
TFAP2C Knockdown Sensitizes Bladder Cancer Cells to Cisplatin Treatment via Regulation of EGFR and NF-κB

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Bioinformatics
2.3. Tissue Samples
2.4. RNA Isolation and qRT–PCR
2.5. Immunohistochemistry (IHC)
2.6. Cell Culture
2.7. Cell Transfection
2.8. CCK-8 Cell Proliferation Assay and IC50 Determination
2.9. Transwell Cell Invasion Assay and Scratch Assay
2.10. Colony-Formation Assay
2.11. Immunofluorescence Staining
2.12. Western Blot Analysis
2.13. Cell Cycle and Apoptosis Assay
2.14. In Vivo Xenograft Experiments In Vivo
2.15. TUNEL Detection
2.16. Statistical Analysis
3. Results
3.1. TFAP2C Is Highly Expressed in Tumor Tissues and Is Associated with Patient Prognosis
3.2. TFAP2C Knockdown Enhances the Inhibitory Effect of Cisplatin on BCa Cells Proliferation In Vitro
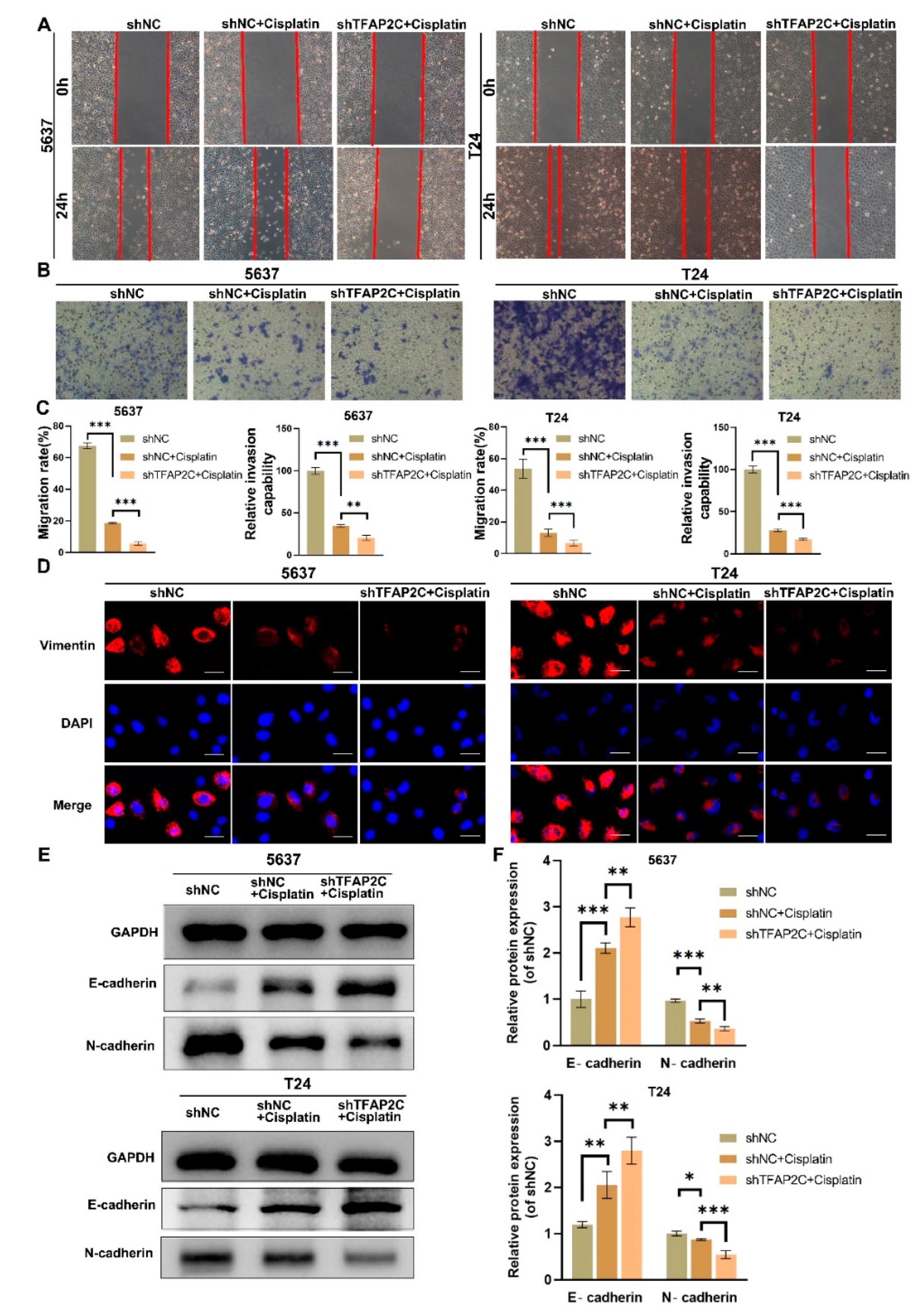
3.3. TFAP2C Knockdown Enhances the Inhibitory Effect of Cisplatin on BCa Cells Migration and Invasion In Vitro
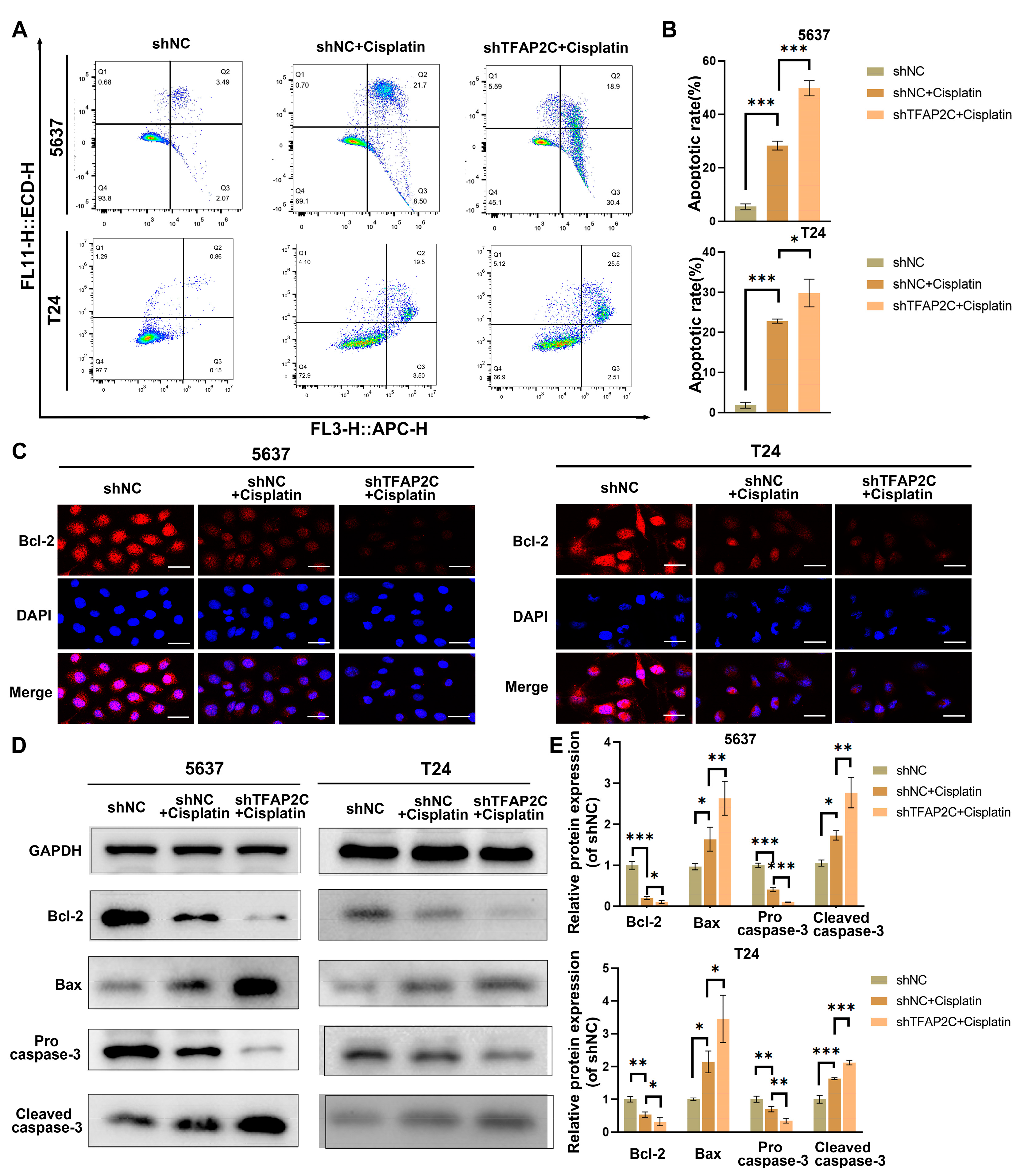
3.4. TFAP2C Knockdown Enhances Cisplatin-Induced Apoptosis In Vitro
3.5. TFAP2C Knockdown Enhances Cisplatin-Induced Cell Cycle Arrest In Vitro
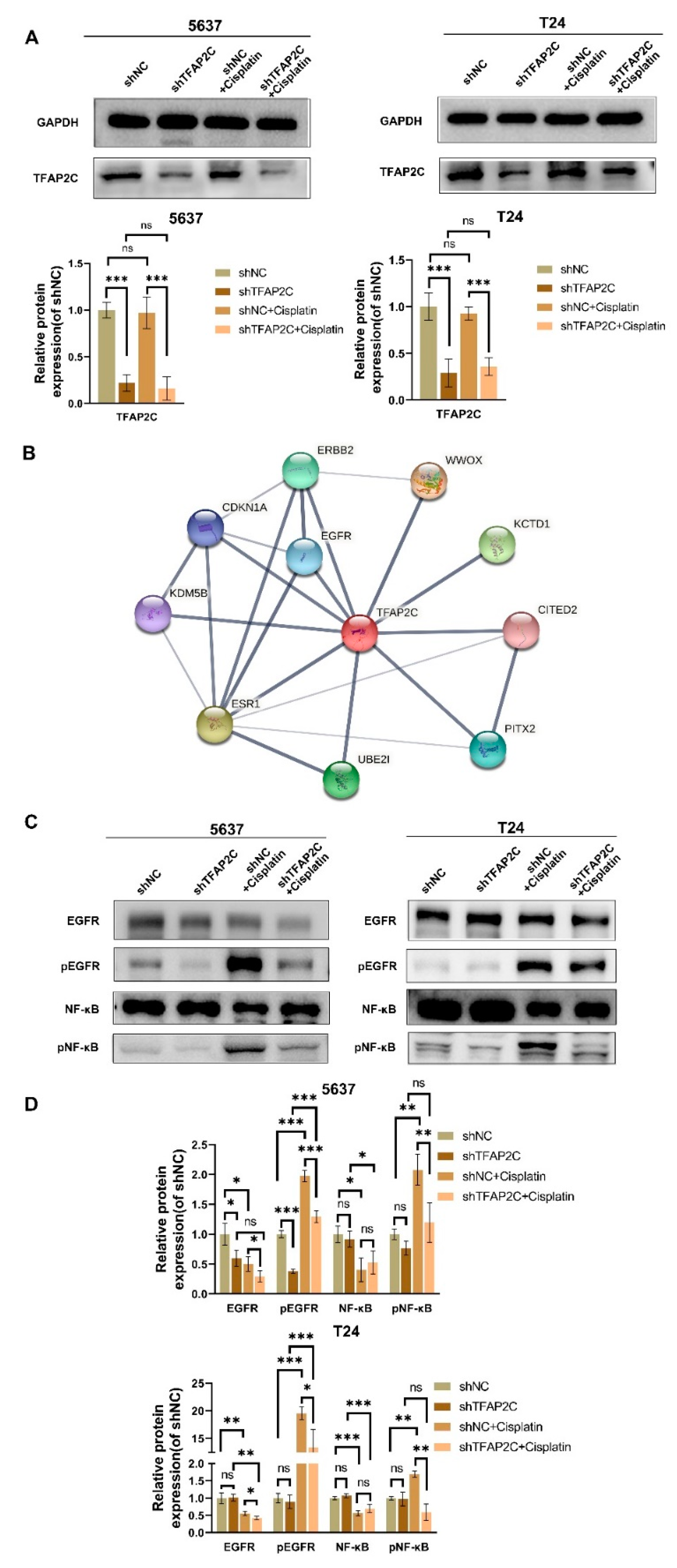
3.6. TFAP2C Knockdown Inhibits Cisplatin-Induced NF-κB and EGFR Activation In Vitro
3.7. TFAP2C Affects the Sensitivity of BCa Cells to Cisplatin by Regulating the Activation of EGFR In Vitro
