
Colorectal Cancer and Purinergic Signalling: An Overview

 ,
,
Abstract
Highlights
- CD39-CD73 axis in tumor-associated immune cells promotes immune exhaustion, impairment of antitumor immune activity, and increased CRC progression.
- CD73 overexpression in cancer cells is associated with tumor growth, chemoresistance, and decreased patient survival.
- Extracellular ATP exhibits dual effects, either reducing tumor cell proliferation or favoring chemoresistance in a concentration-dependent fashion.
- Purinergic signaling components exhibit prognostic value and have the potential to be utilized as therapeutic targets.
- CD73 induces colorectal cancer progression.
- CD39 contributes to immunosuppressive activity of T cells and poor prognosis.
- P2 and P1 purinoceptors were mainly associated with poor prognosis for patients.
- Ectonucleotidases and purinoceptors have potential as therapeutic targets in CRC.
Simple Summary
Abstract
1. Introduction
2. Methodology
3. Immune Cell Dysfunction Is Associated with CRC Progression
3.1. Influence of the Gut Microbiome on Tumorigenesis
3.2. Macrophages and Neutrophils
3.3. Myeloid-Derived Suppressor Cells
3.4. Lymphocytes
4. Purinergic Signaling in Colorectal Cancer
4.1. Ectonucleotidases—General Aspects
4.1.1. Ectonucleotidases—The Role of CD39 in CRC Progression
4.1.2. Ectonucleotidases—Participation of CD73 in CRC Progression
4.2. Purinoceptors—General Aspects
4.2.1. ATP as an Agonist of Purinoceptor-Mediated Protumor and Antitumor Actions
4.2.2. P1 Receptors and Their Relationship with CRC Progression
4.2.3. P2 Receptors—P2X Participation in CRC Development
4.2.4. P2 receptors—P2Y Participation in CRC Development
5. Concluding Remarks
6. Open Questions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef]
- De’ Angelis, G.L.; Bottarelli, L.; Azzoni, C.; De’ Angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Acta Biomed. 2018, 89, 97–101. [Google Scholar] [CrossRef]
- Buccafusca, G.; Proserpio, I.; Tralongo, A.C.; Rametta Giuliano, S.; Tralongo, P. Early colorectal cancer: Diagnosis, treatment and survivorship care. Crit. Rev. Oncol. Hematol. 2019, 136, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Dzunic, M.; Petkovic, I.; Cvetanovic, A.; Vrbic, S.; Pejcic, I. Current and future targets and therapies in metastatic colorectal cancer. J. BUON 2019, 24, 1785–1792. [Google Scholar] [PubMed]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Piawah, S.; Venook, A.P. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer 2019, 125, 4139–4147. [Google Scholar] [CrossRef]
- Arnold, D.; Lueza, B.; Douillard, J.-Y.; Peeters, M.; Lenz, H.-J.; Venook, A.; Heinemann, V.; Van Cutsem, E.; Pignon, J.-P.; Tabernero, J.; et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann. Oncol. 2017, 28, 1713–1729. [Google Scholar] [CrossRef]
- Robinson, J.R.; Newcomb, P.A.; Hardikar, S.; Cohen, S.A.; Phipps, A.I. Stage IV colorectal cancer primary site and patterns of distant metastasis. Cancer Epidemiol. 2017, 48, 92–95. [Google Scholar] [CrossRef]
- JIN, K.; GAO, W.; LU, Y.; LAN, H.; TENG, L.; CAO, F. Mechanisms regulating colorectal cancer cell metastasis into liver (Review). Oncol. Lett. 2012, 3, 11–15. [Google Scholar] [CrossRef][Green Version]
- American Cancer Society American Cancer Society. Cancer Facts & Figures 2021. American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- Liu, Q.; Ma, Y.; Luo, D.; Cai, S.; Li, Q.; Li, X. Real-world study of a novel prognostic scoring system: For a more precise prognostication and better clinical treatment guidance in stages II and III colon cancer. Int. J. Colorectal Dis. 2018, 33, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Puppa, G.; Sonzogni, A.; Colombari, R.; Pelosi, G. TNM Staging System of Colorectal Carcinoma: A Critical Appraisal of Challenging Issues. Arch. Pathol. Lab. Med. 2010, 134, 837–852. [Google Scholar] [CrossRef]
- Marks, K.M.; West, N.P.; Morris, E.; Quirke, P. Clinicopathological, genomic and immunological factors in colorectal cancer prognosis. Br. J. Surg. 2018, 105, e99–e109. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Herrinton, L.; Liu, L.; Levin, T.; Allison, J.; Lewis, J.; Velayos, F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology 2012, 143, 382–389. [Google Scholar] [CrossRef]
- Sillo, T.O.; Beggs, A.D.; Morton, D.G.; Middleton, G. Mechanisms of immunogenicity in colorectal cancer. Br. J. Surg. 2019, 106, 1283–1297. [Google Scholar] [CrossRef]
- Koi, M.; Carethers, J.M. The colorectal cancer immune microenvironment and approach to immunotherapies. Futur. Oncol. 2017, 13, 1633–1647. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.-S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef]
- Allard, D.; Chrobak, P.; Allard, B.; Messaoudi, N.; Stagg, J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol. Lett. 2019, 205, 31–39. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Braganhol, E.; Whiteside, T.L. Inhibition of the Adenosinergic Pathway in Cancer Rejuvenates Innate and Adaptive Immunity. Int. J. Mol. Sci. 2019, 20, 5698. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling: Past, present and future. Brazilian J. Med. Biol. Res. 2009, 42, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Sévigny, J.; Zimmermann, H. The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal. 2006, 2, 409–430. [Google Scholar] [CrossRef]
- Burnstock, G.; Boeynaems, J.-M. Purinergic signalling and immune cells. Purinergic Signal. 2014, 10, 529–564. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; McNamee, E.N.; Idzko, M.; Gambari, R.; Eltzschig, H.K. Purinergic Signaling During Immune Cell Trafficking. Trends Immunol. 2016, 37, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.; Schreiber, H.; Fu, Y. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Strasser, K.; Birnleitner, H.; Beer, A.; Pils, D.; Gerner, M.C.; Schmetterer, K.G.; Bachleitner-Hofmann, T.; Stift, A.; Bergmann, M.; Oehler, R. Immunological differences between colorectal cancer and normal mucosa uncover a prognostically relevant immune cell profile. Oncoimmunology 2019, 8, e1537693. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef]
- Lam, K.C.; Araya, R.E.; Huang, A.; Chen, Q.; Di Modica, M.; Rodrigues, R.R.; Lopès, A.; Johnson, S.B.; Schwarz, B.; Bohrnsen, E.; et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 2021, 184, 5338–5356.e21. [Google Scholar] [CrossRef]
- Snoderly, H.; Boone, B.; Bennewitz, M. Neutrophil extracellular traps in breast cancer and beyond: Current perspectives on NET stimuli, thrombosis and metastasis, and clinical utility for diagnosis and treatment. Breast Cancer Res. 2019, 21, 145. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The role of tumor-associated neutrophils in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [PubMed]
