
Development of a Neurotensin-Derived 68Ga-Labeled PET Ligand with High In Vivo Stability for Imaging of NTS1 Receptor-Expressing Tumors

 , , , , , and
, , , , , and
Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. General Experimental Conditions
2.2. Cell Culture and Preparation of HEK293T Cells Stably Expressing the Human NTS2R
2.3. Radiochemical Binding Assays
2.3.1. NTS1R Binding
2.3.2. NTS2R Binding
2.4. Fura-2 Ca2+-Assay
2.5. Investigation of the Stability of 8, 9, 11, 12, 14–23, 38–49 and 54–57 in Human Plasma
2.6. Circular Dichroism (CD) Analysis
2.7. Synthesis, In Vitro and In Vivo Characterization of PET Tracers [68Ga]21, [68Ga]33, [68Ga]37 and [68Ga]56
2.7.1. PET Tracer Synthesis
2.7.2. Determination of the Distribution Coefficient logD7.4 of PET Ligands [68Ga]21, [68Ga]33, [68Ga]37 and [68Ga]56
2.7.3. Mouse Xenograft Model
2.7.4. Animal Anesthetization
2.7.5. Biodistribution Studies
2.7.6. HPLC Analysis of Urine from Mice Injected with [68Ga]21, [68Ga]33, [68Ga]37 or [68Ga]56
2.7.7. HPLC Analysis of Blood Plasma from Mice Injected with [68Ga]56
2.7.8. Determination of the Internalization of [68Ga]56 in HT-29 Tumor Cells
2.7.9. PET/CT Imaging with [68Ga]56
2.7.10. Tracer Administration
2.7.11. Imaging Analysis
3. Results and Discussion
3.1. Chemistry
3.2. Circular Dichoism (CD) Analysis
3.3. Peptide Stability in Human Plasma
3.4. In Vitro Binding Studies at the NTS1R and NTS2R and NTS1R Agonistic Activities
3.5. Radiosynthesis and Distribution Coefficients
3.6. Biodistribution of PET Ligands [68Ga]21, [68Ga]33, [68Ga]37 and [68Ga]56, and Cellular Uptake of [68Ga]56
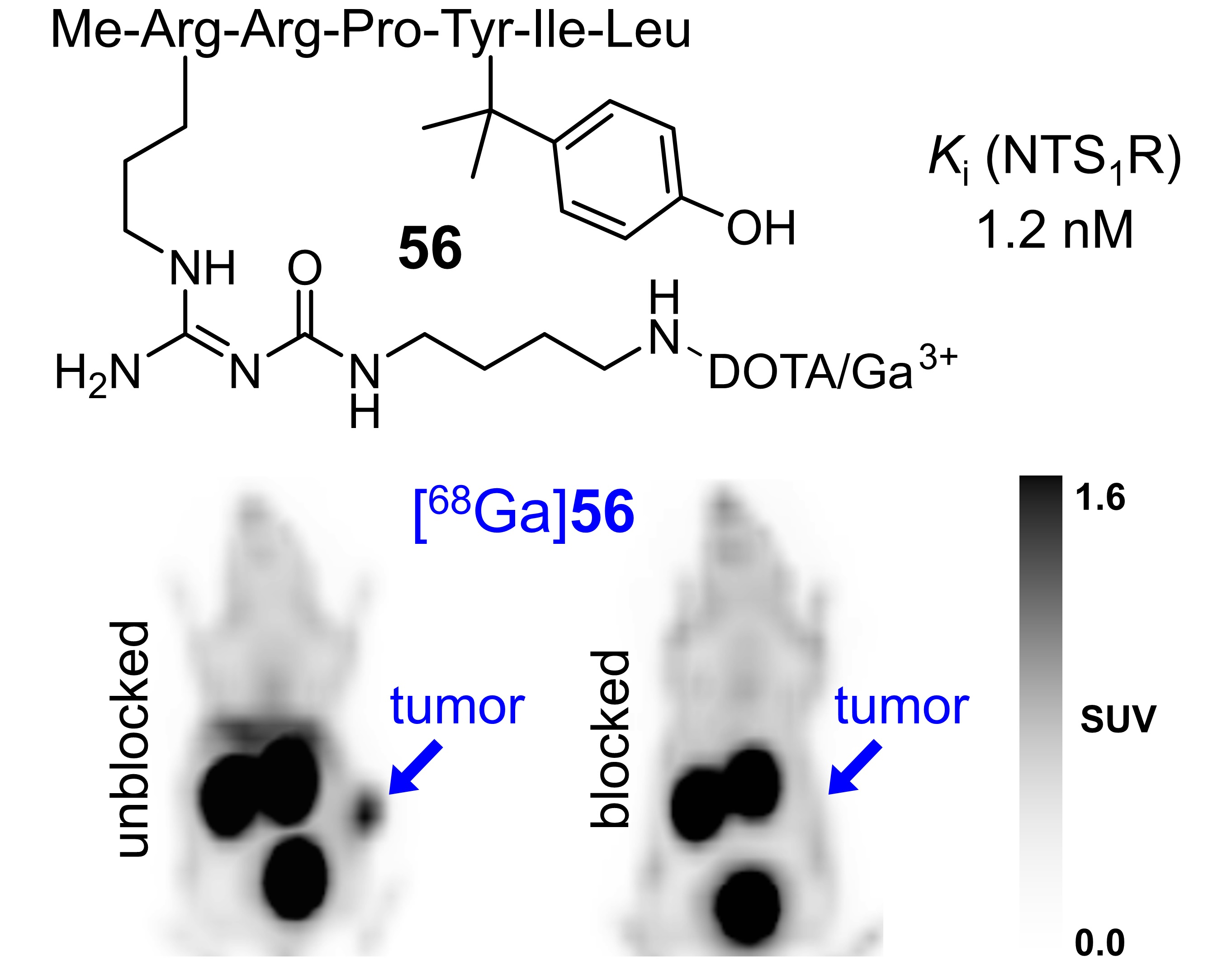
3.7. PET/CT Imaging with [68Ga]56
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bugni, J.M.; Pothoulakis, C. Neurotensin. In Handbook of Biologically Active Peptides (Section XIII: Gastrointestinal Peptides), 2nd ed.; Kastin, A.J., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 1265–1270. [Google Scholar]
- St-Gelais, F.; Jomphe, C.; Trudeau, L.E. The role of neurotensin in central nervous system pathophysiology: What is the evi-dence? J. Psychiatry Neurosci. 2006, 31, 229–245. [Google Scholar] [PubMed]
- Mazella, J.; Vincent, J.-P. Functional roles of the NTS2 and NTS3 receptors. Peptides 2006, 27, 2469–2475. [Google Scholar] [CrossRef]
- Boules, M.; Li, Z.; Smith, K.; Fredrickson, P.; Richelson, E. Diverse Roles of Neurotensin Agonists in the Central Nervous System. Front. Endocrinol. 2013, 4, 36. [Google Scholar] [CrossRef]
- Xiao, Z.Y.; Cilz, N.I.; Kurada, L.; Hu, B.Q.; Yang, C.X.; Wada, E.; Combs, C.K.; Porter, J.E.; Lesage, F.; Lei, S.B. Activation of Neurotensin Receptor 1 Facilitates Neuronal Excitability and Spatial Learning and Memory in the Entorhinal Cortex: Beneficial Actions in an Alzheimer’s Disease Model. J. Neurosci. 2014, 34, 7027–7042. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, L.E.; Leinninger, G.M. Role of central neurotensin in regulating feeding: Implications for the development and treatment of body weight disorders. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 900–916. [Google Scholar] [CrossRef] [PubMed]
