
PRELP Regulates Cell–Cell Adhesion and EMT and Inhibits Retinoblastoma Progression
 , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Human RB Data
2.3. Mouse Experiments
2.4. Cell Culture Conditions
2.5. Viability Assays
2.6. Proliferation and Adhesion Assays
2.7. Anchorage-Independent Growth Assays
2.8. Immunostaining
2.9. mRNA Expression Profiling
2.10. Quantification and Statistical Analysis
3. Results
3.1. PRELP Expression Is Strongly Suppressed in RB
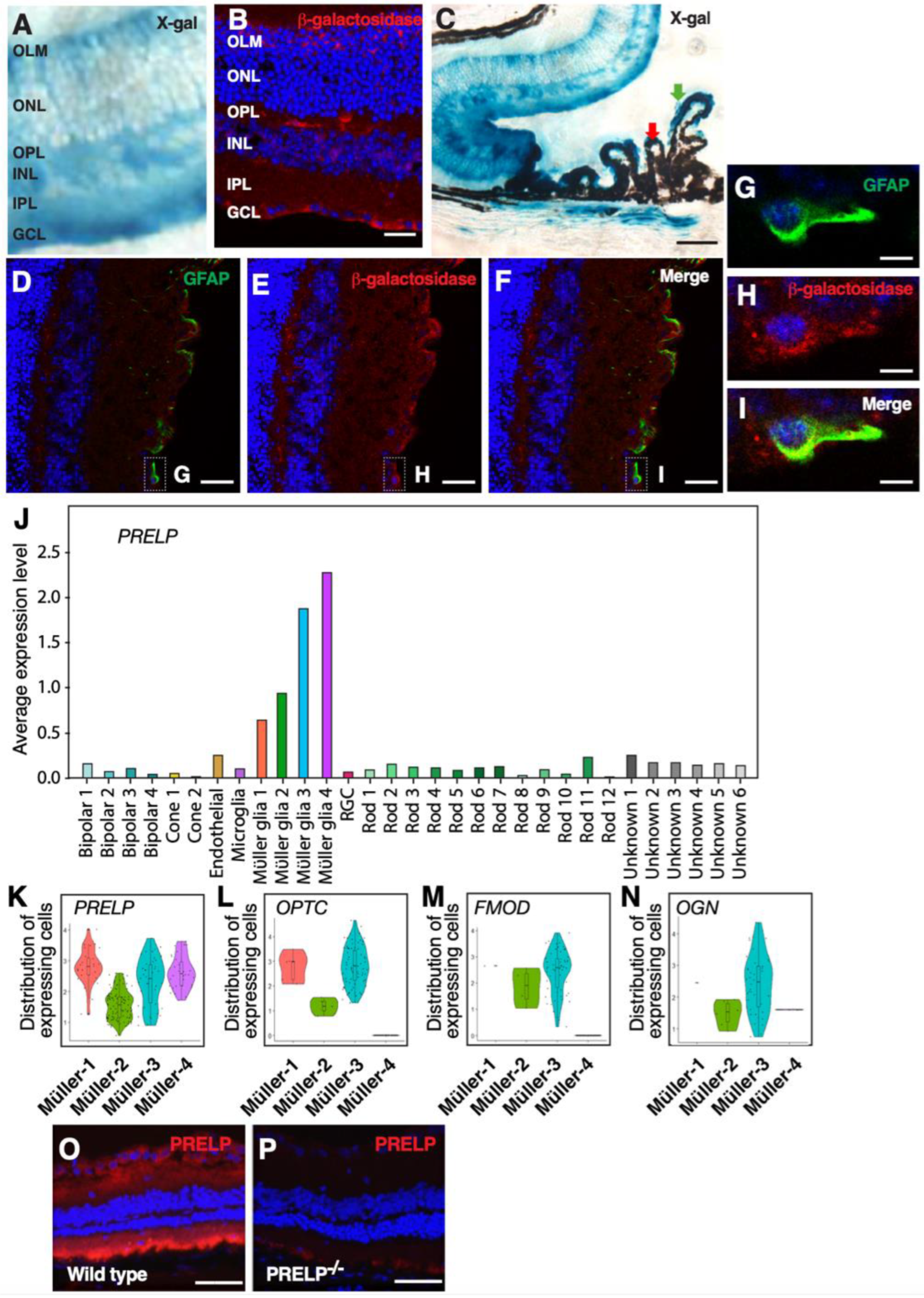
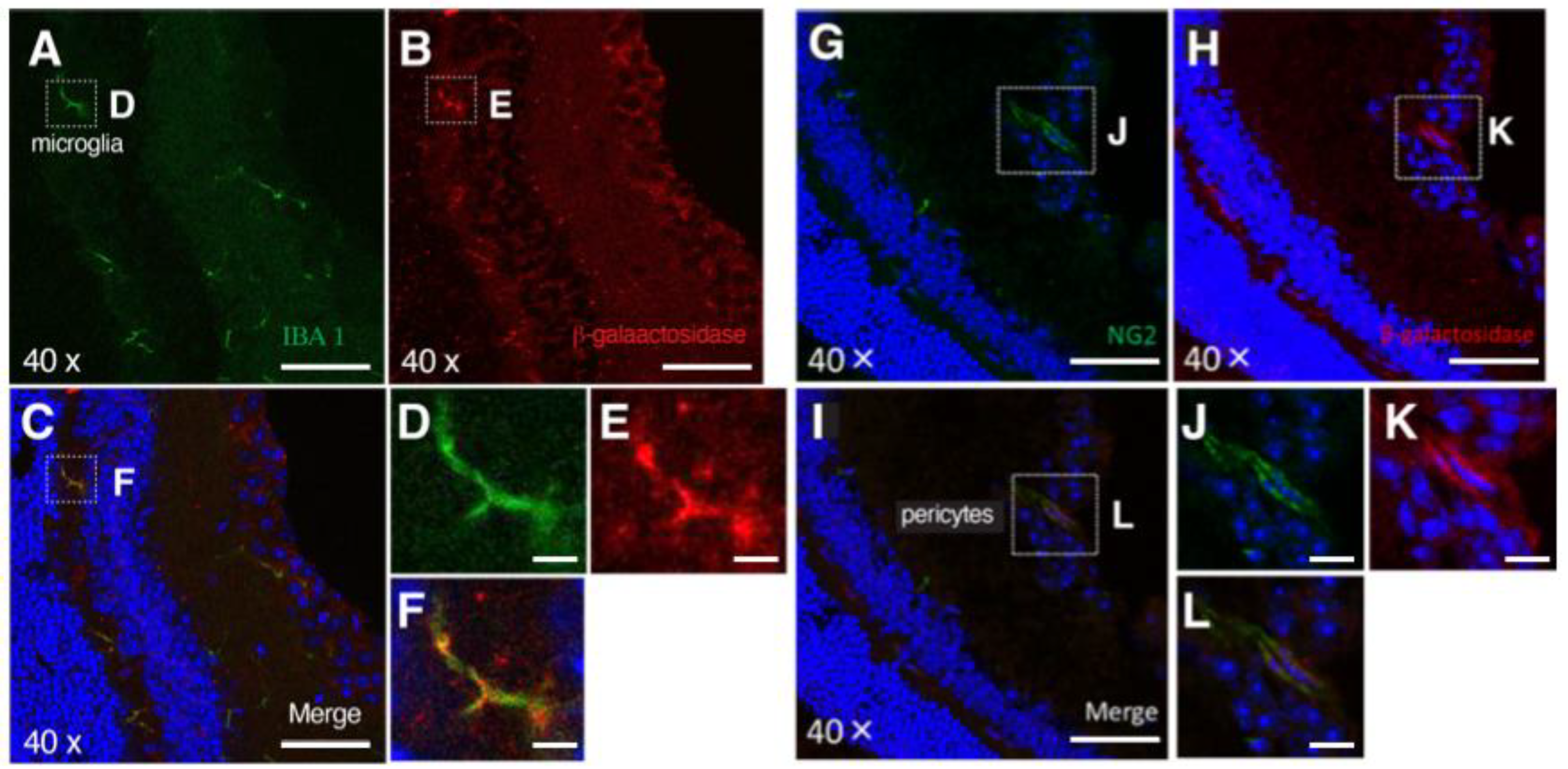
3.2. PRELP Is Highly Expressed by Glial Cells in the Retina
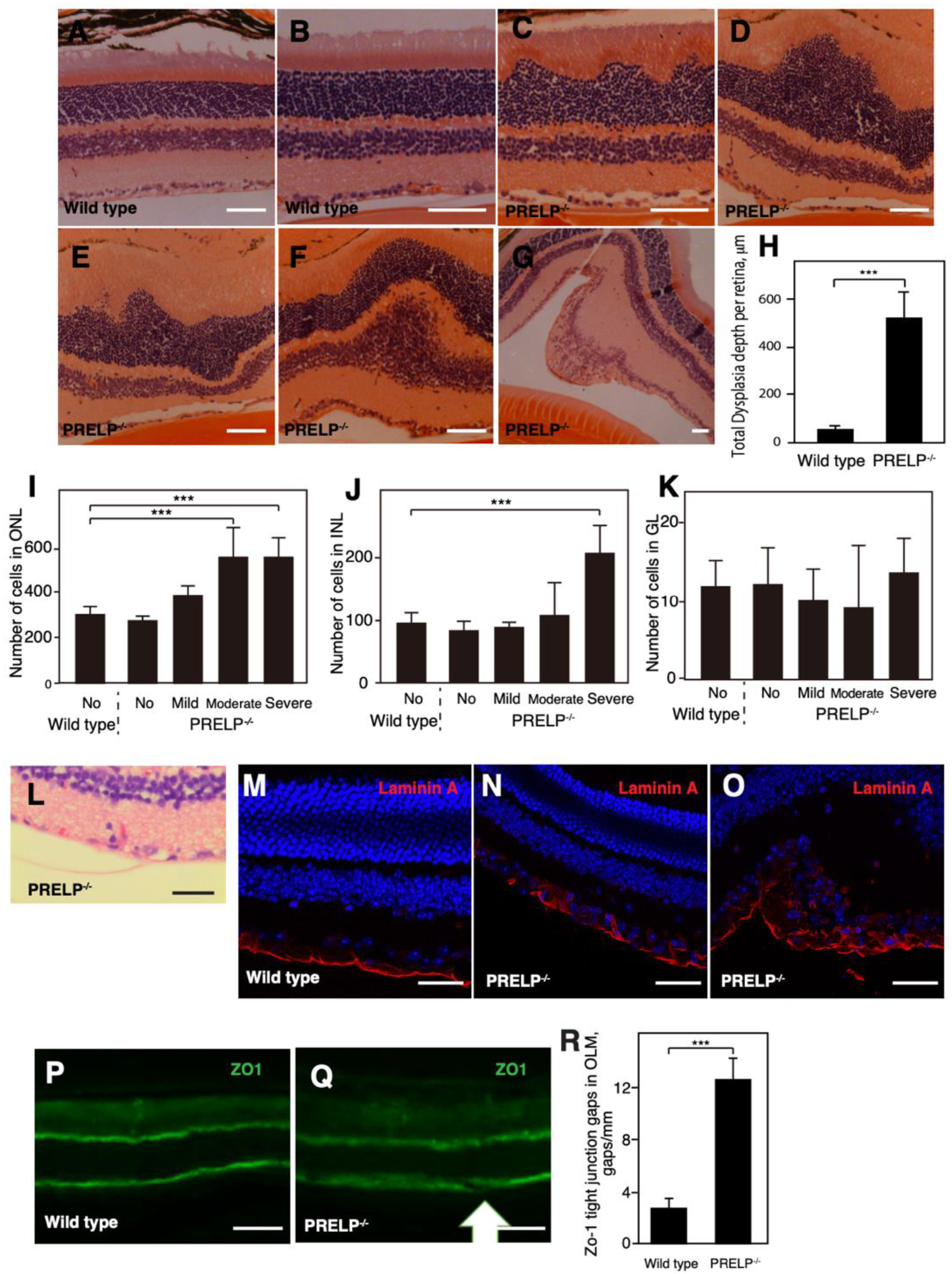
3.3. PRELP−/− Mice Exhibit Dysplasia in the Retinal Cell Layers
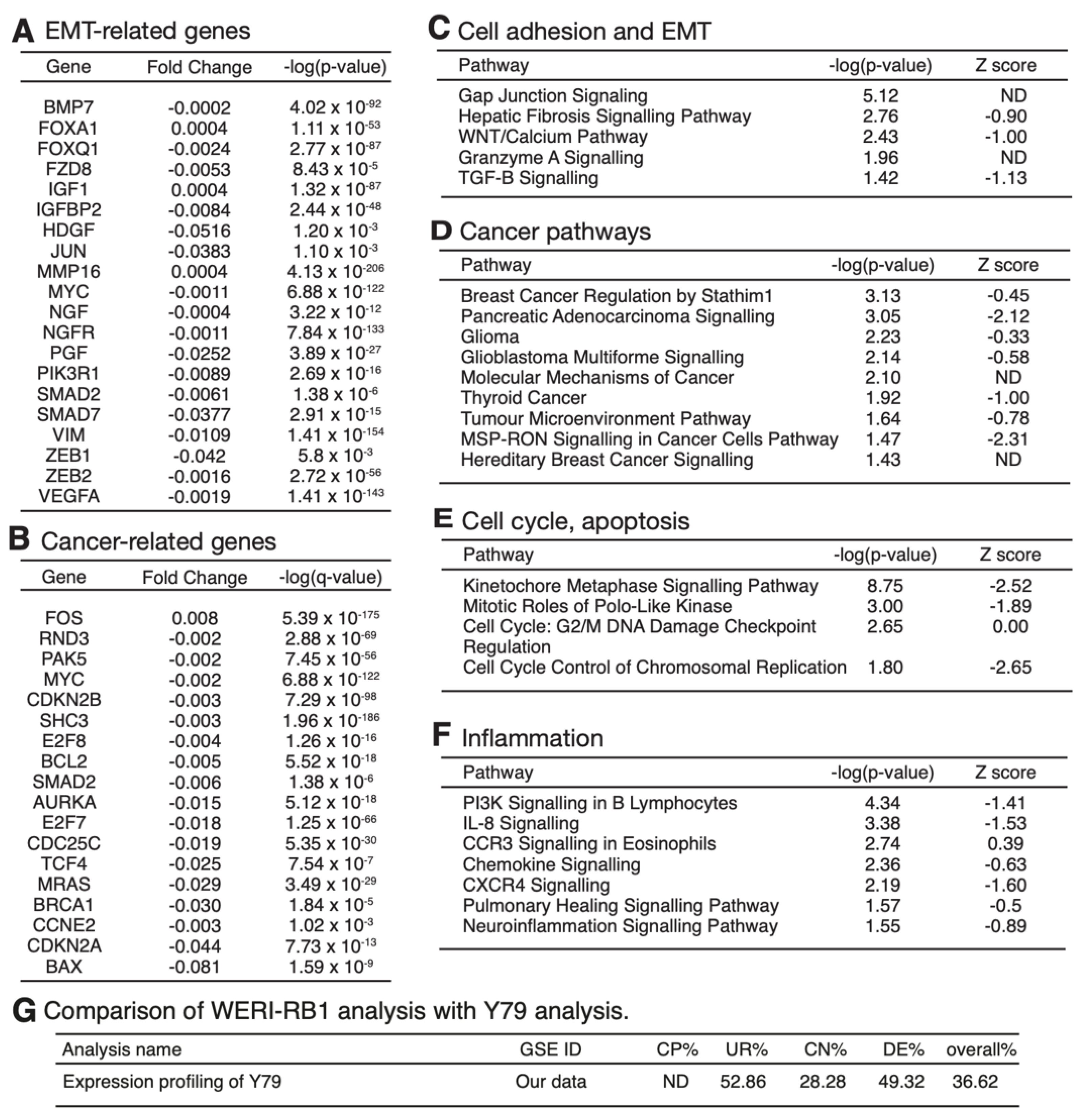
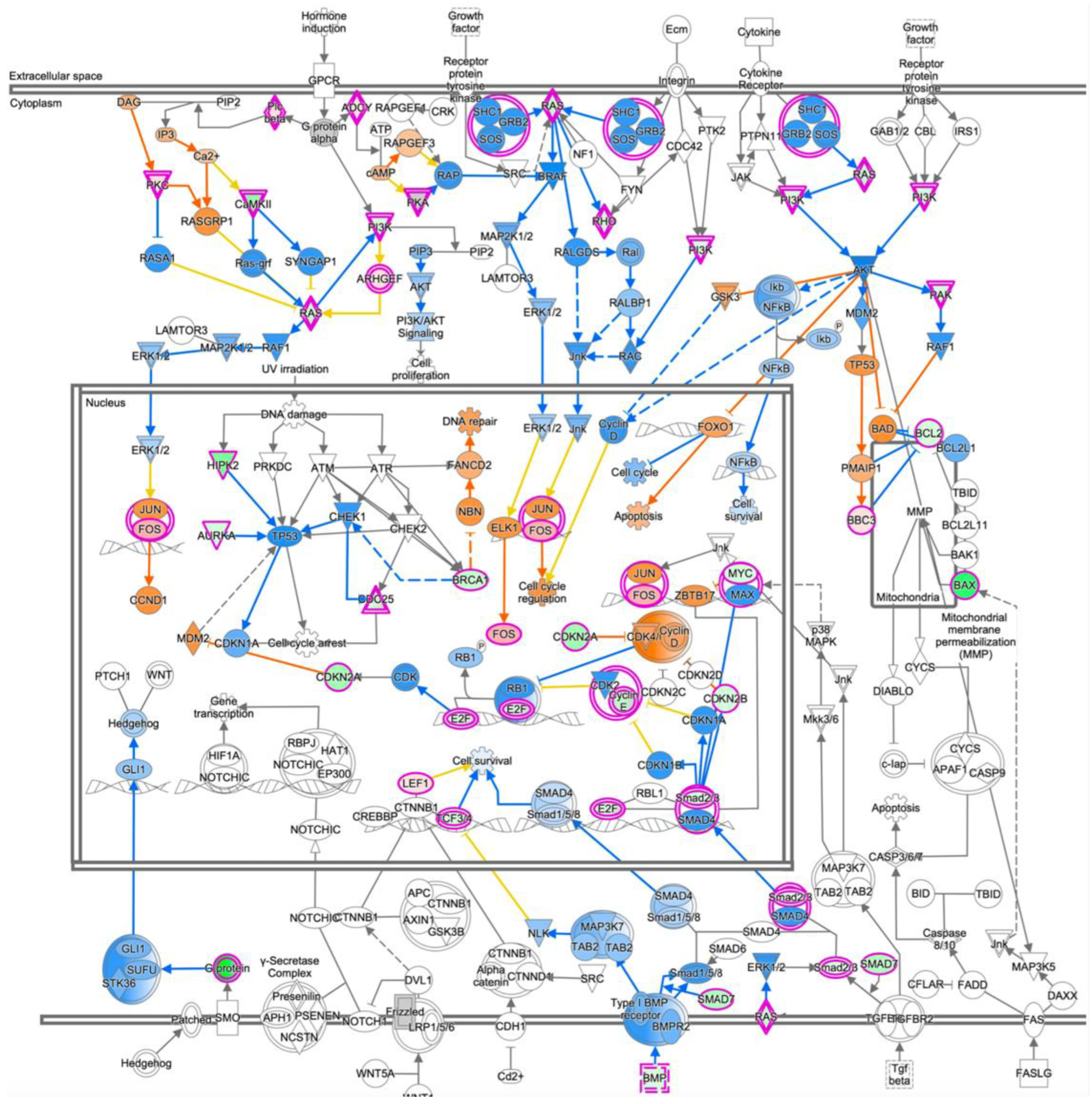
3.4. mRNA Expression Profiling Analysis of PRELP−/− Mouse Retina Revealed That PRELP Is Involved in Cancer, Adhesion, and Inflammation
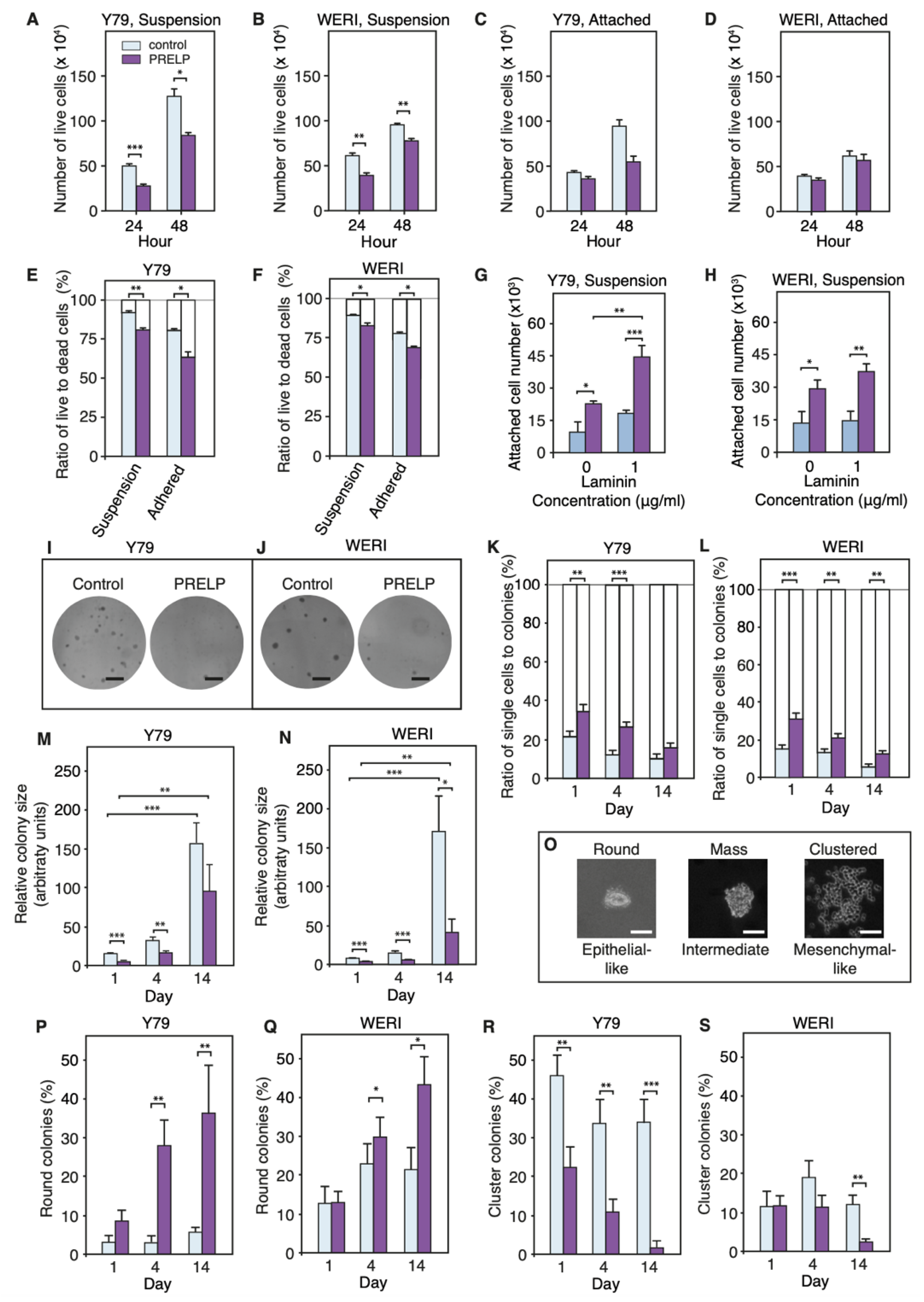
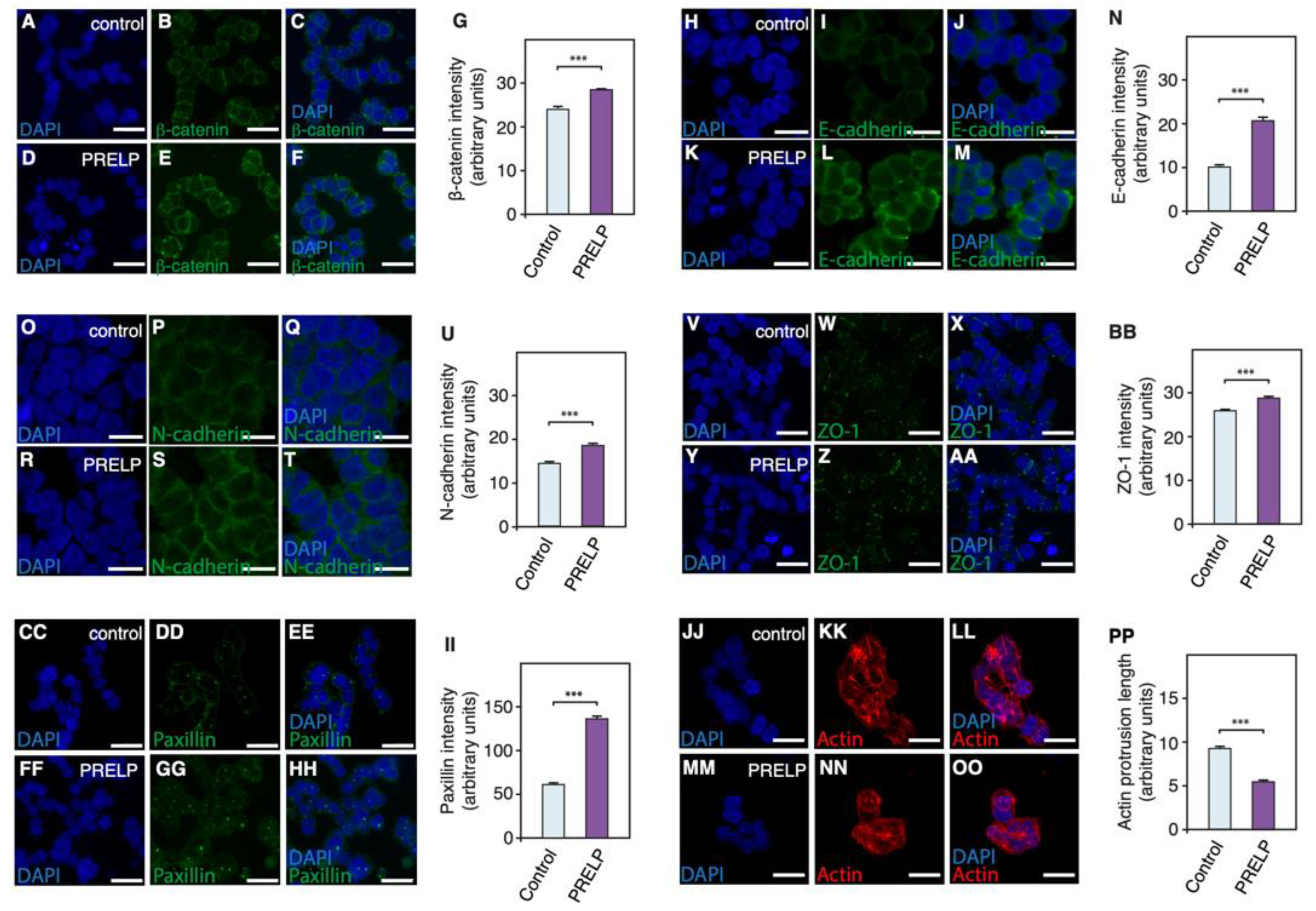
3.5. PRELP Application Reduces RB Cancer Cell Viability in Association with Enhanced Cell Adhesion, Inhibition of Anchorage Independent Growth, and Facilitation of MET
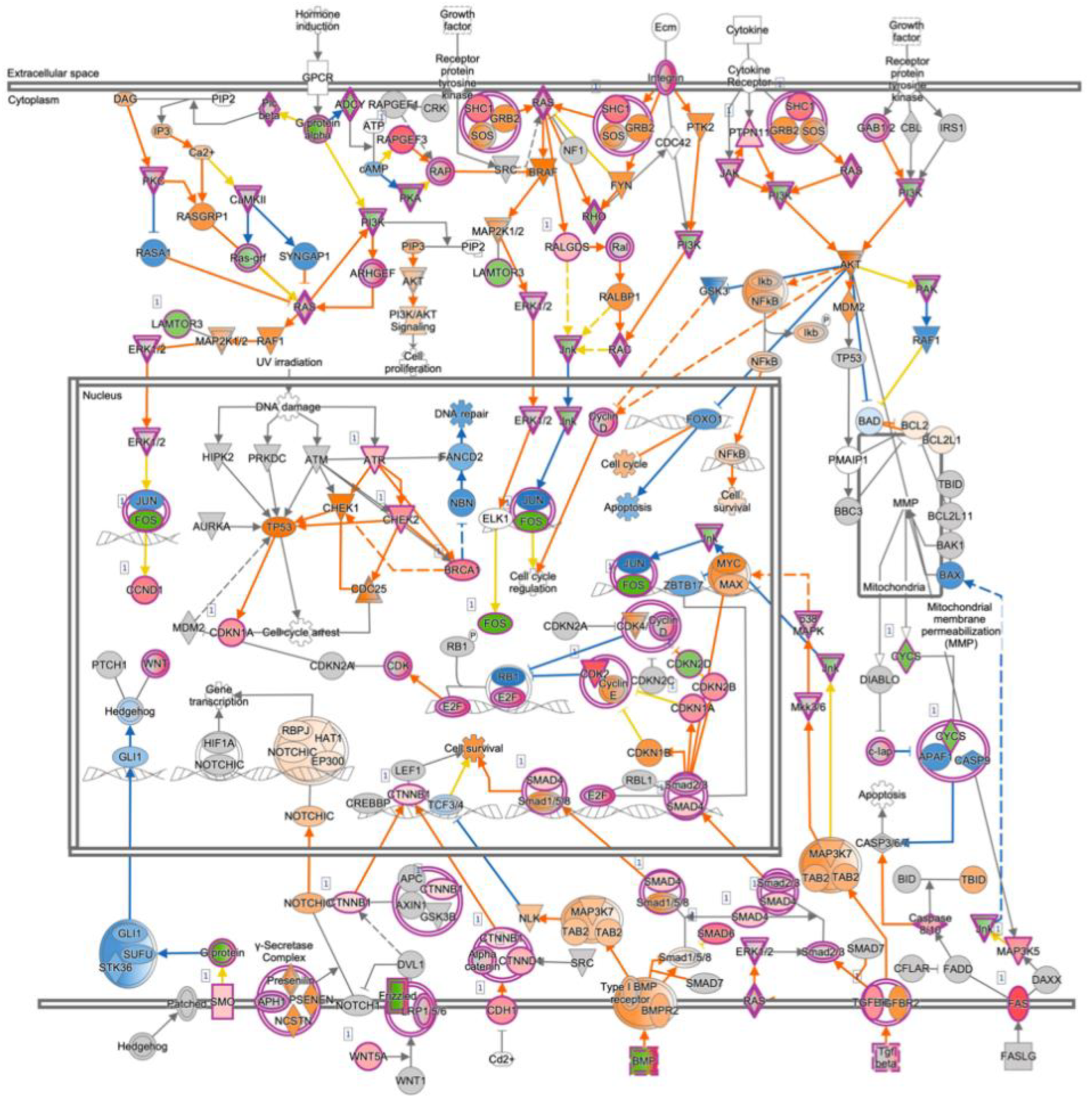
3.6. Expression Profiling after Application of PRELP Revealed That PRELP Suppresses Various Tumor Related Pathways and Enhances Cell-Cell Adhesion
4. Discussion
4.1. PRELP Secreted from Müller Glial Cells Has Function to Maintain Rigid Retinal Structure
4.2. Role of PRELP-Depleted Microenvironment in RB Progression
4.3. Difference between PRELP−/− Mouse Retina and Human RB
4.4. Generation of PRELP-Depleted Microenvironment in RB
