
Bacterial Involvement in Progression and Metastasis of Adenocarcinoma of the Stomach

 ,
,  and
and
Simple Summary
Abstract
1. Introduction
2. Colorectal Neoplasia and Metastasis
3. Metastasis Sites of Gastric Neoplasia
3.1. Krukenberg Tumors and Diffuse-Type Gastric Adenocarcinoma
3.2. Epithelial-Mesenchymal Transition in Gastric Adenocarcinoma Metastasis
4. Tumor Microenvironments
5. Bacterial Involvement in Gastric Carcinogenesis
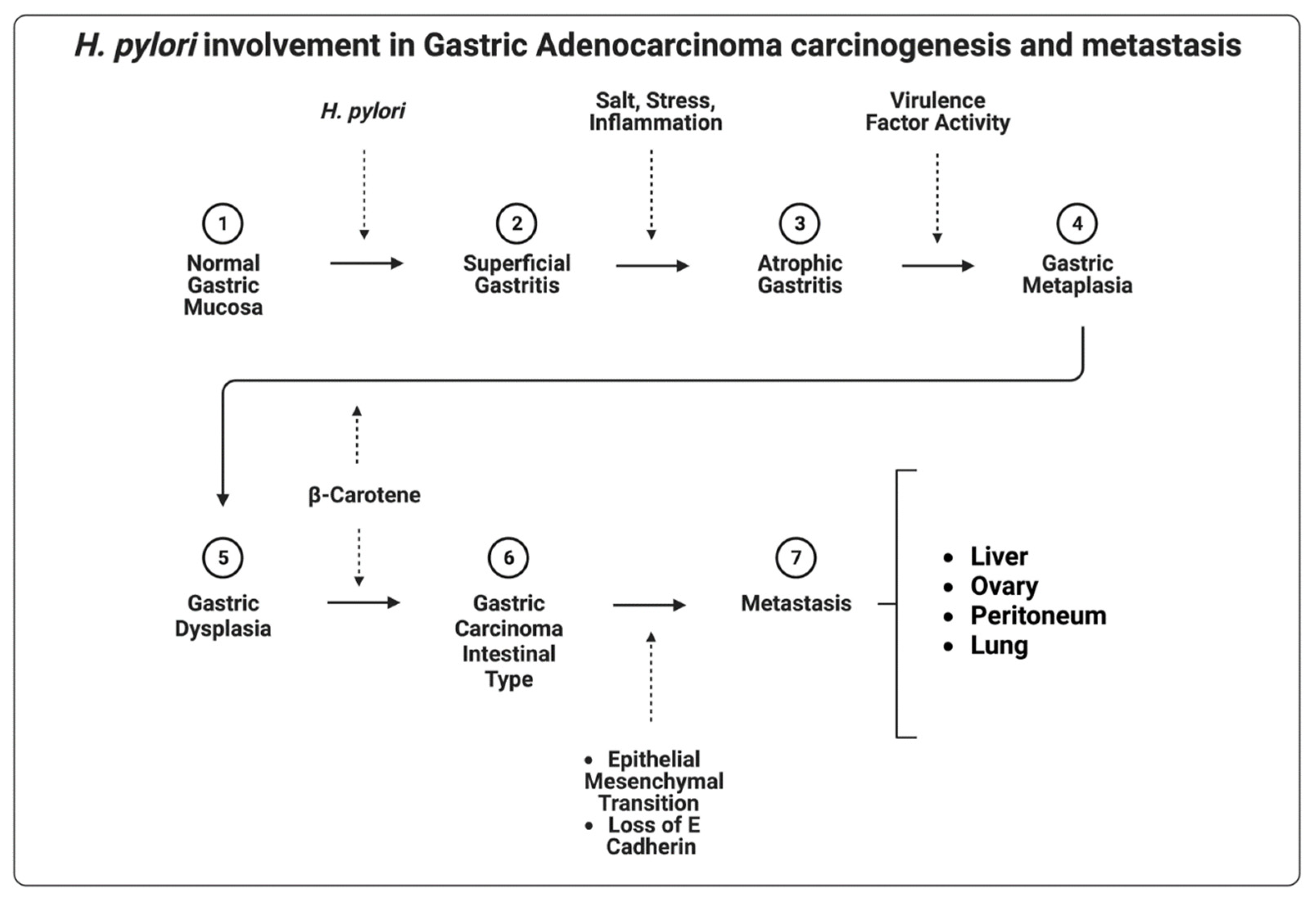
5.1. Helicobacter pylori
5.2. Mycoplasma hyorhinis
5.3. Fusobacterium nucleatum
6. Considering the Microbiome in Gastrointestinal Cancer Treatment
Microbiome after Gastrectomy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seely, K.D.; Morgan, A.D.; Hagenstein, L.D.; Florey, G.M.; Small, J.M. Bacterial Involvement in Progression and Metastasis of Colorectal Neoplasia. Cancers 2022, 14, 1019. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The Current and Future Incidence and Mortality of Gastric Cancer in 185 Countries, 2020–40: A Population-Based Modelling Study. eClinicalMedicine 2022, 47, 101404. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massagué, J. Targeting Metastatic Cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mukaisho, K.-I.; Nakayama, T.; Hagiwara, T.; Hattori, T.; Sugihara, H. Two Distinct Etiologies of Gastric Cardia Adenocarcinoma: Interactions among PH, Helicobacter Pylori, and Bile Acids. Front. Microbiol. 2015, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Turner, E.S.; Turner, J.R. Expanding the Lauren Classification: A New Gastric Cancer Subtype? Gastroenterology 2013, 145, 505–508. [Google Scholar] [CrossRef]
- Compton, C.; Sobin, L.H. Protocol for the Examination of Specimens Removed from Patients with Gastric Carcinoma: A Basis for Checklists. Members of the Cancer Committee, College of American Pathologists, and the Task Force for Protocols on the Examination of Specimens From Patients With Gastric Cancer. Arch. Pathol. Lab. Med. 1998, 122, 9–14. [Google Scholar]
- Budczies, J.; von Winterfeld, M.; Klauschen, F.; Bockmayr, M.; Lennerz, J.K.; Denkert, C.; Wolf, T.; Warth, A.; Dietel, M.; Anagnostopoulos, I.; et al. The Landscape of Metastatic Progression Patterns across Major Human Cancers. Oncotarget 2015, 6, 570–583. [Google Scholar] [CrossRef]
- Riihimäki, M.; Hemminki, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. Metastatic Spread in Patients with Gastric Cancer. Oncotarget 2016, 7, 52307–52316. [Google Scholar] [CrossRef]
- Shi, Z.; Ding, H.; Shen, Q.W.; Lu, X.G.; Chen, J.Y.; Chen, X.; Tang, X. The Clinical Manifestation, Survival Outcome and Predictive Prognostic Factors of 137 Patients with Primary Gastrointestinal Lymphoma (PGIL): Strobe Compliant. Medicine 2018, 97, e9583. [Google Scholar] [CrossRef]
- Patnaik, S.; Jyotsnarani, Y.; Rammurti, S. Radiological Features of Metastatic Gastrointestinal Stromal Tumors. J. Clin. Imaging Sci. 2012, 2, 43. [Google Scholar] [CrossRef]
- Riihimäki, M.; Hemminki, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. The Epidemiology of Metastases in Neuroendocrine Tumors. Int. J. Cancer 2016, 139, 2679–2686. [Google Scholar] [CrossRef]
- Hansen, S.; Melby, K.K.; Aase, S.; Jellum, E.; Vollset, S.E. Helicobacter Pylori Infection and Risk of Cardia Cancer and Non-Cardia Gastric Cancer. A Nested Case-Control Study. Scand. J. Gastroenterol. 1999, 34, 353–360. [Google Scholar] [CrossRef]
- Li, Y.; Gong, J.; Zhang, Q.; Lu, Z.; Gao, J.; Li, Y.; Cao, Y.; Shen, L. Dynamic Monitoring of Circulating Tumour Cells to Evaluate Therapeutic Efficacy in Advanced Gastric Cancer. Br. J. Cancer 2016, 114, 138–145. [Google Scholar] [CrossRef]