- Sarret, P.; Cavelier, F. Neurotensin and its receptors. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Maoret, J.J.; Pospai, D.; Rouyerfessard, C.; Couvineau, A.; Laboisse, C.; Voisin, T.; Laburthe, M. Neurotensin Receptor and Its mRNA Are Expressed in Many Human Colon Cancer Cell Lines but Not in Normal Colonic Epithelium: Binding Studies and RT-PCR Experiments. Biochem. Biophys. Res. Commun. 1994, 203, 465–471. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Friess, H.; Buchler, M.; Laissue, J. Neurotensin receptors: A new marker for human ductal pancreatic adenocarcinoma. Gut 1998, 42, 546–550. [Google Scholar] [CrossRef]
- Souazé, F.; Dupouy, S.; Viardot-Foucault, V.; Bruyneel, E.; Attoub, S.; Gespach, C.; Gompel, A.; Forgez, P. Expression of Neurotensin and NT1 Receptor in Human Breast Cancer: A Potential Role in Tumor Progression. Cancer Res. 2006, 66, 6243–6249. [Google Scholar] [CrossRef]
- Carraway, R.; Leeman, S.E. The amino acid sequence of a hypothalamic peptide, neurotensin. J. Biol. Chem. 1975, 250, 1907–1911. [Google Scholar] [CrossRef]
- Tyler, B.M.; Douglas, C.L.; Fauq, A.; Pang, Y.-P.; Stewart, J.A.; Cusack, B.; McCormick, D.J.; Richelson, E. In vitro binding and CNS effects of novel neurotensin agonists that cross the blood–brain barrier. Neuropharmacology 1999, 38, 1027–1034. [Google Scholar] [CrossRef]
- Vincent, J.-P.; Mazella, J.; Kitabgi, P. Neurotensin and neurotensin receptors. Trends Pharmacol. Sci. 1999, 20, 302–309. [Google Scholar] [CrossRef]
- Bergmann, R.; Scheunemann, M.; Heichert, C.; Mäding, P.; Wittrisch, H.; Kretzschmar, M.; Rodig, H.; Tourwé, D.; Iterbeke, K.; Chavatte, K.; et al. Biodistribution and catabolism of 18F-labeled neurotensin (8–13) analogs. Nucl. Med. Biol. 2002, 29, 61–72. [Google Scholar] [CrossRef]
- García-Garayoa, E.; Bläuenstein, P.; Blanc, A.; Maes, V.; Tourwé, D.; Schubiger, P.A. A stable neurotensin-based radiopharmaceutical for targeted imaging and therapy of neurotensin receptor-positive tumours. Eur. J. Pediatr. 2008, 36, 37–47. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Hocke, C.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Synthesis of a 68Ga-Labeled Peptoid−Peptide Hybrid for Imaging of Neurotensin Receptor Expression in Vivo. ACS Med. Chem. Lett. 2010, 1, 224–228. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Haubner, R.; Hocke, C.; Ocker, M.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Labeling and Glycosylation of Peptides Using Click Chemistry: A General Approach to 18F-Glycopeptides as Effective Imaging Probes for Positron Emission Tomography. Angew. Chem. Int. Ed. 2009, 49, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Ruckdeschel, T.; Tripal, P.; Haubner, R.; Einsiedel, J.; Hübner, H.; Gmeiner, P.; Kuwert, T.; Prante, O. In Vivo Monitoring of the Antiangiogenic Effect of Neurotensin Receptor-Mediated Radiotherapy by Small-Animal Positron Emission Tomography: A Pilot Study. Pharmaceuticals 2014, 7, 464–481. [Google Scholar] [CrossRef]
- Jia, Y.; Shi, W.; Zhou, Z.; Wagh, N.K.; Fan, W.; Brusnahan, S.K.; Garrison, J.C. Evaluation of DOTA-chelated neurotensin analogs with spacer-enhanced biological performance for neurotensin-receptor-1-positive tumor targeting. Nucl. Med. Biol. 2015, 42, 816–823. [Google Scholar] [CrossRef]
- Deng, H.; Wang, H.; Zhang, H.; Wang, M.; Giglio, B.; Ma, X.; Jiang, G.; Yuan, H.; Wu, Z.; Li, Z. Imaging Neurotensin Receptor in Prostate Cancer With 64Cu-Labeled Neurotensin Analogs. Mol. Imaging 2017, 16, 1536012117711369. [Google Scholar] [CrossRef] [PubMed]