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 2017, 140, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Granot, Z.; Jablonska, J. Distinct Functions of Neutrophil in Cancer and Its Regulation. Mediators Inflamm. 2015, 2015. [Google Scholar] [CrossRef]
- Germann, M.; Zangger, N.; Sauvain, M.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G.; et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGF β. EMBO Mol. Med. 2020, 12, e10681. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, K.; Zhou, H.; Peng, L.; You, W.; Fu, Z. Profiles of immune infiltration in colorectal cancer and their clinical significant: A gene expression-based study. Cancer Med. 2018, 7, 4496–4508. [Google Scholar] [CrossRef]
- Gouveia-Fernandes, S. Monocytes and Macrophages in Cancer: Unsuspected Roles. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1219, pp. 161–185. [Google Scholar]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 4709–4721. [Google Scholar] [CrossRef] [PubMed]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology 2017, 6, e1342918. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, T.; Kang, Z.; Guo, G.; Sun, Y.; Lin, K.; Huang, Q.; Shi, X.; Ni, Z.; Ding, N.; et al. Tumor-infiltrating immune cells act as a marker for prognosis in colorectal cancer. Front. Immunol. 2019, 10, 2368. [Google Scholar] [CrossRef]
- Popěna, I.; Abols, A.; Saulite, L.; Pleiko, K.; Zandberga, E.; Jěkabsons, K.; Endzeliņš, E.; Llorente, A.; Lině, A.; Riekstiņa, U. Effect of colorectal cancer-derived extracellular vesicles on the immunophenotype and cytokine secretion profile of monocytes and macrophages. Cell Commun. Signal. 2018, 16, 17. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Bonavita, E.; Galdiero, M.R.; Jaillon, S.; Mantovani, A. Phagocytes as Corrupted Policemen in Cancer-Related Inflammation, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 128, ISBN 9780128023167. [Google Scholar]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Varricchi, G.; Loffredo, S.; Mantovani, A.; Marone, G. Roles of neutrophils in cancer growth and progression. J. Leukoc. Biol. 2018, 103, 457–464. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Vance, R.E. Self-tolerance of natural killer cells. Nat. Rev. Immunol. 2006, 6, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.M.; Yokoyama, W.M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.; Ma, X.; Kundu, N.; Fulton, A. Prostaglandin E 2 (PGE 2) suppresses natural killer cell function primarily through the PGE 2 receptor EP4. Cancer Immunol. Immunother. 2011, 60, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Pietra, G.; Manzini, C.; Rivara, S.; Vitale, M.; Cantoni, C.; Petretto, A.; Balsamo, M.; Conte, R.; Benelli, R.; Minghelli, S.; et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. 2012, 72, 1407–1415. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.O.; Chang, X.; Blaser, B.W.; Caligiuri, M.A.; Zheng, P.; Liu, Y. Tumor growth impedes natural-killer-cell maturation in the bone marrow. Blood 2006, 108, 246–252. [Google Scholar] [CrossRef]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Braun, M.; Kahlert, C.; Spille, A.; Quack, C.; Rahbari, N.; Koch, M.; Weitz, J.; Kloor, M.; Zoernig, I.; et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin. Cancer Res. 2011, 17, 678–689. [Google Scholar] [CrossRef]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef]
- Speiser, D.E.; Ho, P.C.; Verdeil, G. Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol. 2016, 16, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Malissen, B.; Bongrand, P. Early T cell activation: Integrating biochemical, structural, and biophysical cues. Annu. Rev. Immunol. 2015, 33, 539–561. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3-potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3 + CD4 + T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef]
- Betts, G.; Jones, E.; Junaid, S.; El-Shanawany, T.; Scurr, M.; Mizen, P.; Kumar, M.; Jones, S.; Rees, B.; Williams, G.; et al. Suppression of tumour-specific CD4 + T cells by regulatory T cells is associated with progression of human colorectal cancer. Gut 2012, 61, 1163–1171. [Google Scholar] [CrossRef]
- Frey, D.M.; Droeser, R.A.; Viehl, C.T.; Zlobec, I.; Lugli, A.; Zingg, U.; Oertli, D.; Kettelhack, C.; Terracciano, L.; Tornillo, L. High frequency of tumor-infiltrating FOXP3+ regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int. J. Cancer 2010, 126, 2635–2643. [Google Scholar] [CrossRef]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.D.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.J.; Toma, C.; Yu, B.; Zhang, K.; Omilusik, K.; Phan, A.T.; Wang, D.; Getzler, A.J.; Nguyen, T.; Crotty, S.; et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 2017, 552, 253–257. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Saleh, R.; Taha, R.Z.; Toor, S.M.; Sasidharan Nair, V.; Murshed, K.; Khawar, M.; Al-Dhaheri, M.; Petkar, M.A.; Abu Nada, M.; Elkord, E. Expression of immune checkpoints and T cell exhaustion markers in early and advanced stages of colorectal cancer. Cancer Immunol. Immunother. 2020, 69, 1989–1999. [Google Scholar] [CrossRef]
- Kim, C.G.; Jang, M.; Kim, Y.; Leem, G.; Kim, K.H.; Lee, H.; Kim, T.S.; Choi, S.J.; Kim, H.D.; Han, J.W.; et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci. Immunol. 2019, 4, eaay0555. [Google Scholar] [CrossRef]
- Di, J.; Liu, M.; Fan, Y.; Gao, P.; Wang, Z.; Jiang, B.; Su, X. Phenotype molding of T cells in colorectal cancer by single-cell analysis. Int. J. Cancer 2020, 146, 2281–2295. [Google Scholar] [CrossRef]
- Quinn, D.I.; Shore, N.D.; Egawa, S.; Gerritsen, W.R.; Fizazi, K. Immunotherapy for castration-resistant prostate cancer: Progress and new paradigms. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 245–260. [Google Scholar] [CrossRef]
- Emens, L.A. Breast cancer immunotherapy: Facts and hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef]
- Eroglu, Z.; Zaretsky, J.M.; Hu-Lieskovan, S.; Kim, D.W.; Algazi, A.; Johnson, D.B.; Liniker, E.; Kong, B.; Munhoz, R.; Rapisuwon, S.; et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 2018, 553, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Shin, S.; Dy, G. Advances in cancer immunotherapy in solid tumors. Cancers 2016, 8, 106. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Tintelnot, J.; Stein, A. Immunotherapy in colorectal cancer: Available clinical evidence, challenges and novel approaches. World J. Gastroenterol. 2019, 25, 3920–3928. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. Prostatic acid phosphatase, a neglected ectonucleotidase. Purinergic Signal. 2009, 5, 273–275. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Ferretti, E.; Horenstein, A.; Canzonetta, C.; Costa, F.; Morandi, F. Canonical and non-canonical adenosinergic pathways. Immunol. Lett. 2019, 205, 25–30. [Google Scholar] [CrossRef]
- Allard, D.; Allard, B.; Gaudreau, P.O.; Chrobak, P.; Stagg, J. CD73-adenosine: A next-generation target in immuno-oncology. Immunotherapy 2016, 8, 145–163. [Google Scholar] [CrossRef]