- Necula, L.; Matei, L.; Dragu, D.; Neagu, A.I.; Mambet, C.; Nedeianu, S.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Recent Advances in Gastric Cancer Early Diagnosis. World J. Gastroenterol. 2019, 25, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Hamakawa, T.; Kukita, Y.; Kurokawa, Y.; Miyazaki, Y.; Takahashi, T.; Yamasaki, M.; Miyata, H.; Nakajima, K.; Taniguchi, K.; Takiguchi, S.; et al. Monitoring Gastric Cancer Progression with Circulating Tumour DNA. Br. J. Cancer 2015, 112, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, Z.; Chong, X.; Chen, Y.; Wang, Z.; Yu, R.; Sun, T.; Chen, X.; Shao, Y.; Zhang, X.; et al. Clinical Implications of Plasma CtDNA Features and Dynamics in Gastric Cancer Treated with HER2-targeted Therapies. Clin. Transl. Med. 2020, 10, e254. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.-L.; Lan, Y.-T.; Huang, K.-H.; Liu, C.-A.; Hung, Y.-P.; Lin, C.-H.; Jhang, F.-Y.; Chang, S.-C.; Chen, M.-H.; Chao, Y.; et al. Clinical Significance of Circulating Plasma DNA in Gastric Cancer: Circulating Plasma DNA in Gastric Cancer. Int. J. Cancer 2016, 138, 2974–2983. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, R.; DE Luca, M.; Raffone, A.; Travaglino, A.; Coppellotti, A.; Peltrini, R.; Bracale, U.; D’Ambra, M.; Insabato, L.; Zullo, F.; et al. Clinics and Pathology of Krukenberg Tumor: A Systematic Review and Meta-Analysis. Minerva Obstet. Gynecol. 2022, 74, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Al-Agha, O.M.; Nicastri, A.D. An In-Depth Look at Krukenberg Tumor: An Overview. Arch. Pathol. Lab. Med. 2006, 130, 1725–1730. [Google Scholar] [CrossRef]
- Seow-En, I.; Hwarng, G.; Tan, G.H.C.; Ho, L.M.L.; Teo, M.C.C. Palliative Surgery for Krukenberg Tumors—12-Year Experience and Review of the Literature. World J. Clin. Oncol. 2018, 9, 13–19. [Google Scholar] [CrossRef]
- Aziz, M.; Killeen, R.B.; Kasi, A. Krukenberg Tumor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Agnes, A.; Biondi, A.; Ricci, R.; Gallotta, V.; D’Ugo, D.; Persiani, R. Krukenberg Tumors: Seed, Route and Soil. Surg. Oncol. 2017, 26, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Sun, K.; Zou, Y. Comparison of a Panel of Biomarkers Between Gastric Primary Cancer and the Paired Krukenberg Tumor. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Belardi, B.; Son, S.; Felce, J.H.; Dustin, M.L.; Fletcher, D.A. Cell-Cell Interfaces as Specialized Compartments Directing Cell Function. Nat. Rev. Mol. Cell Biol. 2020, 21, 750–764. [Google Scholar] [CrossRef]
- Zhao, H.; Hu, H.; Chen, B.; Xu, W.; Zhao, J.; Huang, C.; Xing, Y.; Lv, H.; Nie, C.; Wang, J.; et al. Overview on the Role of E-Cadherin in Gastric Cancer: Dysregulation and Clinical Implications. Front. Mol. Biosci. 2021, 8, 689139. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N. Tumor Suppressor Gene E-Cadherin and Its Role in Normal and Malignant Cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed. Biotechnol. 2011, 2011, e567305. [Google Scholar] [CrossRef]
- Nguyen, V.H.L.; Hough, R.; Bernaudo, S.; Peng, C. Wnt/β-Catenin Signalling in Ovarian Cancer: Insights into Its Hyperactivation and Function in Tumorigenesis. J. Ovarian Res. 2019, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aziz, S.G.-G.; Jaghi, N.Z.Z. EMT, Cancer Stem Cells and Autophagy; The Three Main Axes of Metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of Partial EMT in Cancer Progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of Epithelial-Mesenchymal Transition through Epigenetic and Post-Translational Modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.T.; Yang, J. Epithelial–Mesenchymal Transition in Tumor Metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.A.; Shkoda, A.; Kim, S.C.; Sartor, R.B.; Haller, D. IL-10 Gene-Deficient Mice Lack TGF-β/Smad Signaling and Fail to Inhibit Proinflammatory Gene Expression in Intestinal Epithelial Cells after the Colonization with Colitogenic Enterococcus Faecalis. J. Immunol. 2005, 174, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic Inflammation in Cancer Development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Andreu, P.; Coussens, L.M. Interactions between Lymphocytes and Myeloid Cells Regulate Pro-versus Anti-Tumor Immunity. Cancer Metastasis Rev. 2010, 29, 309–316. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Qian, B.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Feng, Z.; Li, X.; Ren, Z.; Feng, J.; He, X.; You, C. Prognostic and Predictive Value of Cadherin 11 for Patients with Gastric Cancer and Its Correlation with Tumor Microenvironment: Results from Microarray Analysis. BioMed Res. Int. 2020, 2020, 8107478. [Google Scholar] [CrossRef] [PubMed]