- Mankoff, D.A.; Link, J.M.; Linden, H.M.; Sundararajan, L.; Krohn, K.A. Tumor Receptor Imaging. J. Nucl. Med. 2008, 49 (Suppl. 2), 149S–163S. [Google Scholar] [CrossRef] [PubMed]
- Correia, J.D.G.; Paulo, A.; Raposinho, P.D.; Santos, I. Radiometallated peptides for molecular imaging and targeted therapy. Dalton Trans. 2011, 40, 6144–6167. [Google Scholar] [CrossRef]
- Morgat, C.; Mishra, A.K.; Varshney, R.; Allard, M.; Fernandez, P.; Hindié, E. Targeting Neuropeptide Receptors for Cancer Imaging and Therapy: Perspectives with Bombesin, Neurotensin, and Neuropeptide-Y Receptors. J. Nucl. Med. 2014, 55, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Lau, W.F.E.; Hicks, R.J. Somatostatin Receptor Imaging with68Ga DOTATATE PET/CT: Clinical Utility, Normal Patterns, Pearls, and Pitfalls in Interpretation. RadioGraphics 2015, 35, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Prante, O. Radiopharmaceuticals for imaging and endoradiotherapy of neurotensin receptor-positive tumors. J. Label. Compd. Radiopharm. 2018, 61, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Desai, H.; Borges-Neto, S.; Wong, T.Z. Molecular Imaging and Therapy for Neuroendocrine Tumors. Curr. Treat. Options Oncol. 2019, 20, 78. [Google Scholar] [CrossRef]
- Damuka, N.; Solingapuram Sai, K.K. Method to development of PET radiopharmaceutical for cancer imaging. In Cancer Biomarkers, 1st ed.; Deep, G., Ed.; Humana: New York, NY, USA, 2022; Volume 2413, pp. 13–22. [Google Scholar]
- Fani, M.; Maecke, H.R. Radiopharmaceutical development of radiolabelled peptides. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. 1), S11–S30. [Google Scholar] [CrossRef]
- Richter, S.; Wuest, F. 18F-Labeled Peptides: The Future Is Bright. Molecules 2014, 19, 20536–20556. [Google Scholar] [CrossRef]
- Kumar, K.; Ghosh, A. 18F-AlF Labeled Peptide and Protein Conjugates as Positron Emission Tomography Imaging Pharmaceuticals. Bioconjugate Chem. 2018, 29, 953–975. [Google Scholar] [CrossRef]
- Schottelius, M.; Wester, H.-J. Molecular imaging targeting peptide receptors. Methods 2009, 48, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Xie, J.; Chen, X. Peptide-Based Probes for Targeted Molecular Imaging. Biochemistry 2010, 49, 1364–1376. [Google Scholar] [CrossRef]
- Fani, M.; Maecke, H.R.; Okarvi, S.M. Radiolabeled Peptides: Valuable Tools for the Detection and Treatment of Cancer. Theranostics 2012, 2, 481–501. [Google Scholar] [CrossRef]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Maschauer, S.; Hübner, H.; Gmeiner, P.; Prante, O. Synthesis and Evaluation of a 18F-Labeled Diarylpyrazole Glycoconjugate for the Imaging of NTS1-Positive Tumors. J. Med. Chem. 2013, 56, 9361–9365. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Rohracker, M.; Stiebler, M.; Goldschmidt, J.; Grosser, O.S.; Osterkamp, F.; Pethe, A.; Reineke, U.; Smerling, C.; Amthauer, H. Comparative Evaluation of the Biodistribution Profiles of a Series of Nonpeptidic Neurotensin Receptor-1 Antagonists Reveals a Promising Candidate for Theranostic Applications. J. Nucl. Med. 2016, 57, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- Renard, E.; Moreau, M.; Bellaye, P.-S.; Guillemin, M.; Collin, B.; Prignon, A.; Denat, F.; Goncalves, V. Positron Emission Tomography Imaging of Neurotensin Receptor-Positive Tumors with 68Ga-Labeled Antagonists: The Chelate Makes the Difference Again. J. Med. Chem. 2021, 64, 8564–8578. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, R.; Chastel, A.; Previti, S.; Hindié, E.; Vimont, D.; Zanotti-Fregonara, P.; Fernandez, P.; Garrigue, P.; Lamare, F.; Schollhammer, R.; et al. Silicon-Containing Neurotensin Analogues as Radiopharmaceuticals for NTS1-Positive Tumors Imaging. Bioconjugate Chem. 2020, 31, 2339–2349. [Google Scholar] [CrossRef]