- Bagheri, S.; Saboury, A.; Haertlé, T. Adenosine deaminase inhibition. Int. J. Biol. Macromol. 2019, 141, 1246–1257. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Wang, X.; Zhang, H.; Lin, F.; Liu, C.; Dong, K. The roles of adenosine deaminase in autoimmune diseases. Autoimmun. Rev. 2021, 20, 102709. [Google Scholar] [CrossRef]
- Sundström, P.; Stenstad, H.; Langenes, V.; Ahlmanner, F.; Theander, L.; Ndah, T.G.; Fredin, K.; Börjesson, L.; Gustavsson, B.; Bastid, J.; et al. Regulatory T cells from colon cancer patients inhibit effector T-cell migration through an adenosine-dependent mechanism. Cancer Immunol. Res. 2016, 4, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wu, P.; Cheng, P.; Zhang, Z.; Wang, Z.; Yu, X.; Shao, X.; Wu, D.; Ye, J.; Zhang, T.; et al. Tumor-infiltrating CD39+ γδTregs are novel immunosuppressive T cells in human colorectal cancer. Oncoimmunology 2017, 6, e1277305. [Google Scholar] [CrossRef]
- Khaja, A.S.S.; Toor, S.M.; Salhat, H.E.; Ali, B.R.; Elkord, E. Intratumoral FoxP3+Helios+ regulatory T Cells upregulating immunosuppressive molecules are expanded in human colorectal cancer. Front. Immunol. 2017, 8, 619. [Google Scholar] [CrossRef]
- Parodi, A.; Battaglia, F.; Kalli, F.; Ferrera, F.; Conteduca, G.; Tardito, S.; Stringara, S.; Ivaldi, F.; Negrini, S.; Borgonovo, G.; et al. CD39 is highly involved in mediating the suppression activity of tumor-infiltrating CD8+ T regulatory lymphocytes. Cancer Immunol. Immunother. 2013, 62, 851–862. [Google Scholar] [CrossRef]
- Scurr, M.; Ladell, K.; Besneux, M.; Christian, A.; Hockey, T.; Smart, K.; Bridgeman, H.; Hargest, R.; Phillips, S.; Davies, M.; et al. Highly prevalent colorectal cancer-infiltrating LAP + Foxp3 - T cells exhibit more potent immunosuppressive activity than Foxp3 + regulatory T cells. Mucosal Immunol. 2014, 7, 428–439. [Google Scholar] [CrossRef]
- Timperi, E.; Pacella, I.; Schinzari, V.; Focaccetti, C.; Sacco, L.; Farelli, F.; Caronna, R.; Del Bene, G.; Longo, F.; Ciardi, A.; et al. Regulatory T cells with multiple suppressive and potentially pro-tumor activities accumulate in human colorectal cancer. Oncoimmunology 2016, 5, e1175800. [Google Scholar] [CrossRef]
- Zhulai, G.; Churov, A.V.; Oleinik, E.K.; Romanov, A.A.; Semakova, P.N.; Oleinik, V.M. Activation of cd4+cd39+ T cells in colorectal cancer. Bull. Russ. State Med. Univ. 2018, 7, 47–53. [Google Scholar] [CrossRef]
- Takatori, H.; Kawashima, H.; Matsuki, A.; Meguro, K.; Tanaka, S.; Iwamoto, T.; Sanayama, Y.; Nishikawa, N.; Tamachi, T.; Ikeda, K.; et al. Helios Enhances Treg Cell Function in Cooperation With FoxP3. Arthritis Rheumatol. 2015, 67, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, M.; Okabayashi, K.; Hasegawa, H.; Tsuruta, M.; Kitagawa, Y. The Worse Prognosis of Right-Sided Compared with Left-Sided Colon Cancers: A Systematic Review and Meta-analysis. J. Gastrointest. Surg. 2015 203 2015, 20, 648–655. [Google Scholar] [CrossRef]
- Liang, L.; Zeng, J.H.; Qin, X.G.; Chen, J.Q.; Luo, D.Z.; Chen, G. Distinguishable Prognostic Signatures of Left- and Right-Sided Colon Cancer: A Study Based on Sequencing Data. Cell. Physiol. Biochem. 2018, 48, 475–490. [Google Scholar] [CrossRef]
- Zhan, Y.; Zheng, L.; Liu, J.; Hu, D.; Wang, J.; Liu, K.; Guo, J.; Zhang, T.; Kong, D. PLA2G4A promotes right-sided colorectal cancer progression by inducing CD39+γδ Treg polarization. JCI Insight 2021, 6, e148028. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Gallerano, D.; Ciminati, S.; Grimaldi, A.; Piconese, S.; Cammarata, I.; Focaccetti, C.; Pacella, I.; Accapezzato, D.; Lancellotti, F.; Sacco, L.; et al. Genetically driven CD39 expression shapes human tumor-infiltrating CD8+ T-cell functions. Int. J. Cancer 2020, 147, 2597–2610. [Google Scholar] [CrossRef] [PubMed]
- Bonnereau, J.; Courau, T.; Asesio, N.; Salfati, D.; Bouhidel, F.; Corte, H.; Hamoudi, S.; Hammoudi, N.; Lavolé, J.; Vivier-Chicoteau, J.; et al. Autologous T cell responses to primary human colorectal cancer spheroids are enhanced by ectonucleotidase inhibition. Gut, 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Rodin, W.; Sundström, P.; Ahlmanner, F.; Szeponik, L.; Zajt, K.K.; Wettergren, Y.; Bexe Lindskog, E.; Quiding Järbrink, M. Exhaustion in tumor-infiltrating Mucosal-Associated Invariant T (MAIT) cells from colon cancer patients. Cancer Immunol. Immunother. 2021, 70, 3461–3475. [Google Scholar] [CrossRef]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rébé, C.; Derangère, V.; Chevriaux, A.; Boidot, R.; Végran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, Z.; Wu, L.; Zhang, M.; Li, W.; Ding, J.; Zhu, J.; Wei, H.; Zhao, K. Circulating and Tumor-Infiltrating Myeloid-Derived Suppressor Cells in Patients with Colorectal Carcinoma. PLoS One 2013, 8, e57114. [Google Scholar] [CrossRef]
- Künzli, B.M.; Bernlochner, M.-I.; Rath, S.; Käser, S.; Csizmadia, E.; Enjyoji, K.; Cowan, P.; D’Apice, A.; Dwyer, K.; Rosenberg, R.; et al. Impact of CD39 and purinergic signalling on the growth and metastasis of colorectal cancer. Purinergic Signal. 2011, 7, 231–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Ding, Z.; He, C.; Gao, G.; Lyu, S.; Gao, Y.; Du, J. Identification of Novel CD39 Inhibitors Based on Virtual Screening and Enzymatic Assays. J. Chem. Inf. Model. 2021; Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Gaibar, M.; Galán, M.; Romero-Lorca, A.; Antón, B.; Malón, D.; Moreno, A.; Fernández-Santander, A.; Novillo, A. Genetic variants of ANGPT1, CD39, FGF2 and MMP9 linked to clinical outcome of bevacizumab plus chemotherapy for metastatic colorectal cancer. Int. J. Mol. Sci. 2021, 22, 1381. [Google Scholar] [CrossRef]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Yu, J.; Zhou, P. Role of CD73 in Disease: Promising Prognostic Indicator and Therapeutic Target. Curr. Med. Chem. 2018, 25, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, M.; Wakui, M.; Monnai, M.; Mizushima, T.; Nishime, C.; Kawai, K.; Ohmura, M.; Suemizu, H.; Hishiki, T.; Suematsu, M.; et al. Reduced CD73 expression and its association with altered purine nucleotide metabolism in colorectal cancer cells robustly causing liver metastases. Oncol. Lett. 2010, 1, 431–436. [Google Scholar] [CrossRef]
- Cushman, S.M.; Jiang, C.; Hatch, A.J.; Shterev, I.; Sibley, A.B.; Niedzwiecki, D.; Venook, A.P.; Owzar, K.; Hurwitz, H.I.; Nixon, A.B. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: Results from CALGB 80203 (Alliance). Clin. Cancer Res. 2014, 21, 1078–1086. [Google Scholar] [CrossRef]
- Hatch, A.J.; Sibley, A.B.; Starr, M.D.; Brady, J.C.; Jiang, C.; Jia, J.; Bowers, D.L.; Pang, H.; Owzar, K.; Niedzwiecki, D.; et al. Blood-based markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: Results from CALGB 80203 (Alliance). Cancer Med. 2016, 5, 2249–2260. [Google Scholar] [CrossRef]