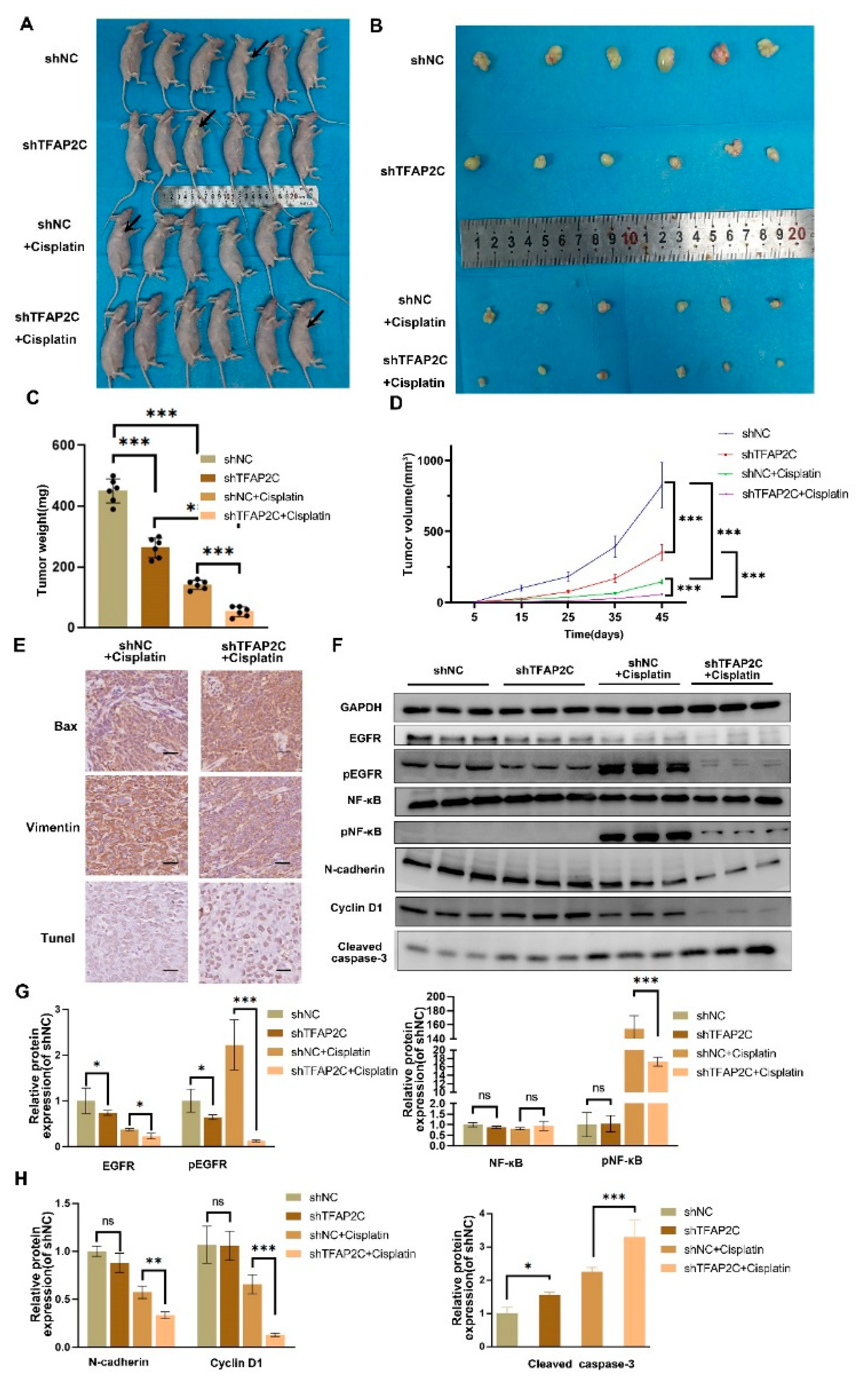
3.8. TFAP2C Enhances the Anti-Tumor Effect of Cisplatin In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Witjes, A.J.; Lebret, T.; Comperat, E.M.; Cowan, N.C.; de Santis, M.; Bruins, H.M.; Hernandez, V.; Espinos, E.L.; Dunn, J.; Rouanne, M.; et al. Updated 2016 Eau Guidelines on Muscle-Invasive and Metastatic Bladder Cancer. Eur. Urol. 2017, 71, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Mazina, O.M.; Phillips, M.A.; Williams, T.; Vines, C.A.; Cherr, G.N.; Rice, R.H. Redistribution of Transcription Factor Ap-2alpha in Differentiating Cultured Human Epidermal Cells. J. Investig. Dermatol. 2001, 117, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Aqeilan, R.I.; Palamarchuk, A.; Weigel, R.J.; Herrero, J.J.; Pekarsky, Y.; Croce, C.M. Physical and Functional Interactions between the Wwox Tumor Suppressor Protein and the Ap-2gamma Transcription Factor. Cancer Res. 2004, 64, 8256–8261. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Kubaczka, C.; Kaiser, S.; Nettersheim, D.; Mughal, S.S.; Riesenberg, S.; Holzel, M.; Winterhager, E.; Schorle, H. Tpbpa-Cre-Mediated Deletion of Tfap2c Leads to Deregulation of Cdkn1a, Akt1 and the Erk Pathway, Causing Placental Growth Arrest. Development 2016, 143, 787–798. [Google