- Keller, M.; Kuhn, K.K.; Einsiedel, J.; Hübner, H.; Biselli, S.; Mollereau, C.; Wifling, D.; Svobodová, J.; Bernhardt, G.; Cabrele, C.; et al. Mimicking of Arginine by Functionalized Nω-Carbamoylated Arginine as a New Broadly Applicable Approach to Labeled Bioactive Peptides: High Affinity Angiotensin, Neuropeptide Y, Neuropeptide FF, and Neurotensin Receptor Ligands As Examples. J. Med. Chem. 2016, 59, 1925–1945. [Google Scholar] [CrossRef]
- Schindler, L.; Bernhardt, G.; Keller, M. Modifications at Arg and Ile Give Neurotensin(8–13) Derivatives with High Stability and Retained NTS1 Receptor Affinity. ACS Med. Chem. Lett. 2019, 10, 960–965. [Google Scholar] [CrossRef]
- Alshoukr, F.; Prignon, A.; Brans, L.; Jallane, A.; Mendes, S.; Talbot, J.-N.; Tourwé, D.; Barbet, J.; Gruaz-Guyon, A. Novel DOTA-Neurotensin Analogues for 111In Scintigraphy and 68Ga PET Imaging of Neurotensin Receptor-Positive Tumors. Bioconjugate Chem. 2011, 22, 1374–1385. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Hübner, H.; Gmeiner, P.; Prante, O. 18F- and 68Ga-Labeled Neurotensin Peptides for PET Imaging of Neurotensin Receptor 1. J. Med. Chem. 2016, 59, 6480–6492. [Google Scholar] [CrossRef]
- García-Garayoa, E.; Allemann-Tannahill, L.; Bläuenstein, P.; Willmann, M.; Carrel-Rémy, N.; Tourwé, D.; Iterbeke, K.; Conrath, P.; Schubiger, P.A. In vitro and in vivo evaluation of new radiolabeled neurotensin(8–13) analogues with high affinity for NT1 receptors. Nucl. Med. Biol. 2001, 28, 75–84. [Google Scholar] [CrossRef]
- Mascarin, A.; Valverde, I.E.; Vomstein, S.; Mindt, T.L. 1,2,3-Triazole Stabilized Neurotensin-Based Radiopeptidomimetics for Improved Tumor Targeting. Bioconjugate Chem. 2015, 26, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Tyler-McMahon, B.M.; Stewart, J.A.; Farinas, F.; McCormick, D.J.; Richelson, E. Highly potent neurotensin analog that causes hypothermia and antinociception. Eur. J. Pharmacol. 2000, 390, 107–111. [Google Scholar] [CrossRef]
- Bruehlmeier, M.; Garayoa, E.G.; Blanc, A.; Holzer, B.; Gergely, S.; Tourwé, D.; Schubiger, P.A.; Bläuenstein, P. Stabilization of neurotensin analogues: Effect on peptide catabolism, biodistribution and tumor binding. Nucl. Med. Biol. 2002, 29, 321–327. [Google Scholar] [CrossRef]
- García-Garayoa, E.; Bläuenstein, P.; Bruehlmeier, M.; Blanc, A.; Iterbeke, K.; Conrath, P.; Tourwé, D.; Schubiger, P.A. Preclin-ical evaluation of a new, stabilized neurotensin(8-13) pseudopeptide radiolabeled with 99mTc. J. Nucl. Med. 2002, 43, 374–383. [Google Scholar]
- Bläuenstein, P.; Garayoa, E.G.; Rüegg, D.; Blanc, A.; Tourwé, D.; Beck-Sickinger, A.; Schubiger, P.A. Improving the Tumor Uptake of 99mTc-Labeled Neuropeptides Using Stabilized Peptide Analogues. Cancer Biother. Radiopharm. 2004, 19, 181–188. [Google Scholar] [CrossRef]
- Maes, V.; Garcia-Garayoa, E.; Bläuenstein, P.; Tourwé, D. Novel 99mTc-Labeled Neurotensin Analogues with Optimized Biodistribution Properties. J. Med. Chem. 2006, 49, 1833–1836. [Google Scholar] [CrossRef]