- Wu, X.R.; He, X.S.; Chen, Y.F.; Yuan, R.X.; Zeng, Y.; Lian, L.; Zou, Y.F.; Lan, N.; Wu, X.J.; Lan, P. High expression of CD73 as a poor prognostic biomarker in human colorectal cancer. J. Surg. Oncol. 2012, 106, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Song, B.; Wang, X.; Chang, X.S.; Pang, T.; Zhang, X.; Yin, K.; Fang, G.E. The expression and clinical significance of CD73 molecule in human rectal adenocarcinoma. Tumor Biol. 2015, 36, 5459–5466. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Chen, Y.; Li, F.; Li, W.; Zhou, H.; Yang, Y.; Pei, Z. Effects of CD73 on human colorectal cancer cell growth in vivo and in vitro. Oncol. Rep. 2016, 35, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fu, S.; Li, D.; Chen, Y. Expression and clinical significance of serum NT5E protein in patients with colorectal cancer. Cancer Biomarkers 2019, 24, 461–468. [Google Scholar] [CrossRef]
- Messaoudi, N.; Cousineau, I.; Arslanian, E.; Henault, D.; Stephen, D.; Vandenbroucke-Menu, F.; Dagenais, M.; Létourneau, R.; Plasse, M.; Roy, A.; et al. Prognostic value of CD73 expression in resected colorectal cancer liver metastasis. Oncoimmunology 2020, 9, 1746138. [Google Scholar] [CrossRef]
- Allard, B.; Turcotte, M.; Stagg, J. CD73-Generated Adenosine: Orchestrating the Tumor-Stroma Interplay to Promote Cancer Growth. J. Biomed. Biotechnol. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Haskó, G. Anti-CD73 in Cancer Immunotherapy: Awakening New Opportunities. Trends in Cancer 2016, 2, 95–109. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Fernandes, M.C.; Figueiró, F.; Battastini, A.M.O.; Scholl, J.N.; de Oliveira, F.H.; Spanevello, R.M.; et al. CD73 Downregulation Decreases In Vitro and In Vivo Glioblastoma Growth. Mol. Neurobiol. 2019, 56, 3260–3279. [Google Scholar] [CrossRef]
- Gao, Z.W.; Wang, H.P.; Lin, F.; Wang, X.; Long, M.; Zhang, H.Z.; Dong, K. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer 2017, 17, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Lv, M.; Qiao, B.; Li, X. Blockade pf CD73/adenosine axis improves the therapeutic efficacy of docetaxel in epithelial ovarian cancer. Arch. Gynecol. Obstet. 2019, 299, 1737–1746. [Google Scholar] [CrossRef]
- Yang, Q.; Du, J.; Zu, L. Overexpression of CD73 in prostate cancer is associated with lymph node metastasis. Pathol. Oncol. Res. 2013, 19, 811–814. [Google Scholar] [CrossRef]
- Terp, M.G.; Gammelgaard, O.L.; Vever, H.; Gjerstorff, M.F.; Ditzel, H.J. Sustained compensatory p38 MAPK signaling following treatment with MAPK inhibitors induces the immunosuppressive protein CD73 in cancer: Combined targeting could improve outcomes. Mol. Oncol. 2021, 15, 3299–3316. [Google Scholar] [CrossRef]
- Xie, M.; Qin, H.; Luo, Q.; Huang, Q.; He, X.; Yang, Z.; Lan, P.; Lian, L. MicroRNA-30a regulates cell proliferation and tumor growth of colorectal cancer by targeting CD73. BMC Cancer 2017, 17, 305. [Google Scholar] [CrossRef]
- Sun, P.; Wang, H.; He, Z.; Chen, X.; Wu, Q.; Chen, W.; Sun, Z.; Weng, M.; Zhu, M.; Ma, D.; et al. Fasting inhibits colorectal cancer growth by reducing M2 polarization of tumor-associated macrophages. Oncotarget 2017, 8, 74649–74660. [Google Scholar] [CrossRef]
- Burnstock, G. Historical review: ATP as a neurotransmitter. Trends Pharmacol. Sci. 2006, 27, 166–176. [Google Scholar] [CrossRef] [PubMed]
- García-Alcocer, G.; Padilla, K.; Rodríguez, A.; Miledi, R.; Berumen, L. Distribution of the purinegic receptors P2X(4) and P2X(6) during rat gut development. Neurosci. Lett. 2012, 509, 92–95. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, D.; Valdebenito, S.; Eugenin, E. The role of Pannexin-1 channels and extracellular ATP in the pathogenesis of the human immunodeficiency virus. Purinergic Signal. 2021, 17, 563–576. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2, 239821281881749. [Google Scholar] [CrossRef]
- Yaguchi, T.; Saito, M.; Yasuda, Y.; Kanno, T.; Nakano, T.; Nishizaki, T. Higher concentrations of extracellular ATP suppress proliferation of Caco-2 human colonic cancer cells via an unknown receptor involving PKC inhibition. Cell. Physiol. Biochem. 2010, 26, 125–134. [Google Scholar] [CrossRef]
- Dillard, C.; Borde, C.; Mohammad, A.; Puchois, V.; Jourdren, L.; Larsen, A.K.; Sabbah, M.; Maréchal, V.; Escargueil, A.E.; Pramil, E. Expression Pattern of Purinergic Signaling Components in Colorectal Cancer Cells and Differential Cellular Outcomes Induced by Extracellular ATP and Adenosine. Int. J. Mol. Sci. 2021, 22, 11472. [Google Scholar] [CrossRef]
- Vinette, V.; Placet, M.; Arguin, G.; Gendron, F.-P. Multidrug Resistance-Associated Protein 2 Expression Is Upregulated by Adenosine 5’-Triphosphate in Colorectal Cancer Cells and Enhances Their Survival to Chemotherapeutic Drugs. PLoS ONE 2015, 10, e0136080. [Google Scholar] [CrossRef]
- Kim, C.D.; Kim, S.H.; Jung, S.H.; Kim, J.H. Clinical value of an adenosine triphosphate-based chemotherapy response assay in resectable stage III colorectal cancer. Ann. Surg. Treat. Res. 2019, 97, 93–102. [Google Scholar] [CrossRef]
- Wen, Y.; Chen, X.; Zhu, X.; Gong, Y.; Yuan, G.; Qin, X.; Liu, J. Photothermal-Chemotherapy Integrated Nanoparticles with Tumor Microenvironment Response Enhanced the Induction of Immunogenic Cell Death for Colorectal Cancer Efficient Treatment. ACS Appl. Mater. Interfaces 2019, 11, 43393–43408. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine Receptors: Expression, Function and Regulation. Int. J. Mol. Sci. 2014, 15, 2024. [Google Scholar] [CrossRef]
- Lan, B.; Zhang, J.; Zhang, P.; Zhang, W.; Yang, S.; Lu, D.; Li, W.; Dai, Q. Metformin suppresses CRC growth by inducing apoptosis via ADORA1. Front. Biosci.-Landmark 2017, 22, 248–257. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Shi, L.; Song, H.; Shi, P.; Yang, T.; Fan, R.; Jiang, T.; Song, J. Prognostic impact of adenosine receptor 2 (A2aR) and programmed cell death ligand 1 (PD-L1) expression in colorectal cancer. Biomed. Res. Int. 2019, 2019, 8014627. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Yang, R.; Li, L.; Xu, X.; Liang, S. Purinergic signaling: A potential therapeutic target for depression and chronic pain. Purinergic Signal. 2021, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.F.; Kondo, T.; Nakazawa, T.; Niu, D.F.; Mochizuki, K.; Kawasaki, T.; Yamane, T.; Katoh, R. Hypoxia-inducible adenosine A2B receptor modulates proliferation of colon carcinoma cells. Hum. Pathol. 2010, 41, 1550–1557. [Google Scholar] [CrossRef]
- Long, J.S.; Crighton, D.; O’Prey, J.; MacKay, G.; Zheng, L.; Palmer, T.M.; Gottlieb, E.; Ryan, K.M. Extracellular adenosine sensing-a metabolic cell death priming mechanism downstream of p53. Mol. Cell 2013, 50, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Kitsou, M.; Ayiomamitis, G.D.; Zaravinos, A. High expression of immune checkpoints is associated with the TIL load, mutation rate and patient survival in colorectal cancer. Int. J. Oncol. 2020, 57, 237–248. [Google Scholar] [CrossRef]