- Rugge, M.; Correa, P.; Dixon, M.F.; Fiocca, R.; Hattori, T.; Lechago, J.; Leandro, G.; Price, A.B.; Sipponen, P.; Solcia, E.; et al. Gastric Mucosal Atrophy: Interobserver Consistency Using New Criteria for Classification and Grading. Aliment. Pharmacol. Ther. 2002, 16, 1249–1259. [Google Scholar] [CrossRef]
- Cook, J.D.; Brown, G.M.; Valberg, L.S. The Effect of Achylia Gastrica on Iron Absorption*. J. Clin. Investig. 1964, 43, 1185–1191. [Google Scholar] [CrossRef]
- Engstrand, L.; Graham, D.Y. The Microbiome and Gastric Cancer. Dig. Dis. Sci. 2020, 65, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for Culture of “unculturable” Bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Annibale, B.; Lahner, E. Autoimmune Gastritis and Gastric Microbiota. Microorganisms 2020, 8, 1827. [Google Scholar] [CrossRef] [PubMed]
- Rajilic-Stojanovic, M.; Figueiredo, C.; Smet, A.; Hansen, R.; Kupcinskas, J.; Rokkas, T.; Andersen, L.; Machado, J.C.; Ianiro, G.; Gasbarrini, A.; et al. Systematic Review: Gastric Microbiota in Health and Disease. Aliment. Pharmacol. Ther. 2020, 51, 582–602. [Google Scholar] [CrossRef]
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric Microbial Community Profiling Reveals a Dysbiotic Cancer-Associated Microbiota. Gut 2018, 67, 226–236. [Google Scholar] [CrossRef]
- Parsons, B.N.; Ijaz, U.Z.; D’Amore, R.; Burkitt, M.D.; Eccles, R.; Lenzi, L.; Duckworth, C.A.; Moore, A.R.; Tiszlavicz, L.; Varro, A.; et al. Comparison of the Human Gastric Microbiota in Hypochlorhydric States Arising as a Result of Helicobacter Pylori-Induced Atrophic Gastritis, Autoimmune Atrophic Gastritis and Proton Pump Inhibitor Use. PLoS Pathog. 2017, 13, e1006653. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.P.; Ali, W.M.; Sivadasan, R.; Rajeeve, K. Bacteria-Cancer Interface: Awaiting the Perfect Storm. Pathog. Basel Switz. 2021, 10, 1321. [Google Scholar] [CrossRef]
- Della Bella, C.; Soluri, M.F.; Puccio, S.; Benagiano, M.; Grassi, A.; Bitetti, J.; Cianchi, F.; Sblattero, D.; Peano, C.; D’Elios, M.M. The Helicobacter Pylori CagY Protein Drives Gastric Th1 and Th17 Inflammation and B Cell Proliferation in Gastric MALT Lymphoma. Int. J. Mol. Sci. 2021, 22, 9459. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Li, N.-S.; Hu, Y.; Lu, N.-H. Impact Factors That Modulate Gastric Cancer Risk in Helicobacter Pylori-Infected Rodent Models. Helicobacter 2019, 24, e12580. [Google Scholar] [CrossRef] [PubMed]
- Mori, G.; Pasca, M.R. Gut Microbial Signatures in Sporadic and Hereditary Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 1312. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Rabkin, C.S.; Turner, N.; Fraumeni, J.F.; Rosenberg, P.S.; Camargo, M.C. The Changing Face of Noncardia Gastric Cancer Incidence Among US Non-Hispanic Whites. J. Natl. Cancer Inst. 2018, 110, 608–615. [Google Scholar] [CrossRef]
- Floch, P.; Mégraud, F.; Lehours, P. Helicobacter Pylori Strains and Gastric MALT Lymphoma. Toxins 2017, 9, 132. [Google Scholar] [CrossRef]
- Ford, A.C.; Yuan, Y.; Moayyedi, P. Helicobacter Pylori Eradication Therapy to Prevent Gastric Cancer: Systematic Review and Meta-Analysis. Gut 2020, 69, 2113–2121. [Google Scholar] [CrossRef]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter Pylori Virulence Factors—Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells 2021, 10, 27. [Google Scholar] [CrossRef]