- Nock, B.A.; Nikolopoulou, A.; Reubi, J.-C.; Maes, V.; Conrath, P.; Tourwé, D.; Maina, T. Toward Stable N4-Modified Neurotensins for NTS1-Receptor-Targeted Tumor Imaging with 99mTc. J. Med. Chem. 2006, 49, 4767–4776. [Google Scholar] [CrossRef]
- Maina, T.; Nikolopoulou, A.; Stathopoulou, E.; Galanis, A.S.; Cordopatis, P.; Nock, B.A. [99mTc]Demotensin 5 and 6 in the NTS1-R-targeted imaging of tumours: Synthesis and preclinical results. Eur. J. Pediatr. 2007, 34, 1804–1814. [Google Scholar] [CrossRef]
- Alshoukr, F.; Rosant, C.; Maes, V.; Abdelhak, J.; Raguin, O.; Burg, S.; Sarda, L.; Barbet, J.; Tourwé, D.; Pelaprat, D.; et al. Novel Neurotensin Analogues for Radioisotope Targeting to Neurotensin Receptor-Positive Tumors. Bioconjugate Chem. 2009, 20, 1602–1610. [Google Scholar] [CrossRef]
- Boules, M.; Liang, Y.; Briody, S.; Miura, T.; Fauq, I.; Oliveros, A.; Wilson, M.; Khaniyev, S.; Williams, K.; Li, Z.; et al. NT79: A novel neurotensin analog with selective behavioral effects. Brain Res. 2010, 1308, 35–46. [Google Scholar] [CrossRef]
- Mascarin, A.; Valverde, I.E.; Mindt, T.L. Structure-Activity Relationship Studies of Amino Acid Substitutions in Radiolabeled Neurotensin Conjugates. ChemMedChem 2015, 11, 102–107. [Google Scholar] [CrossRef]
- Chavatte, K.; Terriere, D.; Jeannin, L.; Iterbeke, K.; Briejer, M.; Schuurkes, J.; Mertens, J.J.R.; Bruyneel, E.; Tourwé, D.; Leysen, J.E.; et al. Labelling and evaluation of new stabilised neurotensin(8-13) analogues for single photon emission tomography (SPET). J. Label. Comp. Radiopharm. 1999, 42, 423–435. [Google Scholar] [CrossRef]
- De León-Rodriguez, L.M.; Kovacs, Z.; Dieckmann, G.R.; Sherry, A.D. Solid-Phase Synthesis of DOTA–Peptides. Chem. A Eur. J. 2004, 10, 1149–1155. [Google Scholar] [CrossRef]
- Guérin, B.; Ait-Mohand, S.; Tremblay, M.-C.; Dumulon-Perreault, V.; Fournier, P.; Bénard, F. Total Solid-Phase Synthesis of NOTA-Functionalized Peptides for PET Imaging. Org. Lett. 2009, 12, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.K.; Ertl, T.; Dukorn, S.; Keller, M.; Bernhardt, G.; Reiser, O.; Buschauer, A. High Affinity Agonists of the Neuropeptide Y (NPY) Y4 Receptor Derived from the C-Terminal Pentapeptide of Human Pancreatic Polypeptide (hPP): Synthesis, Stereochemical Discrimination, and Radiolabeling. J. Med. Chem. 2016, 59, 6045–6058. [Google Scholar] [CrossRef]
- Keller, M.; Mahuroof, S.A.; Yee, V.H.; Carpenter, J.; Schindler, L.; Littmann, T.; Pegoli, A.; Hübner, H.; Bernhardt, G.; Gmeiner, P.; et al. Fluorescence Labeling of Neurotensin(8–13) via Arginine Residues Gives Molecular Tools with High Receptor Affinity. ACS Med. Chem. Lett. 2019, 11, 16–22. [Google Scholar] [CrossRef]
- Schindler, L.; Wohlfahrt, K.; von Krüchten, L.G.; Prante, O.; Keller, M.; Maschauer, S. Neurotensin analogs by fluoroglycosylation at Nω-carbamoylated arginines for PET imaging of NTS1-positive tumors. Sci. Rep. 2022, 12, 15028. [Google Scholar] [CrossRef]
- Grätz, L.; Laasfeld, T.; Allikalt, A.; Gruber, C.G.; Pegoli, A.; Tahk, M.-J.; Tsernant, M.-L.; Keller, M.; Rinken, A. BRET- and fluorescence anisotropy-based assays for real-time monitoring of ligand binding to M2 muscarinic acetylcholine receptors. Biochim. Biophys. Acta 2020, 1868, 118930. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Müller, M.; Knieps, S.; Geßele, K.; Dove, S.; Bernhardt, G.; Buschauer, A. Synthesis and Neuropeptide Y Y1 Receptor Antagonistic Activity ofN,N-Disubstituted ω-Guanidino- and ω-Aminoalkanoic Acid Amides. Arch. Pharm. 1997, 330, 333–342. [Google Scholar] [CrossRef]