- Balber, T.; Singer, J.; Berroterán-Infante, N.; Dumanic, M.; Fetty, L.; Fazekas-Singer, J.; Vraka, C.; Nics, L.; Bergmann, M.; Pallitsch, K.; et al. Preclinical in Vitro and in Vivo Evaluation of [18F]FE@SUPPY for Cancer PET Imaging: Limitations of a Xenograft Model for Colorectal Cancer. Contrast Media Mol. Imaging 2018, 2018, 1269830. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Santinelli, C.; Buccioni, M.; Navia, A.M.; Lambertucci, C.; Zhurina, A.; Yli-Harja, O.; Volpini, R.; Kandhavelu, M. Anticancer activity study of A3 adenosine receptor agonists. Life Sci. 2018, 205, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Gusic, M.; Benndorf, K.; Sattler, C. Dissecting activation steps in P2X7 receptors. Biochem. Biophys. Res. Commun. 2021, 569, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Oury, C.; Wéra, O. P2X1: A unique platelet receptor with a key role in thromboinflammation. Platelets 2021, 32, 902–908. [Google Scholar] [CrossRef]
- Fong, Z.; Griffin, C.; Large, R.; Hollywood, M.; Thornbury, K.; Sergeant, G. Regulation of P2X1 receptors by modulators of the cAMP effectors PKA and EPAC. Proc. Natl. Acad. Sci. USA 2021, 118, e2108094118. [Google Scholar] [CrossRef]
- Grohmann, M.; Schumacher, M.; Günther, J.; Singheiser, S.; Nußbaum, T.; Wildner, F.; Gerevich, Z.; Jabs, R.; Hirnet, D.; Lohr, C.; et al. BAC transgenic mice to study the expression of P2X2 and P2Y 1 receptors. Purinergic Signal. 2021, 17, 449–465. [Google Scholar] [CrossRef]
- Schneider, R.; Leven, P.; Glowka, T.; Kuzmanov, I.; Lysson, M.; Schneiker, B.; Miesen, A.; Baqi, Y.; Spanier, C.; Grants, I.; et al. A novel P2X2-dependent purinergic mechanism of enteric gliosis in intestinal inflammation. EMBO Mol. Med. 2021, 13, e12724. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, Y.; Wu, D.; Hu, J.; Zang, W.; Li, X.; Yang, J.; Gao, T. Involvement of P2X2 receptor in the medial prefrontal cortex in ATP modulation of the passive coping response to behavioral challenge. Genes. Brain. Behav. 2020, 19, e12691. [Google Scholar] [CrossRef]
- King, B. P2X3 receptors participate in purinergic inhibition of gastrointestinal smooth muscle. Auton. Neurosci. 2021, 234, 102830. [Google Scholar] [CrossRef]
- Xia, L.; Luo, H.; Ma, Q.; Xie, Y.; Li, W.; Hu, H.; Xu, Z. GPR151 in nociceptors modulates neuropathic pain via regulating P2X3 function and microglial activation. Brain 2021, 144, 3405–3420. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Metzner, K.; Zhou, F.; Flauaus, C.; Balzulat, A.; Engel, P.; Petersen, J.; Ehinger, R.; Bausch, R.; Ruth, P.; et al. Functional Coupling of Slack Channels and P2X3 Receptors Contributes to Neuropathic Pain Processing. Int. J. Mol. Sci. 2021, 22, 405. [Google Scholar] [CrossRef] [PubMed]
- de Baaij, J.; Blanchard, M.; Lavrijsen, M.; Leipziger, J.; Bindels, R.; Hoenderop, J. P2X4 receptor regulation of transient receptor potential melastatin type 6 (TRPM6) Mg2+ channels. Pflugers Arch. 2014, 466, 1941–1952. [Google Scholar] [CrossRef]
- Padilla, K.; Gonzalez-Mendoza, D.; Berumen, L.; Escobar, J.; Miledi, R.; García-Alcocer, G. Differential gene expression patterns and colocalization of ATP-gated P2X6/P2X4 ion channels during rat small intestine ontogeny. Gene Expr. Patterns 2016, 21, 81–88. [Google Scholar] [CrossRef]
- Yu, Q.; Zhao, Z.; Sun, J.; Guo, W.; Fu, J.; Burnstock, G.; He, C.; Xiang, Z. Expression of P2X6 receptors in the enteric nervous system of the rat gastrointestinal tract. Histochem. Cell Biol. 2010, 133, 177–188. [Google Scholar] [CrossRef]
- Mishra, A.; Behura, A.; Kumar, A.; Naik, L.; Swain, A.; Das, M.; Sarangi, S.; Dokania, P.; Dirisala, V.; Bhutia, S.; et al. P2X7 receptor in multifaceted cellular signalling and its relevance as a potential therapeutic target in different diseases. Eur. J. Pharmacol. 2021, 906, 174235. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F.; De Ponte Conti, B. The P2X7 Receptor in Tumor Immunity. Front. cell Dev. Biol. 2021, 9, 694831. [Google Scholar] [CrossRef]
- Rabelo, I.; Arnaud-Sampaio, V.; Adinolfi, E.; Ulrich, H.; Lameu, C. Cancer Metabostemness and Metabolic Reprogramming via P2X7 Receptor. Cells 2021, 10, 1782. [Google Scholar] [CrossRef]
- Campagno, K.; Lu, W.; Jassim, A.; Albalawi, F.; Cenaj, A.; Tso, H.; Clark, S.; Sripinun, P.; Gómez, N.; Mitchell, C. Rapid morphologic changes to microglial cells and upregulation of mixed microglial activation state markers induced by P2X7 receptor stimulation and increased intraocular pressure. J. Neuroinflammation 2021, 18, 217. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk-Węgrzynowicz, M.; Strużyńska, L. Astroglial and Microglial Purinergic P2X7 Receptor as a Major Contributor to Neuroinflammation during the Course of Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 8404. [Google Scholar] [CrossRef] [PubMed]
- Purohit, R.; Bera, A. Pannexin 1 plays a pro-survival role by attenuating P2X7 receptor-mediated Ca 2+ influx. Cell Calcium 2021, 99, 102216. [Google Scholar] [CrossRef]
- Hirayama, Y.; Anzai, N.; Koizumi, S. Mechanisms underlying sensitization of P2X7 receptors in astrocytes for induction of ischemic tolerance. Glia 2021, 69, 2100–2110. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Grassi, F. Modulation of innate and adaptive immunity by P2X ion channels. Curr. Opin. Immunol. 2018, 52, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. P2X receptors and inflammation. Curr. Med. Chem. 2015, 22, 5–24. [Google Scholar] [CrossRef]
- Qian, F.; Xiao, J.; Hu, B.; Sun, N.; Yin, W.; Zhu, J. High expression of P2X7R is an independent postoperative indicator of poor prognosis in colorectal cancer. Hum. Pathol. 2017, 64, 61–68. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, J.; Wang, L. The role of P2X7 receptor in prognosis and metastasis of colorectal cancer. Adv. Med. Sci. 2019, 64, 388–394. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, M.; Peng, R.; Liu, J.; Wang, F.; Li, Y.; Zhao, Q.; Liu, J. The prognostic and clinicopathological value of tumor-associated macrophages in patients with colorectal cancer: A systematic review and meta-analysis. Int. J. Colorectal Dis. 2020, 35, 1651–1661. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Wang, L.; Lou, Y. A438079 affects colorectal cancer cell proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. Biochem. Biophys. Res. Commun. 2021, 558, 147–153. [Google Scholar] [CrossRef]
- Bernardazzi, C.; Castelo-Branco, M.T.L.; Pêgo, B.; Ribeiro, B.E.; Rosas, S.L.B.; Santana, P.T.; Machado, J.C.; Leal, C.; Thompson, F.; Coutinho-Silva, R.; et al. The P2X7 Receptor Promotes Colorectal Inflammation and Tumorigenesis by Modulating Gut Microbiota and the Inflammasome. Int. J. Mol. Sci. 2022, 23, 4616. [Google Scholar] [CrossRef]
- Hofman, P.; Cherfils-Vicini, J.; Bazin, M.; Ilie, M.; Juhel, T.; Hébuterne, X.; Gilson, E.; Schmid-Alliana, A.; Boyer, O.; Adriouch, S.; et al. Genetic and pharmacological inactivation of the purinergic P2RX7 receptor dampens inflammation but increases tumor incidence in a mouse model of colitis-associated cancer. Cancer Res. 2015, 75, 835–845. [Google Scholar] [CrossRef]