Scholar]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.; Jiang, Q.; Deng, J.; Cheng, F.; Lin, Y.; Cheng, L.; Ye, Y.; Chen, X.; Yao, Y.; et al. Tfap2c Facilitates Somatic Cell Reprogramming by Inhibiting C-Myc-Dependent Apoptosis and Promoting Mesenchymal-to-Epithelial Transition. Cell Death Dis. 2020, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Eckert, D.; Buhl, S.; Weber, S.; Jager, R.; Schorle, H. The Ap-2 Family of Transcription Factors. Genome Biol. 2005, 6, 246. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.; Drechsel, D.; Schmid, M.T.; Ninkovic, J.; Irmler, M.; Brill, M.S.; Restani, L.; Gianfranceschi, L.; Cerri, C.; Weber, S.N.; et al. Ap2gamma Regulates Basal Progenitor Fate in a Region- and Layer-Specific Manner in the Developing Cortex. Nat. Neurosci. 2009, 12, 1229–1237. [Google Scholar] [CrossRef]
- De Andrade, J.P.; Park, J.M.; Gu, V.W.; Woodfield, G.W.; Kulak, M.V.; Lorenzen, A.W.; Wu, V.T.; van Dorin, S.E.; Spanheimer, P.M.; Weigel, R.J. Egfr Is Regulated by Tfap2c in Luminal Breast Cancer and Is a Target for Vandetanib. Mol. Cancer Ther. 2016, 15, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, E.; Lee, S.; Kim, D.; Chun, J.; Park, K.H.; Youn, H.; Youn, B. Tfap2c-Mediated Upregulation of Tgfbr1 Promotes Lung Tumorigenesis and Epithelial-Mesenchymal Transition. Exp. Mol. Med. 2016, 48, e273. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, D.; Tai, J.; Chen, S.; Yu, M.; Ren, D.; Wang, L. Tfap2c Promotes Stemness and Chemotherapeutic Resistance in Colorectal Cancer Via Inactivating Hippo Signaling Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 27. [Google Scholar] [CrossRef]
- Wei, J.; Yin, Y.; Zhou, J.; Chen, H.; Peng, J.; Yang, J.; Tang, Y. Mettl3 Potentiates Resistance to Cisplatin through M(6) a Modification of Tfap2c in Seminoma. J. Cell Mol. Med. 2020, 24, 11366–11380. [Google Scholar] [CrossRef]
- Villares, G.J.; Zigler, M.; Blehm, K.; Bogdan, C.; McConkey, D.; Colin, D.; Bar-Eli, M. Targeting Egfr in Bladder Cancer. World J. Urol. 2007, 25, 573–579. [Google Scholar] [CrossRef]
- Neal, D.E.; Mellon, K. Epidermal Growth Factor Receptor and Bladder Cancer: A Review. Urol. Int. 1992, 48, 365–371. [Google Scholar] [CrossRef]
- Kassouf, W.; Black, P.C.; Tuziak, T.; Bondaruk, J.; Lee, S.; Brown, G.A.; Adam, L.; Wei, C.; Baggerly, K.; Bar-Eli, M.; et al. Distinctive Expression Pattern of Erbb Family Receptors Signifies an Aggressive Variant of Bladder Cancer. J. Urol. 2008, 179, 353–358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Colquhoun, A.J.; Mellon, J.K. Epidermal Growth Factor Receptor and Bladder Cancer. Postgrad. Med. J. 2002, 78, 584–589. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neal, D.E.; Sharples, L.; Smith, K.; Fennelly, J.; Hall, R.R.; Harris, A.L. The Epidermal Growth Factor Receptor and the Prognosis of Bladder Cancer. Cancer 1990, 65, 1619–1625. [Google Scholar] [CrossRef]
- Wang, W.J.; Li, C.F.; Chu, Y.Y.; Wang, Y.H.; Hour, T.C.; Yen, C.J.; Chang, W.C.; Wang, J.M. Inhibition of the Egfr/Stat3/Cebpd Axis Reverses Cisplatin Cross-Resistance with Paclitaxel in the Urothelial Carcinoma of the Urinary Bladder. Clin. Cancer Res. 2017, 23, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear Factor-Kappab in Cancer Development and Progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Basseres, D.S.; Baldwin, A.S. Nuclear Factor-Kappab and Inhibitor of Kappab Kinase Pathways in Oncogenic Initiation and Progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of Nf-Kappab and Ikappab Proteins: Implications in Cancer and Inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guan, Z.; Liang, L.; Cheng, Y.; Zhou, J.; Li, J.; Xu, Y. Nf-Kappab Signaling Plays Irreplaceable Roles in Cisplatin-Induced Bladder Cancer Chemoresistance and Tumor Progression. Int. J. Oncol. 2016, 48, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. Egfr and Nf-Kappab: Partners in Cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Homepage. Available online: http://cancergenome.nih.gov/abouttcga (accessed on 3 April 2022).