- Jakoby, B.W.; Bercier, Y.; Conti, M.; Casey, M.E.; Bendriem, B.; Townsend, D.W. Physical and clinical performance of the mCT time-of-flight PET/CT scanner. Phys. Med. Biol. 2011, 56, 2375–2389. [Google Scholar] [CrossRef]
- DiFilippo, F.P.; Patel, S.; Asosingh, K.; Erzurum, S.C. Small-animal imaging using clinical positron emission tomogra-phy/computed tomography and super-resolution. Mol. Imaging 2012, 11, 210–219. [Google Scholar] [CrossRef]
- Miller, S.C.; Scanlan, T.S. Site-Selective N-Methylation of Peptides on Solid Support. J. Am. Chem. Soc. 1997, 119, 2301–2302. [Google Scholar] [CrossRef]
- Müller, C.; Gleixner, J.; Tahk, M.-J.; Kopanchuk, S.; Laasfeld, T.; Weinhart, M.; Schollmeyer, D.; Betschart, M.U.; Lüdeke, S.; Koch, P.; et al. Structure-Based Design of High-Affinity Fluorescent Probes for the Neuropeptide Y Y1 Receptor. J. Med. Chem. 2022, 65, 4832–4853. [Google Scholar] [CrossRef]
- Khatun, U.L.; Goswami, S.K.; Mukhopadhyay, C. Modulation of the neurotensin solution structure in the presence of ganglioside GM1 bicelle. Biophys. Chem. 2012, 168–169, 48–59. [Google Scholar] [CrossRef]
- Vita, N.; Laurent, P.; Lefort, S.; Chalon, P.; Dumont, X.; Kaghad, M.; Gully, D.; Le Fur, G.; Ferrara, P.; Caput, D. Cloning and expression of a complementary DNA encoding a high affinity human neurotensin receptor. FEBS Lett. 1993, 317, 139–142. [Google Scholar] [CrossRef]
- Kokko, K.P.; Hadden, M.K.; Orwig, K.S.; Mazella, J.; Dix, T.A. In Vitro Analysis of Stable, Receptor-Selective Neurotensin[8–13] Analogues. J. Med. Chem. 2003, 46, 4141–4148. [Google Scholar] [CrossRef]
- Richelson, E.; McCormick, D.J.; Pang, Y.P.; Phillips, K.S. Peptide analogs that are potent and selective for human neurotensin receptor subtype 2. U.S. Patent Application US2009/0062212A1, 5 March 2009. [Google Scholar]
- Cusack, B.; McCormick, D.J.; Pang, Y.-P.; Souder, T.; Garcia, R.; Fauq, A.; Richelson, E. Pharmacological and Biochemical Profiles of Unique Neurotensin 8-13 Analogs Exhibiting Species Selectivity, Stereoselectivity, and Superagonism. J. Biol. Chem. 1995, 270, 18359–18366. [Google Scholar] [CrossRef]
- Barroso, S.; Richard, F.; Nicolas-Ethève, D.; Reversat, J.-L.; Bernassau, J.-M.; Kitabgi, P.; Labbé-Jullié, C. Identification of Residues Involved in Neurotensin Binding and Modeling of the Agonist Binding Site in Neurotensin Receptor 1. J. Biol. Chem. 2000, 275, 328–336. [Google Scholar] [CrossRef]
- Pang, Y.-P.; Cusack, B.; Groshan, K.; Richelson, E. Proposed Ligand Binding Site of the Transmembrane Receptor for Neurotensin(8–13). J. Biol. Chem. 1996, 271, 15060–15068. [Google Scholar] [CrossRef]
- Härterich, S.; Koschatzky, S.; Einsiedel, J.; Gmeiner, P. Novel insights into GPCR—Peptide interactions: Mutations in extracellular loop 1, ligand backbone methylations and molecular modeling of neurotensin receptor 1. Bioorganic Med. Chem. 2008, 16, 9359–9368. [Google Scholar] [CrossRef]
- Einsiedel, J.; Held, C.; Hervet, M.; Plomer, M.; Tschammer, N.; Hübner, H.; Gmeiner, P. Discovery of Highly Potent and Neurotensin Receptor 2 Selective Neurotensin Mimetics. J. Med. Chem. 2011, 54, 2915–2923. [Google Scholar] [CrossRef] [PubMed]