- Liu, X.; Rong, Z.; Shou, C. Mycoplasma Hyorhinis Infection Promotes Gastric Cancer Cell Motility via β-Catenin Signaling. Cancer Med. 2019, 8, 5301–5312. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Meng, L.; Jiang, B.; Zhang, J.; Yang, H.; Wu, J.; Shou, C. P37 from Mycoplasma Hyorhinis Promotes Cancer Cell Invasiveness and Metastasis through Activation of MMP-2 and Followed by Phosphorylation of EGFR. Mol. Cancer Ther. 2008, 7, 530–537. [Google Scholar] [CrossRef]
- Boehm, E.T.; Thon, C.; Kupcinskas, J.; Steponaitiene, R.; Skieceviciene, J.; Canbay, A.; Malfertheiner, P.; Link, A. Fusobacterium Nucleatum Is Associated with Worse Prognosis in Lauren’s Diffuse Type Gastric Cancer Patients. Sci. Rep. 2020, 10, 16240. [Google Scholar] [CrossRef] [PubMed]
- Cheok, Y.Y.; Lee, C.Y.Q.; Cheong, H.C.; Vadivelu, J.; Looi, C.Y.; Abdullah, S.; Wong, W.F. An Overview of Helicobacter Pylori Survival Tactics in the Hostile Human Stomach Environment. Microorganisms 2021, 9, 2502. [Google Scholar] [CrossRef]
- Schistosomes, Liver Flukes and Helicobacter Pylori. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1994; Volume 61, pp. 1–241.
- Tang, L.; Tang, B.; Lei, Y.; Yang, M.; Wang, S.; Hu, S.; Xie, Z.; Liu, Y.; Vlodavsky, I.; Yang, S. Helicobacter Pylori-Induced Heparanase Promotes H. pylori Colonization and Gastritis. Front. Immunol. 2021, 12, 2352. [Google Scholar] [CrossRef]
- Narayanan, M.; Reddy, K.M.; Marsicano, E. Peptic Ulcer Disease and Helicobacter Pylori Infection. Mo. Med. 2018, 115, 219–224. [Google Scholar]
- Uotani, T.; Murakami, K.; Uchida, T.; Tanaka, S.; Nagashima, H.; Zeng, X.-L.; Akada, J.; Estes, M.K.; Graham, D.Y.; Yamaoka, Y. Changes of Tight Junction and Interleukin-8 Expression Using a Human Gastroid Monolayer Model of Helicobacter Pylori Infection. Helicobacter 2019, 24, e12583. [Google Scholar] [CrossRef]
- Jain, U.; Saxena, K.; Chauhan, N. Helicobacter Pylori Induced Reactive Oxygen Species: A New and Developing Platform for Detection. Helicobacter 2021, 26, e12796. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Tanaka, M.; Soma, Y.; Shimoyama, T.; Mikami, T.; Crabtree, J.E.; Saito, H.; Munakata, A.; Yoshida, Y. Histological Analysis of Gastritis and Helicobacter pylori Infection in Patients with Early Gastric Cancer: A Case-Control Study. J. Gastroenterol. Hepatol. 2000, 15, 1370–1376. [Google Scholar] [CrossRef]
- Imai, S.; Ooki, T.; Murata-Kamiya, N.; Komura, D.; Tahmina, K.; Wu, W.; Takahashi-Kanemitsu, A.; Knight, C.T.; Kunita, A.; Suzuki, N.; et al. Helicobacter Pylori CagA Elicits BRCAness to Induce Genome Instability That May Underlie Bacterial Gastric Carcinogenesis. Cell Host Microbe 2021, 29, 941–958.e10. [Google Scholar] [CrossRef]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating Cytotoxin A (VacA), a Key Toxin for Helicobacter Pylori Pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef]
- Farzi, N.; Yadegar, A.; Aghdaei, H.A.; Yamaoka, Y.; Zali, M.R. Genetic Diversity and Functional Analysis of OipA Gene in Association with Other Virulence Factors among Helicobacter Pylori Isolates from Iranian Patients with Different Gastric Diseases. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 60, 26–34. [Google Scholar] [CrossRef]
- Yamaoka, Y. Mechanisms of Disease: Helicobacter Pylori Virulence Factors. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 629–641. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter Pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Diaz, M.I.; Valdivia, A.; Godoy, A.; Peña, A.; Rollan, A.; Kirberg, A.; Hebel, E.; Fierro, J.; Klapp, G.; et al. Relationship between Helicobacter Pylori Virulence Factors and Regulatory Cytokines as Predictors of Clinical Outcome. Microbes Infect. 2007, 9, 428–434. [Google Scholar] [CrossRef]