4.5. ECM in Cancer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fabian, I.D.; Onadim, Z.; Karaa, E.; Duncan, C.; Chowdhury, T.; Scheimberg, I.; Ohnuma, S.I.; Reddy, M.A.; Sagoo, M.S. The management of retinoblastoma. Oncogene 2018, 37, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W. Role of MYCN in retinoblastoma. Lancet Oncol. 2013, 14, 270–271. [Google Scholar] [CrossRef]
- Wu, N.; Jia, D.; Bates, B.; Basom, R.; Eberhart, C.G.; MacPherson, D. A mouse model of MYCN-driven retinoblastoma reveals MYCN-independent tumor reemergence. J. Clin. Investig. 2017, 127, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.R.; Broad, K.D.; Onadim, Z.; Price, E.A.; Zou, X.; Sheriff, I.; Karaa, E.K.; Scheimberg, I.; Reddy, M.A.; Sagoo, M.S.; et al. Whole-Genome Sequencing of Retinoblastoma Reveals the Diversity of Rearrangements Disrupting RB1 and Uncovers a Treatment-Related Mutational Signature. Cancers 2021, 13, 754. [Google Scholar] [CrossRef]
- Zielinski, B.; Gratias, S.; Toedt, G.; Mendrzyk, F.; Stange, D.E.; Radlwimmer, B.; Lohmann, D.R.; Lichter, P. Detection of chromosomal imbalances in retinoblastoma by matrix-based comparative genomic hybridization. Genes Chromosomes Cancer 2005, 43, 294–301. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Dellett, M.; Hu, W.; Papadaki, V.; Ohnuma, S. Small leucine rich proteoglycan family regulates multiple signalling pathways in neural development and maintenance. Dev. Growth Differ. 2012, 54, 327–340. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Oncosuppressive functions of decorin. Mol. Cell. Oncol. 2015, 2, e975645. [Google Scholar] [CrossRef]
- Bengtsson, E.; Morgelin, M.; Sasaki, T.; Timpl, R.; Heinegard, D.; Aspberg, A. The leucine-rich repeat protein PRELP binds perlecan and collagens and may function as a basement membrane anchor. J. Biol. Chem. 2002, 277, 15061–15068. [Google Scholar] [CrossRef]