- Eiselt, E.; Gonzalez, S.; Martin, C.; Chartier, M.; Betti, C.; Longpré, J.-M.; Cavelier, F.; Tourwè, D.; Gendron, L.; Ballet, S.; et al. Neurotensin Analogues Containing Cyclic Surrogates of Tyrosine at Position 11 Improve NTS2 Selectivity Leading to Analgesia without Hypotension and Hypothermia. ACS Chem. Neurosci. 2019, 10, 4535–4544. [Google Scholar] [CrossRef]
- Chalon, P.; Vita, N.; Kaghad, M.; Guillemot, M.; Bonnin, J.; Delpech, B.; Le Fur, G.; Ferrara, P.; Caput, D. Molecular cloning of a levocabastine-sensitive neurotensin binding site. FEBS Lett. 1996, 386, 91–94. [Google Scholar] [CrossRef]
- Mazella, J.; Botto, J.-M.; Guillemare, E.; Coppola, T.; Sarret, P.; Vincent, J.-P. Structure, Functional Expression, and Cerebral Localization of the Levocabastine-Sensitive Neurotensin/Neuromedin N Receptor from Mouse Brain. J. Neurosci. 1996, 16, 5613–5620. [Google Scholar] [CrossRef]
- Vita, N.; Oury-Donat, F.; Chalon, P.; Guillemot, M.; Kaghad, M.; Bachy, A.; Thurneyssen, O.; Garcia, S.; Poinot-Chazel, C.; Casellas, P.; et al. Neurotensin is an antagonist of the human neurotensin NT2 receptor expressed in Chinese hamster ovary cells. Eur. J. Pharmacol. 1998, 360, 265–272. [Google Scholar] [CrossRef]
- Kayed, H.; Meyer, P.; He, Y.; Kraenzlin, B.; Fink, C.; Gretz, N.; Schoenberg, S.O.; Sadick, M. Evaluation of the Metabolic Response to Cyclopamine Therapy in Pancreatic Cancer Xenografts Using a Clinical PET-CT System. Transl. Oncol. 2012, 5, 335–343. [Google Scholar] [CrossRef]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of Somatostatin Receptor–Positive Tumors Using 64Cu- and 68Ga-Somatostatin Antagonists: The Chelate Makes the Difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef]
- Schulz, J.; Rohracker, M.; Stiebler, M.; Goldschmidt, J.; Stöber, F.; Noriega, M.; Pethe, A.; Lukas, M.; Osterkamp, F.; Reineke, U.; et al. Proof of Therapeutic Efficacy of a 177Lu-Labeled Neurotensin Receptor 1 Antagonist in a Colon Carcinoma Xenograft Model. J. Nucl. Med. 2017, 58, 936–941. [Google Scholar] [CrossRef]
- Osterkamp, F.; Smerling, C.; Reineke, U.; Haase, C.; Ungewiss, J. Neurotensin receptor ligands. US10961199B2, 30 March 2021. [Google Scholar]
- Baum, R.P.; Singh, A.; Schuchardt, C.; Kulkarni, H.R.; Klette, I.; Wiessalla, S.; Osterkamp, F.; Reineke, U.; Smerling, C. 177Lu-3BP-227 for Neurotensin Receptor 1–Targeted Therapy of Metastatic Pancreatic Adenocarcinoma: First Clinical Results. J. Nucl. Med. 2018, 59, 809–814. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PET Ligand | Labeling Precursor | Amount of Precursor | Total Product Activity [MBq] a | Decay Corrected Radio-Chemical Yield (%) b | HPLC Analysis: b Purity, tR, k |
|---|---|---|---|---|---|
| [68Ga]21 | 16 | 10–20 µg, 7.5–15.0 nmol | 78.71–111.4 MBq | 67–77 | 97–99%, 8.1–8.2 min, 4.8–4.9 |
| [68Ga]33 | 32 | 20 µg, 11.5 nmol | 93.51 MBq | 75 | 95%, 8.1 min, 4.8 |
| [68Ga]37 | 35 | 20 µg, 12.2 nmol | 69.05 MBq | 77 | 99%, 7.8 min, 4.6 |
| [68Ga]56 | 54 | 20 µg, 14.7 nmol | 95.80–168.6 MBq | 67–80 | 92–99%, 7.3–7.4 min, 4.2–4.3 |
| cpd. | pKi ± SD/Ki [nM] NTS1R a | pKi ± SD/Ki [nM] NTS2R b | NTS1R Selectivity (Ratio Ki (NTS2R)/Ki (NTS1R)) | % Intact Peptide in Plasma after the Given Incubation Time c | ||||
|---|---|---|---|---|---|---|---|---|
| 10 min | 1 h | 6 h | 24 h | 48 h | ||||
| 1 | 9.49/0.33 d | 8.61 ± 0.09/2.5 | 7.6 | 23 d | <1 d | n.d. | <1 d | <1 d |
| 2 | 8.93/1.2 d | n.d. | - | 11 d | <1 d | n.d. | <1 d | <1 d |