- Gao, P.; He, M.; Zhang, C.; Geng, C. Integrated analysis of gene expression signatures associated with colon cancer from three datasets. Gene 2018, 654, 95–102. [Google Scholar] [CrossRef]
- von Kügelgen, I. Molecular pharmacology of P2Y receptor subtypes. Biochem. Pharmacol. 2021, 187, 114361. [Google Scholar] [CrossRef]
- Limami, Y.; Pinon, A.; Leger, D.Y.; Pinault, E.; Delage, C.; Beneytout, J.L.; Simon, A.; Liagre, B. The P2Y 2/Src/p38/COX-2 pathway is involved in the resistance to ursolic acid-induced apoptosis in colorectal and prostate cancer cells. Biochimie 2012, 94, 1754–1763. [Google Scholar] [CrossRef]
- Girard, M.; Dagenais Bellefeuille, S.; Eiselt, É.; Brouillette, R.; Placet, M.; Arguin, G.; Longpré, J.M.; Sarret, P.; Gendron, F.P. The P2Y6 receptor signals through Gαq/Ca2+/PKCα and Gα13/ROCK pathways to drive the formation of membrane protrusions and dictate cell migration. J. Cell. Physiol. 2020, 235, 9676–9690. [Google Scholar] [CrossRef]
- Placet, M.; Arguin, G.; Molle, C.M.; Babeu, J.P.; Jones, C.; Carrier, J.C.; Robaye, B.; Geha, S.; Boudreau, F.; Gendron, F.P. The G protein-coupled P2Y6 receptor promotes colorectal cancer tumorigenesis by inhibiting apoptosis. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1539–1551. [Google Scholar] [CrossRef]
- Husted, S.; van Giezen, J.; Steen Husted, C. Ticagrelor: The First Reversibly Binding Oral P2Y 12 Receptor Antagonist. Cardiovasc. Ther. 2009, 27, 259–274. [Google Scholar] [CrossRef]
- Wright, J.R.; Chauhan, M.; Shah, C.; Ring, A.; Thomas, A.L.; Goodall, A.H.; Adlam, D. The TICONC (Ticagrelor-Oncology) Study: Implications of P2Y12 Inhibition for Metastasis and Cancer-Associated Thrombosis. JACC CardioOncol. 2020, 2, 236–250. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
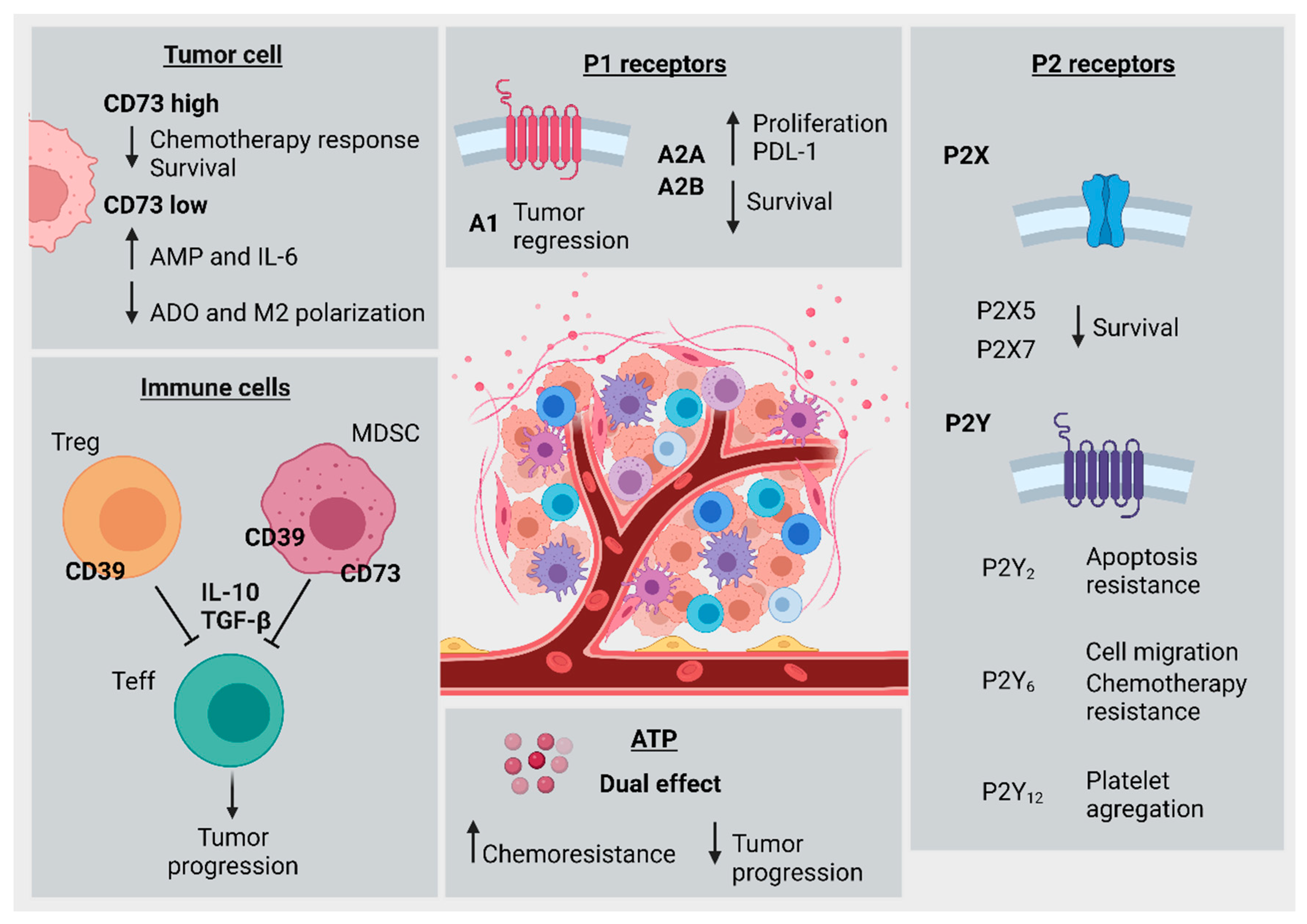{kind=link}
| Reference | Type of Study | Murine or Cell Model | Target | Results |
|---|---|---|---|---|
| Wen et al., 2019 | In vitro/In vivo | CT29/BALB/c | ATP | Increase of ATP levels in the microenvironment |
| Yagushi et al., 2010 | In vitro | Caco-2 | ATP | ATP-mediated PKC inhibition via P2R sensitization |
| Vinette et al., 2015 | In vitro | Caco-2 | ATP | ATP-mediated increase of MRP2 expression via P2R sensitization |
| Kim et al., 2019 | Human tumor (N = 136) | CRC (stage III) | ATP | ATP sensitivity of patients is directly correlated with better response to chemotherapy |
| Dillard et al., 2021 | In vitro | HT29, HCT116, LS513 and LS174T | ATP and ADO | ATP induces cell death in CRC cell lines |
| Kunzli et al., 2011 | In vivo | CD39 transgenic, CD39+/−, and CD39 wild-type mice | CD39 | Increased CD39 expression in endothelium, stromal and mononuclear infiltrating tumor cells High P2Y2 expression in metastatic liver tumors |
| Tumor (N = 63)/adjacent tissue (N = 13) | CRC stage | Lower expression of CD39 and P2Y2 in tumor tissue at initial stages of CRC compared to metastatic tumors | ||
| McCarthy et al., 2013 | In vivo/In situ | HCT116 and HCT15 | CD39L4 | Lower levels of ATP in transfected mt-PCPH and myc-tagged PCPH cells. However, the NTPDase activity of mt-PCPH was undetectable. |
| Parodi et al., 2013 | Tumor (N = 2 CRC/blood donor (13) | Renal, Bladder, CRC stage (III) | CD39 | High CD39 expression in circulating cells from patientsHigh CD39 expression in intratumoral CD8+ Tregs |
| Zhang et al., 2013 | Blood (N = 64)/tumor (N = 5)/in vitro | CRC (I, II, III, IV) cells from blood | CD39 | Increased MDSCs correlates with tumor metastasis and increased stage MDSCs CD39high inhibits CD3+ T cell proliferation |
| Scurr et al., 2014 | Tumor/ adjacent tissue, blood/blood healthy donor (N = 40) | CRC stage I, II (majority), III | CD39 | Increased expression of CD39 in both FOXP3+ and FOXP3− tumor-associated Tregs when compared to circulating Tregs from healthy donors or CRC patients |
| Sundstrom, 2016 | Blood (N = 45) and tumor (N = 7) | - | CD39 | Higher expression of CD39 in circulating and tumor-associated Tregs when compared to healthy donors Adenosine reduces T cell migration by impairing the capacity of monocytes to activate the endothelium |
| Limagne et al., 2016 | Blood mCRC (N = 25) and blood healthy donor (N = 20) | CRC stage IV | CD39 | PD-L1high CD39high CD73high gMDSC levels are associated with poor prognosis FOLFOX plus bevacizumab decreased gMDSC levels |
| Timperi et al., 2016 | Tumor/adjacent tissue, blood (N = 34) | CD39 | Presence of CD39high Tregs with production of IL-17 and IL-1β increased in tumor site The ENPD1 SNP rs10748643 contributes to CD39 expression | |
| Hu et al., 2017 | in vitro | SW480 | CD39 | A2AR and A2BR antagonists blocked the activity of γδTregs CD39+ γδTreg inhibited CD3+ T cell proliferation |
| Tumor/ adjacent tissue (N = 109), blood donors | - | Tumor-associated CD39high γδTreg is correlated to high CTLA-4, PD-1, FOXP3, IL-10, IL-17A, GM-CSF, TGF-β1, and TNFα production. TGF-β1 induced γδTreg to produce more adenosine | ||
| Khaja et al., 2017 | Tumor/ adjacent tissue (N = 12) | CRC stages (I,II,III and IV) | CD39 | CD4+FOXP3+ T cells demonstrated high co-expression of PD-1/CTLA-4 and PD-1/CD39; CD39 was overexpressed in tumors |
| Zhulai et al., 2018 | Blood (N = 42)/blood healthy donors (N = 30)/tumor (N = 5) stage III | Initial (I and II) and advanced (III and IV) staged CRC | CD39 | Advanced stage CRC demonstrated increased CD4+CD39+ lymphocytes in blood and tumor tissue, as well as a negative correlation with CD3+CD4+ T helper cells and CD3−CD19+ B cells. Association between FOXP3 and CD39 in CD4+CD25high T cells. |
| Simoni et al., 2018 | Tumor/ adjacent tissue (N = 94) | Stage I, II, III and IV | CD39 | Lower CD39 expression in CD8+ bystander TILs than tumor-specific CD8+ TILs. CD39+ was correlated with genes associated with T cell proliferation and exhaustion. |
| Strasser et al., 2019 | Tumor/ adjacent tissue (N = 29), gene data (N = 298) | - | CD39 | Higher levels of CD39+Helios+ T cells and pro-inflammatory IFNγ -producing T cells in CRC tissue |
| Gaibar et al., 2021 | Tumor mCRC (N = 57, paraffin) | Stage IV | CD39 | Variant allele CD39 patients demonstrated better response to bevacizumab plus chemotherapy, but no changes to OS or PSF |
| Gallerano et al., 2020 | Tumor/ adjacent tissue, blood (N = 60) | Stage I, II, III and IV | CD39 | CD8+CD39high T lymphocytes were expressed at higher levels within the tumor at initial stage of CRC (I-II), with high PD-1 expression and lower INF-y production. These were correlated with exhausted T cells and suppressed CD4+ T cell proliferation. |