- Papadaki, V.; Asada, K.; Watson, J.K.; Tamura, T.; Leung, A.; Hopkins, J.; Dellett, M.; Sasai, N.; Davaapil, H.; Nik-Zainal, S.; et al. Two Secreted Proteoglycans, Activators of Urothelial Cell-Cell Adhesion, Negatively Contribute to Bladder Cancer Initiation and Progression. Cancers 2020, 12, 3362. [Google Scholar] [CrossRef]
- Kosuge, H.; Nakakido, M.; Nagatoishi, S.; Fukuda, T.; Bando, Y.; Ohnuma, S.I.; Tsumoto, K. Proteomic identification and validation of novel interactions of the putative tumor suppressor PRELP with membrane proteins including IGFI-R and p75NTR. J. Biol. Chem. 2021, 296, 100278. [Google Scholar] [CrossRef] [PubMed]
- Lun, A.T.; Bach, K.; Marioni, J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Assefa, A.T.; Vandesompele, J.; Thas, O. Correction to: On the utility of RNA sample pooling to optimize cost and statistical power in RNA sequencing experiments. BMC Genom. 2020, 21, 384. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, K.; Lapan, S.W.; Whitney, I.E.; Tran, N.M.; Macosko, E.Z.; Kowalczyk, M.; Adiconis, X.; Levin, J.Z.; Nemesh, J.; Goldman, M.; et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 2016, 166, 1308–1323.e1330. [Google Scholar] [CrossRef]
- Ishida, S.; Yamazaki, K.; Shinoda, K.; Kawashima, S.; Oguchi, Y. Macular hole retinal detachment in highly myopic eyes: Ultrastructure of surgically removed epiretinal membrane and clinicopathologic correlation. Retina 2000, 20, 176–183. [Google Scholar] [CrossRef]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef]
- Kanda, A.; Noda, K.; Hirose, I.; Ishida, S. TGF-beta-SNAIL axis induces Muller glial-mesenchymal transition in the pathogenesis of idiopathic epiretinal membrane. Sci. Rep. 2019, 9, 673. [Google Scholar] [CrossRef]
- MacDonald, R.B.; Randlett, O.; Oswald, J.; Yoshimatsu, T.; Franze, K.; Harris, W.A. Muller glia provide essential tensile strength to the developing retina. J. Cell Biol. 2015, 210, 1075–1083. [Google Scholar] [CrossRef]