- Carithers, L.J.; Moore, H.M. The Genotype-Tissue Expression (Gtex) Project. Biopreserv. Biobank. 2015, 13, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (Kmplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Kim, E.J.; Kim, S.K.; Kim, Y.J.; Ha, Y.S.; Jeong, P.; Kim, M.J.; Yun, S.J.; Lee, K.M.; Moon, S.K.; et al. Predictive Value of Progression-Related Gene Classifier in Primary Non-Muscle Invasive Bladder Cancer. Mol. Cancer 2010, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. Ncbi Geo: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative Pcr and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih Image to Imagej: 25 Years of Image Analysis. Nat Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Luo, C.; Zhang, X.; Guo, M.; Sun, M.; Yu, H.; Chen, Q.; Yang, W.; Wang, M.; Zuo, S.; et al. Probing the Impact of Sulfur/Selenium/Carbon Linkages on Prodrug Nanoassemblies for Cancer Therapy. Nat. Commun. 2019, 10, 3211. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.T.; Kim, J.; Yan, C.; Jeong, P.; Choi, S.Y.; Lee, O.J.; Chae, Y.B.; Yun, S.J.; Lee, S.C.; Kim, W.J. S100a9 and Egfr Gene Signatures Predict Disease Progression in Muscle Invasive Bladder Cancer Patients after Chemotherapy. Ann. Oncol. 2014, 25, 974–979. [Google Scholar] [CrossRef]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-Dependent Egfr Activation Induces the Co-Expression of Il-6 and Pai-1 Via the Nfkb Pathway in Advanced-Stage Epithelial Ovarian Cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, M.; Chen, G.; Wang, W.; Zhang, P.; Yue, Y.; Guan, Z.; Wang, X.; Fan, J. Bladder Cancer Cells Interact with Vascular Endothelial Cells Triggering Egfr Signals to Promote Tumor Progression. Int. J. Oncol. 2019, 54, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; de Jesus, J.; et al. Oncogenic Egfr Signaling Activates an Mtorc2-Nf-Kappab Pathway That Promotes Chemotherapy Resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef]
- Patel, V.G.; Oh, W.K.; Galsky, M.D. Treatment of Muscle-Invasive and Advanced Bladder Cancer in 2020. CA Cancer J. Clin. 2020, 70, 404–423. [Google Scholar] [CrossRef] [PubMed]
- Lenis, A.T.; Lec, P.M.; Chamie, K.; Mshs, M.D. Bladder Cancer: A Review. JAMA 2020, 324, 1980–1991. [Google Scholar] [CrossRef]
- Xia, Y.; Yu, W.; Cheng, F.; Rao, T.; Ruan, Y.; Yuan, R.; Ning, J.; Zhou, X.; Lin, F.; Zheng, D. Photobiomodulation with Blue Laser Inhibits Bladder Cancer Progression. Front. Oncol. 2021, 11, 701122. [Google Scholar] [CrossRef]
- Milowsky, M.I.; Rumble, R.B.; Booth, C.M.; Gilligan, T.; Eapen, L.J.; Hauke, R.J.; Boumansour, P.; Lee, C.T. Guideline on Muscle-Invasive and Metastatic Bladder Cancer (European Association of Urology Guideline): American Society of Clinical Oncology Clinical Practice Guideline Endorsement. J. Clin. Oncol. 2016, 34, 1945–1952. [Google Scholar] [CrossRef]
- Wenmaekers, S.; Viergever, B.J.; Kumar, G.; Kranenburg, O.; Black, P.C.; Daugaard, M.; Meijer, R.P. A Potential Role for Huwe1 in Modulating Cisplatin Sensitivity. Cells 2021, 10, 1262. [Google Scholar] [CrossRef]
- Yoon, S.J.; Park, I.; Kwak, C.; Lee, E. Ultrastructural Change Due to Acquired Cisplatin Resistance in Human Bladder Cancer Cells. Oncol. Rep. 2003, 10, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Drayton, R.M.; Catto, J.W. Molecular Mechanisms of Cisplatin Resistance in Bladder Cancer. Expert Rev. Anticancer Ther. 2012, 12, 271–281. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Kalantari, M.; Mohammadinejad, R.; Javaheri, T.; Sethi, G. Association of the Epithelial-Mesenchymal Transition (Emt) with Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 4002. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Karnitz, L.M. Cisplatin-Induced DNA Damage Activates Replication Checkpoint Signaling Components That Differentially Affect Tumor Cell Survival. Mol. Pharmacol. 2009, 76, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F. Emt and Back Again: Visualizing the Dynamic Phenotypes of Metastasis. Cancer Res. 2020, 80, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ma, Z.; Chen, Z.; Li, M.; Wu, Z.; Hong, M.; Wang, H.; Svatek, R.; Rodriguez, R.; Wang, Z. Clinicopathological and Prognostic Value of Ki-67 Expression in Bladder Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158891. [Google Scholar] [CrossRef]