- Yea, S.S.; Yang, Y.I.; Jang, W.H.; Lee, Y.J.; Bae, H.S.; Paik, K.H. Association between TNF-Alpha Promoter Polymorphism and Helicobacter Pylori CagA Subtype Infection. J. Clin. Pathol. 2001, 54, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.; Machado, J.C.; Pharoah, P.; Seruca, R.; Sousa, S.; Carvalho, R.; Capelinha, A.F.; Quint, W.; Caldas, C.; van Doorn, L.-J.; et al. Helicobacter Pylori and Interleukin 1 Genotyping: An Opportunity to Identify High-Risk Individuals for Gastric Carcinoma. JNCI J. Natl. Cancer Inst. 2002, 94, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, Y.A.; Choi, I.J.; Lee, Y.-S.; Kim, S.-Y.; Shin, A.; Cho, S.-J.; Kook, M.-C.; Nam, J.H.; Ryu, K.W.; et al. Effects of Interleukin-10 Polymorphisms, Helicobacter Pylori Infection, and Smoking on the Risk of Noncardia Gastric Cancer. PLoS ONE 2012, 7, e29643. [Google Scholar] [CrossRef]
- Takahashi-Kanemitsu, A.; Knight, C.T.; Hatakeyama, M. Molecular Anatomy and Pathogenic Actions of Helicobacter Pylori CagA That Underpin Gastric Carcinogenesis. Cell. Mol. Immunol. 2020, 17, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, K.; Salinas, R.C. Peptic Ulcer Disease. Am. Fam. Physician 2007, 76, 1005–1012. [Google Scholar]
- NIH Consensus Conference. Helicobacter Pylori in Peptic Ulcer Disease. NIH Consensus Development Panel on Helicobacter Pylori in Peptic Ulcer Disease. JAMA 1994, 272, 65–69. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, L.-Y.; Lu, H.-P.; Liu, J.-Z. Recurrence of Helicobacter Pylori Infection: Incidence and Influential Factors. Chin. Med. J. 2019, 132, 765–771. [Google Scholar] [CrossRef]
- Correa, P. Chronic Gastritis: A Clinico-Pathological Classification. Am. J. Gastroenterol. 1988, 83, 504–509. [Google Scholar]
- Park, Y.H.; Kim, N. Review of Atrophic Gastritis and Intestinal Metaplasia as a Premalignant Lesion of Gastric Cancer. J. Cancer Prev. 2015, 20, 25–40. [Google Scholar] [CrossRef]
- Kirikoshi, H.; Sekihara, H.; Katoh, M. Up-Regulation of WNT10A by Tumor Necrosis Factor Alpha and Helicobacter Pylori in Gastric Cancer. Int. J. Oncol. 2001, 19, 533–536. [Google Scholar] [PubMed]
- Saitoh, T.; Kirikoshi, H.; Mine, T.; Katoh, M. Proto-Oncogene WNT10B Is up-Regulated by Tumor Necrosis Factor Alpha in Human Gastric Cancer Cell Line MKN45. Int. J. Oncol. 2001, 19, 1187–1192. [Google Scholar] [CrossRef]
- Li, N.; Xu, X.; Yang, H.; Wang, H.; Ouyang, Y.; Zhou, Y.; Peng, C.; Yuan, Z.; He, C.; Zeng, C.; et al. Activation of Aquaporin 5 by Carcinogenic Helicobacter Pylori Infection Promotes Epithelial-Mesenchymal Transition via the MEK/ERK Pathway. Helicobacter 2021, 26, e12842. [Google Scholar] [CrossRef] [PubMed]
- Biernat, M.M.; Wróbel, T. Bacterial Infection and Non-Hodgkin B-Cell Lymphoma: Interactions between Pathogen, Host and the Tumor Environment. Int. J. Mol. Sci. 2021, 22, 7372. [Google Scholar] [CrossRef] [PubMed]
- Hamoudi, R.A.; Appert, A.; Ye, H.; Ruskone-Fourmestraux, A.; Streubel, B.; Chott, A.; Raderer, M.; Gong, L.; Wlodarska, I.; De Wolf-Peeters, C.; et al. Differential Expression of NF-ΚB Target Genes in MALT Lymphoma with and without Chromosome Translocation: Insights into Molecular Mechanism. Leukemia 2010, 24, 1487–1497. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-KappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.-H.; Kuo, S.-H.; Chen, L.-T.; Mao, T.-L.; Doong, S.-L.; Wu, M.-S.; Hsu, H.-C.; Tzeng, Y.-S.; Chen, C.-L.; Lin, J.-T.; et al. Nuclear Expression of BCL10 or Nuclear Factor Kappa B Helps Predict Helicobacter Pylori–Independent Status of Low-Grade Gastric Mucosa–Associated Lymphoid Tissue Lymphomas with or without t(11;18)(Q21;Q21). Blood 2005, 106, 1037–1041. [Google Scholar] [CrossRef]
- He, M.; Gao, L.; Zhang, S.; Tao, L.; Wang, J.; Yang, J.; Zhu, M. Prognostic Significance of MiR-34a and Its Target Proteins of FOXP1, P53, and BCL2 in Gastric MALT Lymphoma and DLBCL. Gastric Cancer 2014, 17, 431–441. [Google Scholar] [CrossRef]
- Best, L.M.; Takwoingi, Y.; Siddique, S.; Selladurai, A.; Gandhi, A.; Low, B.; Yaghoobi, M.; Gurusamy, K.S. Non-Invasive Diagnostic Tests for Helicobacter Pylori Infection. Cochrane Database Syst. Rev. 2018, 3, CD012080. [Google Scholar] [CrossRef]