- Dorrell, M.I.; Friedlander, M. Mechanisms of endothelial cell guidance and vascular patterning in the developing mouse retina. Prog. Retin Eye Res. 2006, 25, 277–295. [Google Scholar] [CrossRef]
- Johnson, J.M.; Young, T.L.; Rada, J.A. Small leucine rich repeat proteoglycans (SLRPs) in the human sclera: Identification of abundant levels of PRELP. Mol. Vis. 2006, 12, 1057–1066. [Google Scholar] [PubMed]
- Jacobi, F.K.; Pusch, C.M. A decade in search of myopia genes. Front. Biosci. 2010, 15, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Too, L.K.; Ball, H.J.; McGregor, I.S.; Hunt, N.H. The pro-inflammatory cytokine interferon-gamma is an important driver of neuropathology and behavioural sequelae in experimental pneumococcal meningitis. Brain Behav. Immun. 2014, 40, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zheng, S.; Pan, C.T.; Yuan, M.; Chang, L.; Yao, Y.; Zhao, M.; Liang, J. Analysis of aqueous humor concentrations of cytokines in retinoblastoma. PLoS ONE 2017, 12, e0177337. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Agrawal, A.; Bhushan, R.; Chavalmane, A.K.; Kalathur, R.K.; Takahashi, T.; Kondaiah, P. Expression profiling of genes regulated by TGF-beta: Differential regulation in normal and tumour cells. BMC Genom. 2007, 8, 98. [Google Scholar] [CrossRef]
- Gao, Y.; Lu, X. Decreased expression of MEG3 contributes to retinoblastoma progression and affects retinoblastoma cell growth by regulating the activity of Wnt/beta-catenin pathway. Tumour Biol. 2016, 37, 1461–1469. [Google Scholar] [CrossRef]
- Wu, T.; Wang, L.N.; Tang, D.R.; Sun, F.Y. SOST silencing promotes proliferation and invasion and reduces apoptosis of retinoblastoma cells by activating Wnt/beta-catenin signaling pathway. Gene Ther. 2017, 24, 399–407. [Google Scholar] [CrossRef]