- Rosenblatt, R.; Jonmarker, S.; Lewensohn, R.; Egevad, L.; Sherif, A.; Kalkner, K.M.; Nilsson, S.; Valdman, A.; Ullen, A. Current Status of Prognostic Immunohistochemical Markers for Urothelial Bladder Cancer. Tumour. Biol. 2008, 29, 311–322. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell Cycle, Cdks and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Cavallo, F.; Feldman, D.R.; Barchi, M. Revisiting DNA Damage Repair, P53-Mediated Apoptosis and Cisplatin Sensitivity in Germ Cell Tumors. Int. J. Dev. Biol. 2013, 57, 273–280. [Google Scholar] [CrossRef]
- Brozovic, A.; Ambriovic-Ristov, A.; Osmak, M. The Relationship between Cisplatin-Induced Reactive Oxygen Species, Glutathione, and Bcl-2 and Resistance to Cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, R.; Chen, Q.; Chang, H. Inhibition of Autophagy Using 3-Methyladenine Increases Cisplatin-Induced Apoptosis by Increasing Endoplasmic Reticulum Stress in U251 Human Glioma Cells. Mol. Med. Rep. 2015, 12, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Tait, S.W. Mitochondrial Apoptosis: Killing Cancer Using the Enemy Within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, S.; Kim, S.J.; Shao, F.; Ho, J.W.K.; Wong, K.U.; Miao, Z.; Hao, D.; Zhao, M.; Xu, J.; et al. Cisplatin Prevents Breast Cancer Metastasis through Blocking Early Emt and Retards Cancer Growth Together with Paclitaxel. Theranostics 2021, 11, 2442–2459. [Google Scholar] [CrossRef]
- Hayakawa, J.; Depatie, C.; Ohmichi, M.; Mercola, D. The Activation of C-Jun Nh2-Terminal Kinase (Jnk) by DNA-Damaging Agents Serves to Promote Drug Resistance Via Activating Transcription Factor 2 (Atf2)-Dependent Enhanced DNA Repair. J. Biol. Chem. 2003, 278, 20582–20592. [Google Scholar] [CrossRef]
- Tanabe, M.; Izumi, H.; Ise, T.; Higuchi, S.; Yamori, T.; Yasumoto, K.; Kohno, K. Activating Transcription Factor 4 Increases the Cisplatin Resistance of Human Cancer Cell Lines. Cancer Res. 2003, 63, 8592–8595. [Google Scholar]
- Dempke, W.; Voigt, W.; Grothey, A.; Hill, B.T.; Schmoll, H.J. Cisplatin Resistance and Oncogenes—A Review. Anticancer Drugs 2000, 11, 225–236. [Google Scholar] [CrossRef]
- Kolat, D.; Kaluzinska, Z.; Bednarek, A.K.; Pluciennik, E. The Biological Characteristics of Transcription Factors Ap-2alpha and Ap-2gamma and Their Importance in Various Types of Cancers. Biosci. Rep. 2019, 39, BSR20181928. [Google Scholar] [CrossRef]
- Pastor, W.A.; Liu, W.; Chen, D.; Ho, J.; Kim, R.; Hunt, T.J.; Lukianchikov, A.; Liu, X.; Polo, J.M.; Jacobsen, S.E.; et al. Tfap2c Regulates Transcription in Human Naive Pluripotency by Opening Enhancers. Nat. Cell Biol. 2018, 20, 553–564. [Google Scholar] [CrossRef]
- Williams, C.M.; Scibetta, A.G.; Friedrich, J.K.; Canosa, M.; Berlato, C.; Moss, C.H.; Hurst, H.C. Ap-2gamma Promotes Proliferation in Breast Tumour Cells by Direct Repression of the Cdkn1a Gene. EMBO J. 2009, 28, 3591–3601. [Google Scholar] [CrossRef]
- Hoei-Hansen, C.E.; Nielsen, J.E.; Almstrup, K.; Sonne, S.B.; Graem, N.; Skakkebaek, N.E.; Leffers, H.; Rajpert-De Meyts, E. Transcription Factor Ap-2gamma Is a Developmentally Regulated Marker of Testicular Carcinoma in Situ and Germ Cell Tumors. Clin. Cancer Res. 2004, 10, 8521–8530. [Google Scholar] [CrossRef]
- Chang, T.H.; Tsai, M.F.; Gow, C.H.; Wu, S.G.; Liu, Y.N.; Chang, Y.L.; Yu, S.L.; Tsai, H.C.; Lin, S.W.; Chen, Y.W.; et al. Upregulation of Microrna-137 Expression by Slug Promotes Tumor Invasion and Metastasis of Non-Small Cell Lung Cancer Cells through Suppression of Tfap2c. Cancer Lett. 2017, 402, 190–202. [Google Scholar] [CrossRef]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing Egfr Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the Erbb Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Cruz, A.P.; Gansberger, E.; Pardee, A.B. Epidermal Growth Factor-Induced Nuclear Factor Kappa B Activation: A Major Pathway of Cell-Cycle Progression in Estrogen-Receptor Negative Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 8542–8547. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Levitzki, A.; Gazit, A.; Cavenee, W.K.; Huang, H.J. Drug Resistance of Human Glioblastoma Cells Conferred by a Tumor-Specific Mutant Epidermal Growth Factor Receptor through Modulation of Bcl-Xl and Caspase-3-Like Proteases. Proc. Natl. Acad. Sci. USA 1998, 95, 5724–5729. [Google Scholar] [CrossRef]