| 3 | 9.07/0.88 d | 8.01 ± 0.07/9.9 | 11 | n.d. | >99 d | >99 d | 98 d | 87 d |
| 19 | 7.80 ± 0.03/16 | 7.16 ± 0.18/73 | 4.6 | n.d. | >99 | >99 | 36 ± 1 | 4.2 ± 0.6 |
| 20 | 8.70 ± 0.10/2.0 | 7.70 ± 0.09/20 | 10 | n.d. | >99 | 77 ± 2 | 15 ± 1 | 4.6 ± 0.1 |
| 21 | 8.01 ± 0.08/9.9 | 7.25 ± 0.15/59 | 6.0 | n.d. | >99 | >99 | 26 ± 1 | 4.1 ± 0.1 |
| 22 | 7.70 ± 0.07/20 | 7.08 ± 0.16/88 | 4.4 | n.d. | >99 | 87 ± 2 | 30 ± 1 | 7.7 ± 1.0 |
| 23 | 8.13 ± 0.11/7.5 | n.d. | - | n.d. | >99 | 98 ± 6 | 46 ± 2 | 7.8 ± 0.6 |
| 33 | 8.61 ± 0.07/2.5 | n.d. | - | n.d. | n.d. | n.d. | n.d. | n.d. |
| 36 | 8.53 ± 0.02/3.0 | n.d. | - | n.d. | n.d. | n.d. | n.d. | n.d. |
| 37 | 8.38 ± 0.03/4.2 | n.d. | - | n.d. | n.d. | n.d. | n.d. | n.d. |
| 56 | 8.93 ± 0.17/1.2 | 8.35 ± 0.27/5.2 | 4.3 | n.d. | >99 | >99 | 77 ± 1 | <1 |
| 57 | 7.67 ± 0.04/21 | n.d. | - | n.d. | >99 | >99 | 68 ± 1 | <1 |
| Tissue | Uptake (%ID/g) at Given Times p.i. | |||
|---|---|---|---|---|
| 10 min | 25 min | 45 min | 45 min (Blocking) | |
| kidney | 47 ± 3.4 | 47 ± 7.0 | 55 ± 28 | 53 ± 27 |
| liver | 7.2 ± 0.83 | 5.7 ± 1.6 | 5.2 ± 2.2 | 0.69 ± 0.39 |
| gall bladder (bile) | 1.1 ± 0.71 | 1.9 ± 0.79 | 1.6 ± 0.99 | 0.59 ± 0.49 |
| blood | 3.1 ± 0.34 | 2.9 ± 1.3 | 1.3 ± 0.33 | 1.4 ± 1.0 |
| spleen | 2.5 ± 0.25 | 2.6 ± 0.60 | 2.1 ± 0.52 | 0.56 ± 0.35 |
| intestine | 1.8 ± 0.22 | 2.4 ± 0.68 | 2.1 ± 0.49 | 1.0 ± 0.80 |
| heart | 1.2 ± 0.10 | 1.3 ± 0.49 | 0.72 ± 0.36 | 0.60 ± 0.44 |
| lung | 2.4 ± 0.30 | 2.8 ± 1.5 | 1.2 ± 0.34 | 1.3 ± 0.83 |
| brain | 0.079 ± 0.017 | 0.13 ± 0.11 | 0.061 ± 0.049 | 0.10 ± 0.074 |
| pancreas | 1.3 ± 0.83 | 1.1 ± 1.1 | 0.90 ± 0.57 | 0.51 ± 0.37 |
| femur | 0.95 ± 0.25 | 0.78 ± 0.27 | 0.73 ± 0.29 | 0.65 ± 0.43 |
| muscle | 1.1 ± 0.18 | 0.57 ± 0.23 | 0.55 ± 0.23 | 0.68 ± 0.61 |
| tumor | 3.6 ± 0.20 | 7.3 ± 1.8 | 8.4 ± 2.9 | 0.94 ± 0.55 |
| tumor-to-muscle | 3.3 ± 0.46 | 13 ± 3.0 | 16 ± 2.2 | 1.8 ± 0.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schindler, L.; Moosbauer, J.; Schmidt, D.; Spruss, T.; Grätz, L.; Lüdeke, S.; Hofheinz, F.; Meister, S.; Echtenacher, B.; Bernhardt, G.; et al. Development of a Neurotensin-Derived 68Ga-Labeled PET Ligand with High In Vivo Stability for Imaging of NTS1 Receptor-Expressing Tumors. Cancers 2022, 14, 4922. https://doi.org/10.3390/cancers14194922
Schindler L, Moosbauer J, Schmidt D, Spruss T, Grätz L, Lüdeke S, Hofheinz F, Meister S, Echtenacher B, Bernhardt G, et al. Development of a Neurotensin-Derived 68Ga-Labeled PET Ligand with High In Vivo Stability for Imaging of NTS1 Receptor-Expressing Tumors. Cancers. 2022; 14(19):4922. https://doi.org/10.3390/cancers14194922
Chicago/Turabian StyleSchindler, Lisa, Jutta Moosbauer, Daniel Schmidt, Thilo Spruss, Lukas Grätz, Steffen Lüdeke, Frank Hofheinz, Sebastian Meister, Bernd Echtenacher, Günther Bernhardt, and et al. 2022. "Development of a Neurotensin-Derived 68Ga-Labeled PET Ligand with High In Vivo Stability for Imaging of NTS1 Receptor-Expressing Tumors" Cancers 14, no. 19: 4922. https://doi.org/10.3390/cancers14194922
APA StyleSchindler, L., Moosbauer, J., Schmidt, D., Spruss, T., Grätz, L., Lüdeke, S., Hofheinz, F., Meister, S., Echtenacher, B., Bernhardt, G., Pietzsch, J., Hellwig, D., & Keller, M. (2022). Development of a Neurotensin-Derived 68Ga-Labeled PET Ligand with High In Vivo Stability for Imaging of NTS1 Receptor-Expressing Tumors. Cancers, 14(19), 4922. https://doi.org/10.3390/cancers14194922