- Danovi, D.; Cremona, C.A.; Machado-da-Silva, G.; Basu, S.; Noon, L.A.; Parrinello, S.; Lloyd, A.C. A genetic screen for anchorage-independent proliferation in mammalian cells identifies a membrane-bound neuregulin. PLoS ONE 2010, 5, e11774. [Google Scholar] [CrossRef]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Marchong, M.N.; Yurkowski, C.; Ma, C.; Spencer, C.; Pajovic, S.; Gallie, B.L. Cdh11 acts as a tumor suppressor in a murine retinoblastoma model by facilitating tumor cell death. PLoS Genet. 2010, 6, e1000923. [Google Scholar] [CrossRef] [PubMed]
- Satriyo, P.B.; Bamodu, O.A.; Chen, J.H.; Aryandono, T.; Haryana, S.M.; Yeh, C.T.; Chao, T.Y. Cadherin 11 Inhibition Downregulates beta-catenin, Deactivates the Canonical WNT Signalling Pathway and Suppresses the Cancer Stem Cell-Like Phenotype of Triple Negative Breast Cancer. J. Clin. Med. 2019, 8, 148. [Google Scholar] [CrossRef]
- Tatin, F.; Taddei, A.; Weston, A.; Fuchs, E.; Devenport, D.; Tissir, F.; Makinen, T. Planar cell polarity protein Celsr1 regulates endothelial adherens junctions and directed cell rearrangements during valve morphogenesis. Dev. Cell 2013, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Lewis, K.C.; Murray, N.R.; Fields, A.P. Oncogenic Ect2 signaling regulates rRNA synthesis in NSCLC. Small GTPases 2019, 10, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.E.; Haltom, J.A.; Almeida, M.P.; Wierson, W.A.; Solin, S.L.; Weiss, T.J.; Helmer, J.A.; Sandquist, E.J.; Shive, H.R.; McGrail, M. Epigenetic regulators Rbbp4 and Hdac1 are overexpressed in a zebrafish model of RB1 embryonal brain tumor, and are required for neural progenitor survival and proliferation. Dis. Model Mech. 2018, 11, dmm034124. [Google Scholar] [CrossRef]