- Benhar, M.; Engelberg, D.; Levitzki, A. Cisplatin-Induced Activation of the Egf Receptor. Oncogene 2002, 21, 8723–8731. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kim, K.D.; Lee, E.; Lim, J.S.; Cho, H.J.; Yoon, H.K.; Cho, M.Y.; Baek, K.E.; Park, Y.P.; Paik, S.G.; et al. Up-Regulation of Bfl-1/A1 Via Nf-Kappab Activation in Cisplatin-Resistant Human Bladder Cancer Cell Line. Cancer Lett. 2004, 212, 61–70. [Google Scholar] [CrossRef]
- Park, J.M.; Wu, T.; Cyr, A.R.; Woodfield, G.W.; de Andrade, J.P.; Spanheimer, P.M.; Li, T.; Sugg, S.L.; Lal, G.; Domann, F.E.; et al. The Role of Tcfap2c in Tumorigenesis and Cancer Growth in an Activated Neu Model of Mammary Carcinogenesis. Oncogene 2015, 34, 6105–6114. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Company | Catalog Number | Host | Dilution |
|---|---|---|---|---|
| GAPDH | Servicebio | GB15004 | Rabbit | 1:1000 |
| EGFR | Zhengneng Biotechnology | R22778 | Mouse | 1:1000 |
| pEGFR | Cell Signaling Technology | #3777T | Rabbit | 1:1000 |
| NF-κB | Proteintech | 10745-1-AP | Rabbit | 1:1000 |
| pNF-κB | Cell Signaling Technology | #3033 | Rabbit | 1:1000 |
| Bax | Proteintech | 60267-1-Ig | Mouse | 1:5000 |
| Bcl-2 | Cell Signaling Technology | #15071 | Mouse | 1:1000 |
| TFAP2C | Proteintech | 14572-1-AP | Rabbit | 1:1000 |
| Caspase-3 | Proteintech | 19677-1-AP | Rabbit | 1:1000 |
| E-cadherin | Proteintech | 20874-1-AP | Rabbit | 1:5000 |
| N-cadherin | Proteintech | 22018-1-AP | Rabbit | 1:5000 |
| Cyclin D1 | Proteintech | 60186-1-Ig | Mouse | 1:5000 |
| GAPDH | Servicebio | GB15004 | Rabbit | 1:1000 |
| Antibodies | Company | Catalog Number | Host | Dilution |
|---|---|---|---|---|
| TFAP2C | Proteintech | 14572-1-AP | Rabbit | 1:400 |
| Bax | Cell Signaling Technology | 60267-1-Ig | Mouse | 1:1000 |
| Vimentin | Proteintech | 10366-1-AP | Rabbit | 1:5000 |
| Antibodies | Company | Catalog Number | Host | Dilution |
|---|---|---|---|---|
| pEGFR | Cell Signaling Technology | #3777T | Rabbit | 1:800 |
| pNF-κB | Cell Signaling Technology | #3033 | Rabbit | 1:800 |
| Bcl-2 | Proteintech | 60178-1-Ig | Mouse | 1:400 |
| Ki67 | Proteintech | 27309-1-AP | Rabbit | 1:200 |
| Vimentin | Proteintech | 10366-1-AP | Rabbit | 1:50 |
| Variable | Groups | Total |
|---|---|---|
| Sex | Male | 16 |
| Female | 6 | |
| Age (years) | ≥60 | 14 |
| <60 | 8 | |
| Tumor size (cm) | <3 | 7 |
| ≥3 | 15 | |
| Multiplicity of tumor | Single | 10 |
| Multiple | 12 | |
| Tumor grade | Low grade | 6 |
| High grade | 16 | |
| Tumor stage | Ta, T1 | 4 |
| T2–T4 | 18 | |
| Lymph nodes | Negative | 17 |
| Positive | 5 | |
| Distant metastasis | Absent | 22 |
| Present | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, J.; Chen, W.; Chen, K.; Zhu, S.; Lin, F.; Qi, Y.; Zhang, Y.; Han, S.; Rao, T.; Ruan, Y.; et al. TFAP2C Knockdown Sensitizes Bladder Cancer Cells to Cisplatin Treatment via Regulation of EGFR and NF-κB. Cancers 2022, 14, 4809. https://doi.org/10.3390/cancers14194809
Xing J, Chen W, Chen K, Zhu S, Lin F, Qi Y, Zhang Y, Han S, Rao T, Ruan Y, et al. TFAP2C Knockdown Sensitizes Bladder Cancer Cells to Cisplatin Treatment via Regulation of EGFR and NF-κB. Cancers. 2022; 14(19):4809. https://doi.org/10.3390/cancers14194809
Chicago/Turabian StyleXing, Ji, Wu Chen, Kang Chen, Shaoming Zhu, Fangyou Lin, Yucheng Qi, Yunlong Zhang, Shangting Han, Ting Rao, Yuan Ruan, and et al. 2022. "TFAP2C Knockdown Sensitizes Bladder Cancer Cells to Cisplatin Treatment via Regulation of EGFR and NF-κB" Cancers 14, no. 19: 4809. https://doi.org/10.3390/cancers14194809
APA StyleXing, J., Chen, W., Chen, K., Zhu, S., Lin, F., Qi, Y., Zhang, Y., Han, S., Rao, T., Ruan, Y., Zhao, S., Yu, W., & Cheng, F. (2022). TFAP2C Knockdown Sensitizes Bladder Cancer Cells to Cisplatin Treatment via Regulation of EGFR and NF-κB. Cancers, 14(19), 4809. https://doi.org/10.3390/cancers14194809