| Park et al., 2021 | in vitro/in vivo | Balb/c subcutaneously injected in flank with CT26 cells. Intraperitoneally injected POM-1 daily for 2 weeks. | CD39 | CD39 inhibitor increased CD11b and Ly6C expression in M1 TAMs and F4/80+ macrophages in vitro. CD39 inhibitor resulted in smaller tumors, increased Ly6C and MHC II in F4/80+ macrophages, increased CD8+ T cells in the spleen, increased CD4+ T cells in the blood, and increased Caspase-3 expression, compared with the saline treatment (control group) in vivo. |
| Rodin et al., 2021 | Tumor/ adjacent tissue (N = 28 male, N = 19 female) 10 cm away from tumor | Stage I (4), II (17), III (26), IV (1) | CD39 | CRC infiltrating MAITs (mucosal-associated invariant T cells) have a terminally exhausted phenotype (PD-1highTim-3+CD39+). MAIT cells have reduced polyfunctionality with decreased production of antitumor effector molecules, and blocking PD-1 improved activation of tumor-infiltrating MAIT cells in vitro. |
| Zhao et al., 2020 | In vitro | MC38 and HT29 | CD39 | Expression level of CD39 in colorectal tumor tissues was higher than in normal tissues. CD39 was also highly expressed in both human and murine colorectal cancer cell lines MC38 and HT29. CD39 inhibitor decreased MC38 cell growth at 48 and 72 h. CD39 inhibitor reduced cell proliferation in a dose-dependent manner. |
| Zhan et al., 2021 | Tumor/ adjacent tissue (N = 129)/ In vivo/In vitro | Stage I, II, III and IV. In vitro: CT26. In vivo: CT26-Vec/Pla2g4a cells transplanted into the caecal wall of BALB/c. | CD39 | Left-sided CRC had lower frequency of CD39+γδ Tregs than right-sided CRC. Right sided CRC had increase adenosine level, increased IL-17A production, and decreased IFN-γ–production. |
| Bonnereau et al., 2022 | Tumor/adjacent tissue (N = 44) | Right colon (20), left colon (16), rectum (8). Autologous coculture | CD39/CD73 | CD4+/CD8+ T cells in tumors demonstrated increased CD39 expression and decreased CD73 expression in early stage tumors. Conversely, advanced stage tumors demonstrated decreased CD39 expression and increased CD73 expression in T cells. CD39 blockade increased T cell capacity of infiltration tumor spheroid destruction in cocultures |
| Matsuyama et al., 2010 | In vitro | SW48 and SW48LM2 | CD73 | Reduced CD73 expression in highly liver-metastatic cell line |
| Wu et al., 2012 | Tumor (N = 16 fresh, N = 358 paraffin) | CRC: stage I (N = 54), stage II (N = 147), stage III (N = 124), stage IV (N = 30) | CD73 | High CD73 expression in fresh or paraffin CRC tissue |
| Cushman et al., 2014 | Tumor (N = 103) | mCRC and respective primary tumor | CD73 | Higher levels of CD73 expression were predictive of improved PFS following cetuximab treatment |
| Zhang et al., 2015 | Tumor/adjacent tissue (N = 90) | CRC: stage I (N = 11), stage II (N = 38), stage III (N = 40), stage IV (N = 1) | CD73 | Higher expression of CD73 in both tumor and stromal tissue compared to peritumoral tissue. |
| Wu et al., 2016 | In vitro | RKO, SW480, HCT-15, LoVo and KM12 | CD73 | CD73 expressed in five CRC cell lines; overexpression of CD73 promoted β-catenin/cyclin D1 and EGFR expression. |
| In vivo | CRC human with/without CD73 interference | CD73 increased tumor size and weight | ||
| Hatch et al., 2016 | Blood (N = 152)/tumor (N = 71) | - | CD73 | High plasma levels of CD73 were predictive of shorter OS in all patients. However, high CD73 was correlated with PFS benefit in the KRAS-WT group treated with cetuximab |
| Xie et al., 2017 | In vitro | HEK293T cells, SW480, HCT116, LoVo, CaCo2, HT29, RKO, DLD1, HCT8 | CD73 | miR-30a has a negative effect in regulating expression of CD73 mRNA and protein levels, leading to decreased proliferation and increased apoptosis of cancer cells. |
| In vivo | BALB/c nu/nu mice with injection of SW480 in dorsal skin | Decreased in the mean weight of miR-30a-treated group. | ||
| Tumor/adjacent tissue (N = 27) | - | Lower expression levels of miR-30a and higher expression levels of CD73 within CRC than the corresponding adjacent control tissues. | ||
| Sun et al., 2017 | In vitro | CT26, RAW 264.7 | CD73 | CD73 knockdown and ADO receptor antagonists correlated with decreased M2 polarization and decreased tumor cell proliferation |
| In vivo | WT BALB/c mice | Mice under dietary restriction demonstrated reduced tumor growth without body weight reduction, along with reduced M2 macrophage polarization | ||
| Wang et al., 2019 | Blood (N = 232)/Healthy blood donors (N = 158) | Stage I/II (N = 110), stage III/IV (N = 122) | CD73 | Higher CD73 expression in CRC patients compared with healthy donor. Correlation between CD73 expression and several worse clinicopathological features. Shorter OS patients with higher CD73 expression. |
| Liu et al., 2020 | In vivo | Colitis-associated tumorigenesis | CD73 | CD73 inhibitor led to decreased loss of body weight, decreased number of tumors, longer colon, lower histopathological score, and downregulated expression of CRC tumorigenesis-associated genes. ADO agonist (NECA) demonstrated the opposite effects, and increased TNF-α and IL-6 production. |
| Yu et al., 2020 | In vivo | CD73 null/A2Bnull | CD73 | CD73 promoted tumor progression, along with suppression of antitumor immunity. ADO released during cell death binds to A2B, leading to increased CD73, and binds to A2A, leading to immune suppression. |
| In vitro | EG7.OVA and MC38 | |||
| Tumor (N = 25) | - | |||
| Messaoudi et al., 2020 | Microarray CRCm (N = 251), blood (N = 193) | CRC stage IV | CD73 | CD73high tumors were associated with more aggressive CRC metastasis to liver, poorer response to preoperative chemotherapy presence to mutation in KRAS, shorter time to recurrence, and reduced disease-specific survival. |
| Kim et al., 2021 | In vitro/In vivo | CT26 implantation in BALB/C; colitis-associated cancer model mouse by injection of azoxymethane and Dextran Sulfate Sodium | CD73 | Nt5e and Entpd1 expression affects TCR diversity and transcriptional profiles of T cells; CD73 inhibitor (AB680) improved the anticancer functions of immunosuppressed cells, including Treg and exhausted T cells, and caused increased activation of CD8+ T cells. |
| Lai et al., 2021 | In vitro/In vivo Tumor and blood | Male C57BL/6 and P14 TCR transgenic, CD28−/−, P14CD28−/− and CD73−/− mice | CD73 | CD28−/− mice increase CD73 expression in CD8+ T cells, without differences in CD39 expression, and with increased adenosine level in culture supernatant. CRC tumor and PBL demonstrated CD73 upregulation in Cd28−/−/CD8+ T cells. There was reduced cytolytic activity of CD8+ T cells following treatment with supernatant from CD28−/− cells |
| Ploeg et al., 2021 | In vitro | H292, OvCAR3, DLD1, PC-3M and CHO–K1 | CD73 | Extracellular vesicles derived from cancer cells lines and patients are enriched in CD73. CD73 inhibition in extracellular vesicles leads to reactivate proliferative and cytotoxic capacity of T cells. |
| Terp et al. 2021 | In vitro/human samples (dataset) | HCT116, SKBr3, CT26.CL25 (CT26), A549, PC9, MC38 and 4T1.2 (4T1) | CD73 | CD73 expression was significantly higher in tumors of nonresponders vs. responders to anti-EGFR treatment. Decreased PFS in patients with CD73high vs. CD73low tumors. |