- Li, Y.D.; Lv, Z.; Zhu, W.F. RBBP4 promotes colon cancer malignant progression via regulating Wnt/beta-catenin pathway. World J. Gastroenterol. 2020, 26, 5328–5342. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Mai, M.; Wang, D.; Lv, L.; Rao, J. Effect of RbAp48 knockdown on migration and invasion of human cervical cancer cell line MS751 in vitro. Nan Fang Yi Ke Da Xue Xue Bao 2015, 35, 1564–1569. [Google Scholar]
- Stenfelt, S.; Blixt, M.K.E.; All-Ericsson, C.; Hallbook, F.; Boije, H. Heterogeneity in retinoblastoma: A tale of molecules and models. Clin. Transl. Med. 2017, 6, 42. [Google Scholar] [CrossRef]
- Robanus-Maandag, E.; Dekker, M.; van der Valk, M.; Carrozza, M.L.; Jeanny, J.C.; Dannenberg, J.H.; Berns, A.; te Riele, H. p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev. 1998, 12, 1599–1609. [Google Scholar] [CrossRef]
- MacPherson, D.; Sage, J.; Kim, T.; Ho, D.; McLaughlin, M.E.; Jacks, T. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004, 18, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, D. Insights from mouse models into human retinoblastoma. Cell Div. 2008, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.J.; Reh, T.A. Potential of Muller glia to become neurogenic retinal progenitor cells. Glia 2003, 43, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D. Muller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci. 2014, 15, 431–442. [Google Scholar] [CrossRef]
- Norrie, J.L.; Nityanandam, A.; Lai, K.; Chen, X.; Wilson, M.; Stewart, E.; Griffiths, L.; Jin, H.; Wu, G.; Orr, B.; et al. Retinoblastoma from human stem cell-derived retinal organoids. Nat. Commun. 2021, 12, 4535. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, Y.; Biswas, A.; Mandal, M. Glioma progression through the prism of heat shock protein mediated extracellular matrix remodeling and epithelial to mesenchymal transition. Exp. Cell Res. 2017, 359, 299–311. [Google Scholar] [CrossRef]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sternlicht, M.D.; Lochter, A.; Sympson, C.J.; Huey, B.; Rougier, J.P.; Gray, J.W.; Pinkel, D.; Bissell, M.J.; Werb, Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 1999, 98, 137–146. [Google Scholar] [CrossRef]
- Ma, J.; Zhu, T.P.; Moe, M.C.; Ye, P.; Yao, K. Opticin production is reduced by hypoxia and VEGF in human retinal pigment epithelium via MMP-2 activation. Cytokine 2012, 59, 100–107. [Google Scholar] [CrossRef]
- Pietraszek-Gremplewicz, K.; Karamanou, K.; Niang, A.; Dauchez, M.; Belloy, N.; Maquart, F.X.; Baud, S.; Brezillon, S. Small leucine-rich proteoglycans and matrix metalloproteinase-14: Key partners? Matrix Biol. 2019, 75–76, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Buraschi, S.; Pal, N.; Tyler-Rubinstein, N.; Owens, R.T.; Neill, T.; Iozzo, R.V. Decorin antagonizes Met receptor activity and down-regulates {beta}-catenin and Myc levels. J. Biol. Chem. 2010, 285, 42075–42085. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SLRP Member | Normal Retina Expression (n = 3) | RB Expression (n = 3) | Fold Change | p-Value |
|---|---|---|---|---|
| Osteoglycin (OGN) | 1584.3 | 7.8 | −203.1 | 1.1 × 10−97 |
| Opticin (OPTC) | 5104.3 | 0.6 | −8507.2 | 3.4 × 10−41 |
| Osteomodulin (OMD) | 318.7 | 1.8 | −177.0 | 8.7 × 10−32 |
| Proline/arginine-rich end leucine-rich repeat Protein (PRELP) | 4102.7 | 25.2 | −162.8 | 1.5 × 10−22 |
| Nyctalopin (NYX) | 80.0 | 0.8 | −100.0 | 5.6 × 10−14 |
| Podocan−like protein 1 (PODNL1) | 190.0 | 7.4 | −25.7 | 7.0 × 10−10 |
| Tsukushi (TSKU) | 983.3 | 219.6 | −4.5 | 1.4 × 10−7 |
| Biglycan (BGN) | 1361.7 | 123.0 | −11.1 | 6.1 × 10−7 |