- Kato, S.; Tsukamoto, T.; Mizoshita, T.; Tanaka, H.; Kumagai, T.; Ota, H.; Katsuyama, T.; Asaka, M.; Tatematsu, M. High Salt Diets Dose-Dependently Promote Gastric Chemical Carcinogenesis in Helicobacter Pylori-Infected Mongolian Gerbils Associated with a Shift in Mucin Production from Glandular to Surface Mucous Cells. Int. J. Cancer 2006, 119, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Nascimento Araujo, C.D.; Amorim, A.T.; Barbosa, M.S.; Alexandre, J.C.P.L.; Campos, G.B.; Macedo, C.L.; Marques, L.M.; Timenetsky, J. Evaluating the Presence of Mycoplasma Hyorhinis, Fusobacterium Nucleatum, and Helicobacter Pylori in Biopsies of Patients with Gastric Cancer. Infect. Agent. Cancer 2021, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, J.-Y.; Wu, J.; Meng, L.; Shou, C.-C. Mycoplasma Infections and Different Human Carcinomas. World J. Gastroenterol. 2001, 7, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, H.; Chen, W.; Yao, X.; Xing, Y.; Wang, X.; Zhong, J.; Meng, G. Mycoplasma Hyorhinis Activates the NLRP3 Inflammasome and Promotes Migration and Invasion of Gastric Cancer Cells. PLoS ONE 2013, 8, e77955. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Huang, S.; Wu, J.; Meng, L.; Shou, C. Protein P37 of Mycoplasma Hyorhinis Induces Secretion of TNF-α from Human Peripheral Blood Mononuclear Cells. Chin. Sci. Bull. 2003, 48, 658–662. [Google Scholar] [CrossRef]
- Loozen, G.; Ozcelik, O.; Boon, N.; De Mol, A.; Schoen, C.; Quirynen, M.; Teughels, W. Inter-Bacterial Correlations in Subgingival Biofilms: A Large-Scale Survey. J. Clin. Periodontol. 2014, 41, 1–10. [Google Scholar] [CrossRef]
- Brennan, C.A.; Garrett, W.S. Fusobacterium Nucleatum—Symbiont, Opportunist and Oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.E.M.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon Cancer-Associated Fusobacterium Nucleatum May Originate From the Oral Cavity and Reach Colon Tumors via the Circulatory System. Front. Cell. Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Fu, X. Fusobacterium Nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, Y.; Yu, J.; Chen, T.; Wu, Y.; Shi, L.; Li, Q.; Wu, J.; Fu, X. Invasive Fusobacterium Nucleatum Activates Beta-Catenin Signaling in Colorectal Cancer via a TLR4/P-PAK1 Cascade. Oncotarget 2017, 8, 31802–31814. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium Nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [PubMed]
- Koikawa, K.; Ohuchida, K.; Ando, Y.; Kibe, S.; Nakayama, H.; Takesue, S.; Endo, S.; Abe, T.; Okumura, T.; Iwamoto, C.; et al. Basement Membrane Destruction by Pancreatic Stellate Cells Leads to Local Invasion in Pancreatic Ductal Adenocarcinoma. Cancer Lett. 2018, 425, 65–77. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Cui, C.; Chakraborty, K.; Tang, X.A.; Zhou, G.; Schoenfelt, K.Q.; Becker, K.M.; Hoffman, A.; Chang, Y.-F.; Blank, A.; Reardon, C.A.; et al. Neutrophil Elastase Selectively Kills Cancer Cells and Attenuates Tumorigenesis. Cell 2021, 184, 3163–3177.e21. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Bedogni, B. The Membrane Tethered Matrix Metalloproteinase MT1-MMP at the Forefront of Melanoma Cell Invasion and Metastasis. Pharmacol. Res. 2016, 111, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Sato, H.; Hayashibara, T.; Senba, M.; Geleziunas, R.; Wada, A.; Hirayama, T.; Yamamoto, N. Helicobacter Pylori Induces Matrix Metalloproteinase-9 through Activation of Nuclear Factor ΚB. Gastroenterology 2003, 124, 983–992. [Google Scholar] [CrossRef]
- Lindholm, M.; Manon-Jensen, T.; Madsen, G.I.; Krag, A.; Karsdal, M.A.; Kjeldsen, J.; Mortensen, J.H. Extracellular Matrix Fragments of the Basement Membrane and the Interstitial Matrix Are Serological Markers of Intestinal Tissue Remodeling and Disease Activity in Dextran Sulfate Sodium Colitis. Dig. Dis. Sci. 2019, 64, 3134–3142. [Google Scholar] [CrossRef]
- Zheng, H.; Takahashi, H.; Murai, Y.; Cui, Z.; Nomoto, K.; Niwa, H.; Tsuneyama, K.; Takano, Y. Expressions of MMP-2, MMP-9 and VEGF Are Closely Linked to Growth, Invasion, Metastasis and Angiogenesis of Gastric Carcinoma. Anticancer Res. 2006, 26, 3579–3583. [Google Scholar]
- Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of Gastric Mucosal Microbiota across Different Stomach Microhabitats in a Cohort of 276 Patients with Gastric Cancer. EBioMedicine 2018, 40, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y. Helicobacter Pylori Update: Gastric Cancer, Reliable Therapy, and Possible Benefits. Gastroenterology 2015, 148, 719–731.e3. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jia, K.; Lv, H.; Wang, S.-Q.; Wu, Y.; Lei, H.; Chen, X. EBV-Positive Gastric Cancer: Current Knowledge and Future Perspectives. Front. Oncol. 2020, 10, 583463. [Google Scholar] [CrossRef] [PubMed]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int. J. Mol. Sci. 2020, 21, E4012. [Google Scholar] [CrossRef] [PubMed]
- Marta, Ż.-N.; Agnieszka, W.; Jacek, P.; Jeleń, A.; Adrian, K.; Dagmara, S.-K.; Sałagacka-Kubiak, A.; Balcerczak, E. NFKB2 Gene Expression in Patients with Peptic Ulcer Diseases and Gastric Cancer. Mol. Biol. Rep. 2020, 47, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, W.; Lee, A.; He, J.; Huang, B.; Zheng, W.; Su, T.; Lai, S.; Long, Y.; Chu, H.; et al. The Impact of Helicobacter Pylori Infection, Eradication Therapy and Probiotic Supplementation on Gut Microenvironment Homeostasis: An Open-Label, Randomized Clinical Trial. EBioMedicine 2018, 35, 87–96. [Google Scholar] [CrossRef]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A.; Francois, F.; Perez-Perez, G.; Blaser, M.J.; Relman, D.A. Molecular Analysis of the Bacterial Microbiota in the Human Stomach. Proc. Natl. Acad. Sci. USA 2006, 103, 732–737. [Google Scholar] [CrossRef]
- Montalban-Arques, A.; Wurm, P.; Trajanoski, S.; Schauer, S.; Kienesberger, S.; Halwachs, B.; Högenauer, C.; Langner, C.; Gorkiewicz, G. Propionibacterium Acnes Overabundance and Natural Killer Group 2 Member D System Activation in Corpus-dominant Lymphocytic Gastritis. J. Pathol. 2016, 240, 425–436. [Google Scholar] [CrossRef]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal Microbiome Dysbiosis in Gastric Carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef]
- Dong, T.; Feng, Q.; Liu, F.; Chang, L.K.; Zhou, X.; Han, M.; Tian, X.; Zhong, N.; Liu, S. Alteration of Stomach Microbiota Compositions in the Progression of Gastritis Induces Nitric Oxide in Gastric Cell. Exp. Ther. Med. 2017, 13, 2793–2800. [Google Scholar] [CrossRef][Green Version]
- Bozzetti, F.; Bonfanti, G.; Bufalino, R.; Menotti, V.; Persano, S.; Andreola, S.; Doci, R.; Gennari, L. Adequacy of Margins of Resection in Gastrectomy for Cancer. Ann. Surg. 1982, 196, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Johnston, D.; Sue-Ling, H.; Young, S.; May, J.; Griffith, J.; Miller, G.; Martin, I. Total or Subtotal Gastrectomy for Gastric Carcinoma? A Study of Quality of Life. World J. Surg. 1998, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, J.; Zhang, G.; Kong, D. Individualized Proximal Margin Correlates with Outcomes in Gastric Cancers with Radical Gastrectomy. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317711032. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Kim, B.S.; Yook, J.H.; Kim, B.S. Optimal Proximal Resection Margin Distance for Gastrectomy in Advanced Gastric Cancer. World J. Gastroenterol. 2020, 26, 2232–2246. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Lin, J.-T.; Ho, H.J.; Lai, Z.-L.; Wang, C.-B.; Tang, S.-L.; Wu, C.-Y. Gastric Microbiota and Predicted Gene Functions Are Altered after Subtotal Gastrectomy in Patients with Gastric Cancer. Sci. Rep. 2016, 6, 20701. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-H.; Huang, K.-H.; Chuang, W.-H.; Luo, J.-C.; Lin, C.-C.; Ting, P.-H.; Young, S.-H.; Fang, W.-L.; Hou, M.-C.; Lee, F.-Y. The Long Term Effect of Metabolic Profile and Microbiota Status in Early Gastric Cancer Patients after Subtotal Gastrectomy. PLoS ONE 2018, 13, e0206930. [Google Scholar] [CrossRef]