| Lan et al., 2017 | In vitro | HCT116 and SW480 | A1 | Metformin induced increased A1 expression, suppressed proliferation, and induced apoptosis in both CRC cells in an AMPK-mTOR pathway dependent manner. |
| Wu et al., 2019 | Tumor/adjacent tissue (N = 204) | CRC stage I/II (N = 106) and stage III/IV (N = 98) | A2A | Higher A2A expression in tumor than non-tumor tissue was correlated with tumor size, depth of tumor invasion, and increased TNM stage and PD-L1 expression. |
| Kitsou et al., 2020 | In silico (N = 453) | RNA seq and clinicopathological data | A2A | A2A demonstrated lower expression in CRC compared to normal tissue and was not correlated to OS. In colorectal adenocarcinoma, TIL load was positively correlated to A2A expression. |
| Ma et al., 2010 | In vitro | DLD1, SW480, HCT-15, LOVO, COLO205 | A2B | Higher A2B expression than A1, A2A, and A3 in tissue samples and in cell lines was increased in hypoxic conditions. Inhibition of A2B decreased cell growth. |
| Tumor (N = 88)/adjacent tissue (N = 62) | - | |||
| Long et al., 2013 | In vitro | Saos-2, Phoenix Eco, U2OS, HCT116 | A2B | A2B is upregulated directly by p53, which is activated by cellular stress and can induce cell death by apoptosis. This was demonstrated in hypoxic conditions and during response to chemotherapy. |
| Molck et al., 2016 | In vitro | DLD1, SW480, CPP14, HEK293T | A2B | A2B antagonist increased mitochondrial oxygen consumption and intracellular ROS levels |
| Balber et al., 2017 | In vitro | HT-29 and CHO-K1 | A3 | High A3 expression in HT-29 cells |
| In vivo | Immunodeficient CB17-SCID | No difference between the CHO-K1 and HT-29 cells xenografts. | ||
| Tumor/adjacent tissue (N = 2) | - | [18F]FE@SUPPY accumulation was higher in CRC than in healthy tissue and corresponded to higher expression of A3 | ||
| Marucci et al., 2018 | In vitro | Caco-2, PC3, HepG2, CHO | A3 | A3 agonist inhibited Caco-2 cell growth and migration, promoted apoptosis and increased ROS levels. However, A3 knockdown did not prevent agonist effects. |
| P2X | ||||
| Gao et al., 2018 | In silico (N = 206) | - | P2X5 | P2X5 expression was correlated with worse prognosis and expression levels were higher in the high-risk group. |
| Janakiram et al., 2015 | In vivo | Rag1−/− ApcMin/+ | P2X7 | Increased expression of P2X7R upon Treg transfer and NK cell depletion led to increased tumor cell proliferation, and increased intestinal tumor formation and growth. |
| Hofman et al., 2015 | In vivo | Disrupted P2X7 gene | P2X7 | P2X7blockade stimulated Treg accumulation, reduced colonic inflammation and increased tumor proliferation. This was associated with elevated expression of TGFB1. |
| Quian et al., 2017 | Tumor (N = 12 fresh/N = 116 paraffin) | Stage I (N = 31), stage II (N = 36), stage III (N = 44) and stage IV (N = 5) | P2X7 | P2X7 was increased in tumor tissue and correlated higher TNM stage. |
| Zhang et al., 2019 | In vitro | NCM460, HCT116, SW480, SW620 | P2X7 | Higher expression of P2X7was demonstrated in CRC cell lines, even higher in mCRC cell line, compared to cell lines derived from normal colon cell. |
| Tumor/adjacent tissue (N = 97) | Stage I (N = 16), stage II (N = 30), stage III (N = 41) and stage IV (N = 10) | Higher expression of P2X7in 56 patients with CRC versus no change in 41 patients, compared to adjacent normal column tissues. Higher expression of P2X7in tumor tissue was associated with more advanced disease and shorter survival. P2X7expression was higher in mCRC compared to primary colorectal tissues. | ||
| Zhang et al., 2021 | In vitro/In vivo | HT116, SW620 | P2X7 | P2X7inhibitor (A438079) inhibits CRC cells line proliferation, invasion, and migration and promotes apoptosis by Bcl-2/caspase9/caspase3 pathway. In vivo, P2X7inhibitor inhibits tumor growth. |
| Yang et al., 2020 | In vitro/In vivo | Injected 10 μL of 2 × 106 CT26-Con or CT26-mP2X7R cells into the subserosa of the caecum | P2X7 | P2X7R promoted proliferation, migrated, invasion, and increased the number of tumorspheres of CRC cells in vitro. P2X7overexpression increased the growth and weight of tumors, infiltration of macrophages, TAM recruitment, and stimulation of angiogenesis in vivo. |
| Bernardazzi et al., 2022 | In vivo | P2X7+/+ and P2X7−/− mice AOM/DSS-treated | P2X7 | P2X7+/+ mice demonstrated increased TNF-alpha, IL-17A, and IL-6 following AOM/DSS treatment compared with P2X7−/− mice. The P2X7antagonist (A740003) increased survival and decreased symptoms of tumorigenesis, including body weight loss, shortened colon length, and number of polyps/tumors. Overall, A740003 prevented tumor development in the P2X7+/+ group. No tumor formation was observed in the P2X7−/− group. Dissimilarity in the fecal microbiota was observed between the A740003-treated and untreated AOM/DSS-induced P2X7R+/+ mice, and between the P2X7R+/+ control mice and P2X7−/− control mice. |
| P2Y | ||||
| Limami et al., 2012 | In vitro | HT-29 | P2Y2 | Ursolic acid induced an increase in intracellular ATP and in P2Y2 mRNA. p38 activation was dependent on P2Y2 activation. |
| Placet et al., 2018 | In vitro | HT-29 | P2Y6 | P2Y6R agonist prevents apoptosis. Stimulation of P2Y6R prior to 5-FU treatment provides protection. |
| In vivo | P2Y6−/− or P2Y6+/+ mice | Decreased number and volume of CRC tumors in P2ry6−/− mice P2Y6R increased expression of XIAP and correlated with AKT phosphorylation and resistance to 5-FU | ||
| Girard et al., 2020 | In vitro | Caco-2 | P2Y6 | P2Y6 increased cell migration, through PKCα that stabilizes the actin cytoskeleton |
| Wright et al., 2020 | In vitro | HT-29 | P2Y12 | Reduced cell aggregation and adhesion |
| Oncological patients blood (N = 6)/Healthy donors blood (N = 22) | mCRC | Higher levels of spontaneous platelet aggregation and P-selectin expression in mCRC tissues |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roliano, G.G.; Azambuja, J.H.; Brunetto, V.T.; Butterfield, H.E.; Kalil, A.N.; Braganhol, E. Colorectal Cancer and Purinergic Signalling: An Overview. Cancers 2022, 14, 4887. https://doi.org/10.3390/cancers14194887
Roliano GG, Azambuja JH, Brunetto VT, Butterfield HE, Kalil AN, Braganhol E. Colorectal Cancer and Purinergic Signalling: An Overview. Cancers. 2022; 14(19):4887. https://doi.org/10.3390/cancers14194887
Chicago/Turabian StyleRoliano, Gabriela Gonçalves, Juliana Hofstätter Azambuja, Veronica Toniazzo Brunetto, Hannah Elizabeth Butterfield, Antonio Nochi Kalil, and Elizandra Braganhol. 2022. "Colorectal Cancer and Purinergic Signalling: An Overview" Cancers 14, no. 19: 4887. https://doi.org/10.3390/cancers14194887
APA StyleRoliano, G. G., Azambuja, J. H., Brunetto, V. T., Butterfield, H. E., Kalil, A. N., & Braganhol, E. (2022). Colorectal Cancer and Purinergic Signalling: An Overview. Cancers, 14(19), 4887. https://doi.org/10.3390/cancers14194887