistat + Belinostat | - | 30 | Phase I, Open-Label study | NCT03772925 | Recruiting |
| Pevonedistat/TAK-924/MLN4924 | Naïve high-risk AML with at least 1: adverse genetic features/t-AML/AML with antecedent MDS/≥55 years and considered fit for chemotherapy/AML with MDS-related changes | Pevonedistat + Ara-C + Idarubicin | - | 53 | Phase Ib/II, Open-Label study | NCT03330821 | Active, not recruiting |
| Pevonedistat/TAK-924/MLN4924 | r/r AML or r/r MDS | Pevonedistat + AZA + Ara-C + Fludarabine Phosphate + Methotrexate | - | 12 | Phase I, Open-Label study | NCT03813147 | Active, not recruiting |
| Pevonedistat/TA K-924/MLN4924 | Newly diagnosed s-AML or newly diagnosed CMML/MDS or post-HMA failure CMML/MDS | Pevonedistat +VEN + AZA | - | 40 | Phase I/II, Open-Label study | NCT03862157 | Active, not recruiting |
| Pevonedistat/TAK-924/MLN4924 | Newly diagnosed AML not eligible for intensive chemotherapy | Pevonedistat + AZA | AZA | 466 | Phase III | NCT04090736 | Active, not recruiting |
| Pevonedistat/TAK-924/MLN4924 | r/r AML (dose-escalation phase), newly diagnosed AML or r/r AML (expansion phase) | Pevonedistat +VEN + HMA | - | 24 | Phase I | NCT04172844 | Active, not recruiting |
| Pevonedistat/TAK-924/MLN4924 | AML or MDS after first CR with intensive chemotherapy or CR after allo-HSCT with MRD | Pevonedistat + AZA | AZA | 102 | Phase II | NCT04712942 | Active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krawiec, K.; Strzałka, P.; Czemerska, M.; Wiśnik, A.; Zawlik, I.; Wierzbowska, A.; Pluta, A. Targeting Apoptosis in AML: Where Do We Stand? Cancers 2022, 14, 4995. https://doi.org/10.3390/cancers14204995
Krawiec K, Strzałka P, Czemerska M, Wiśnik A, Zawlik I, Wierzbowska A, Pluta A. Targeting Apoptosis in AML: Where Do We Stand? Cancers. 2022; 14(20):4995. https://doi.org/10.3390/cancers14204995
Chicago/Turabian StyleKrawiec, Kinga, Piotr Strzałka, Magdalena Czemerska, Aneta Wiśnik, Izabela Zawlik, Agnieszka Wierzbowska, and Agnieszka Pluta. 2022. "Targeting Apoptosis in AML: Where Do We Stand?" Cancers 14, no. 20: 4995. https://doi.org/10.3390/cancers14204995
APA StyleKrawiec, K., Strzałka, P., Czemerska, M., Wiśnik, A., Zawlik, I., Wierzbowska, A., & Pluta, A. (2022). Targeting Apoptosis in AML: Where Do We Stand? Cancers, 14(20), 4995. https://doi.org/10.3390/cancers14204995