- Palleja, A.; Kashani, A.; Allin, K.H.; Nielsen, T.; Zhang, C.; Li, Y.; Brach, T.; Liang, S.; Feng, Q.; Jørgensen, N.B.; et al. Roux-En-Y Gastric Bypass Surgery of Morbidly Obese Patients Induces Swift and Persistent Changes of the Individual Gut Microbiota. Genome Med. 2016, 8, 67. [Google Scholar] [CrossRef]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The Role of PH in Determining the Species Composition of the Human Colonic Microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Mashima, I.; Nakazawa, F. Interaction between Streptococcus Spp. and Veillonella Tobetsuensis in the Early Stages of Oral Biofilm Formation. J. Bacteriol. 2015, 197, 2104–2111. [Google Scholar] [CrossRef]
- Martinsen, T.C.; Bergh, K.; Waldum, H.L. Gastric Juice: A Barrier against Infectious Diseases. Basic Clin. Pharmacol. Toxicol. 2005, 96, 94–102. [Google Scholar] [CrossRef]
- Carboni, M.; Guadagni, S.; Pistoia, M.A.; Amicucci, G.; Tuscano, D.; Negro, P.; Smith, P.L.; Walters, C.L. The Microflora of the Gastric Juice after Billroth I and Billroth II Partial Gastrectomy. Scand. J. Gastroenterol. 1986, 21, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, S.; Campisciano, G.; Silvestri, M.; Guerra, M.; Giuricin, M.; Casagranda, B.; Comar, M.; de Manzini, N. Changes in Gut Microbiota Composition after Bariatric Surgery: A New Balance to Decode. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2020, 24, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794. [Google Scholar] [CrossRef] [PubMed]
- Schauer, D.P.; Feigelson, H.S.; Koebnick, C.; Caan, B.; Weinmann, S.; Leonard, A.C.; Powers, J.D.; Yenumula, P.R.; Arterburn, D.E. Bariatric Surgery and the Risk of Cancer in a Large Multisite Cohort. Ann. Surg. 2019, 269, 95–101. [Google Scholar] [CrossRef]
- Adams, T.D.; Hunt, S.C. Cancer and Obesity: Effect of Bariatric Surgery. World J. Surg. 2009, 33, 2028–2033. [Google Scholar] [CrossRef]
- Bergquist, J.R.; Leiting, J.L.; Habermann, E.B.; Cleary, S.P.; Kendrick, M.L.; Smoot, R.L.; Nagorney, D.M.; Truty, M.J.; Grotz, T.E. Early-Onset Gastric Cancer Is a Distinct Disease with Worrisome Trends and Oncogenic Features. Surgery 2019, 166, 547–555. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer Type | Most Common Site | Reference |
|---|---|---|
| Adenocarcinoma | Liver, ovaries | [10] |
| Lymphomas | Spleen, bone, liver | [11] |
| Gastrointestinal stromal tumors | Mesentery, omentum, ovaries | [12] |
| Carcinoids | Liver | [10] |
| Bacteria | Proposed Pathogenesis | References |
|---|---|---|
| H. pylori | Increased cellular proliferation and signaling, loss of E-cadherin, β-catenin stabilization, and subsequent activation of the WNT-signaling pathway via virulence factors | [56,57,58] |
| M. hyorhinis | P37 induction and activation of MMP-2, promotes gastric cancer cell motility via β-catenin stabilization and subsequent activation of the WNT-signaling pathway | [59,60] |
| F. nucleatum | Associated with worse prognosis in Lauren’s diffuse-type gastric cancer patients | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, A.D.; Seely, K.D.; Hagenstein, L.D.; Florey, G.M.; Small, J.M. Bacterial Involvement in Progression and Metastasis of Adenocarcinoma of the Stomach. Cancers 2022, 14, 4886. https://doi.org/10.3390/cancers14194886
Morgan AD, Seely KD, Hagenstein LD, Florey GM, Small JM. Bacterial Involvement in Progression and Metastasis of Adenocarcinoma of the Stomach. Cancers. 2022; 14(19):4886. https://doi.org/10.3390/cancers14194886
Chicago/Turabian StyleMorgan, Amanda D., Kevin D. Seely, Lauren D. Hagenstein, Garrett M. Florey, and James M. Small. 2022. "Bacterial Involvement in Progression and Metastasis of Adenocarcinoma of the Stomach" Cancers 14, no. 19: 4886. https://doi.org/10.3390/cancers14194886
APA StyleMorgan, A. D., Seely, K. D., Hagenstein, L. D., Florey, G. M., & Small, J. M. (2022). Bacterial Involvement in Progression and Metastasis of Adenocarcinoma of the Stomach. Cancers, 14(19), 4886. https://doi.org/10.3390/cancers14194886