| Fibromodulin (FMOD) | 620.3 | 31.8 | −19.5 | 6.5 × 10−7 |
| Asporin (ASPN) | 38.3 | 7.4 | −5.2 | 4.6 × 10−5 |
| Extracellular matrix protein 2 (ECM2) | 203.3 | 14.6 | −13.9 | 1.4 × 10−4 |
| Decorin (DCN) | 1785.7 | 87.0 | −20.5 | 1.8 × 10−4 |
| Chondroadherin (CHAD) | 12.0 | 0.8 | −15.0 | 2.5 × 10−3 |
| Keratocan (KERA) | 12.7 | 1.0 | −12.7 | 5.7 × 10−2 |
| Lumican (LUM) | 98.0 | 20.2 | −4.9 | 1.1 × 10−1 |
| Podocan (PODN) | 163.0 | 123.4 | −1.3 | NA |
| Epiphycan (EPYC) | 0 | 0 | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hopkins, J.; Asada, K.; Leung, A.; Papadaki, V.; Davaapil, H.; Morrison, M.; Orita, T.; Sekido, R.; Kosuge, H.; Reddy, M.A.; et al. PRELP Regulates Cell–Cell Adhesion and EMT and Inhibits Retinoblastoma Progression. Cancers 2022, 14, 4926. https://doi.org/10.3390/cancers14194926
Hopkins J, Asada K, Leung A, Papadaki V, Davaapil H, Morrison M, Orita T, Sekido R, Kosuge H, Reddy MA, et al. PRELP Regulates Cell–Cell Adhesion and EMT and Inhibits Retinoblastoma Progression. Cancers. 2022; 14(19):4926. https://doi.org/10.3390/cancers14194926
Chicago/Turabian StyleHopkins, Jack, Ken Asada, Alex Leung, Vasiliki Papadaki, Hongorzul Davaapil, Matthew Morrison, Tomoko Orita, Ryohei Sekido, Hirofumi Kosuge, M. Ashwin Reddy, and et al. 2022. "PRELP Regulates Cell–Cell Adhesion and EMT and Inhibits Retinoblastoma Progression" Cancers 14, no. 19: 4926. https://doi.org/10.3390/cancers14194926
APA StyleHopkins, J., Asada, K., Leung, A., Papadaki, V., Davaapil, H., Morrison, M., Orita, T., Sekido, R., Kosuge, H., Reddy, M. A., Kimura, K., Mitani, A., Tsumoto, K., Hamamoto, R., Sagoo, M. S., & Ohnuma, S.-i. (2022). PRELP Regulates Cell–Cell Adhesion and EMT and Inhibits Retinoblastoma Progression. Cancers, 14(19), 4926. https://doi.org/10.3390/cancers14194926