A Large Case-Control Study Performed in Spanish Population Suggests That RECQL5 Is the Only RECQ Helicase Involved in Breast Cancer Susceptibility

 , , ,
, , ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. BRCAX Cases
2.2. Controls
2.3. DNA Isolation
2.4. Next-Generation Sequencing
2.5. Bioinformatics Analysis and Variant Filtration

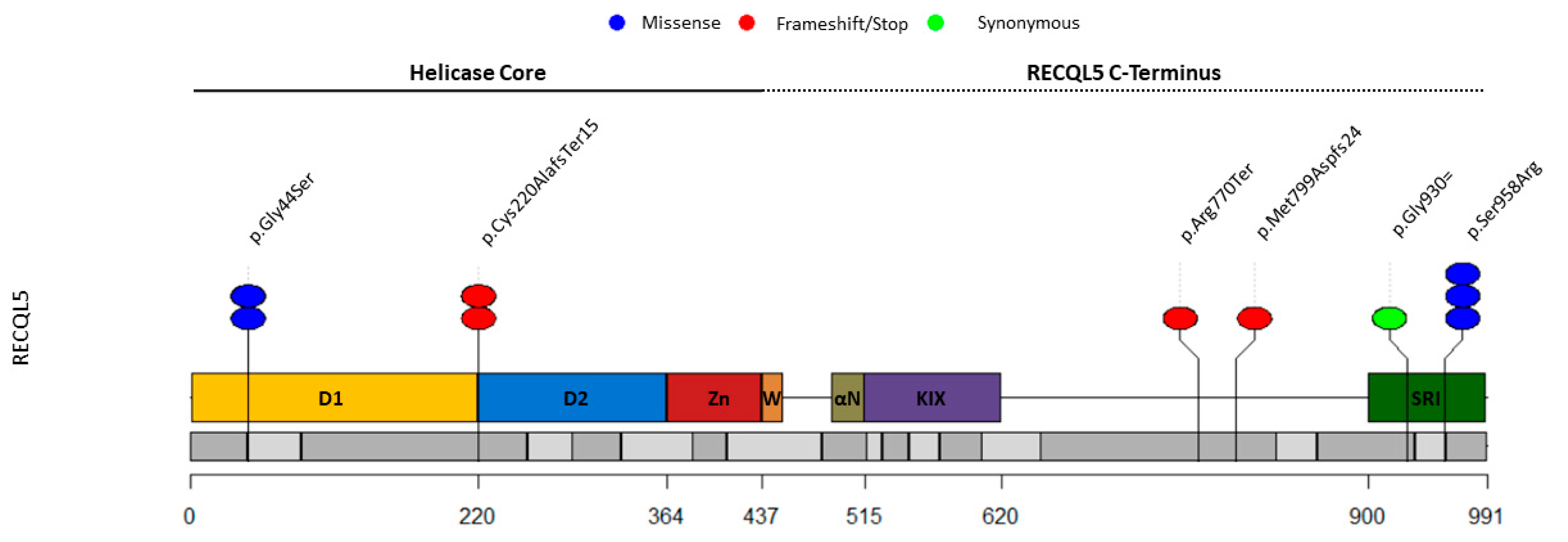{kind=link}
| Gene | Reference | Nucleotide Change a | Protein Change | gnomAD c | CSVS d | Previously Found |
|---|---|---|---|---|---|---|
| RECQL1 | NM_002907.3 | c.84delT | p.Thr29ArgfsTer14 | NR | NR | |
| BLM | NM_000057.2 | c.53_56delCCAG | p.Ala18GlufsTer7 | NR | NR | |
| c.1933C>T | p.Gln645Ter | 0.00008810 | NR | [37] | ||
| WRN | NM_000553.6 | c.205dupA | p.Ile69AsnfsTer2 | NR | NR | |
| c.979G>T | p.Gly327Ter | NR | NR | |||
| c.2604G>A | p.Trp868Ter | NR | NR | |||
| c.4013del | p.Leu1338* | NR | NR | |||
| c.4117_4120dupAGAT | p.Cys1374Ter | NR | NR | |||
| RECQL4 | NM_004260.4 | c.320delA | p.Gln107ArgfsTer7 | NR | NR | |
| c.447dupC | p.Ser150Leufs*8 | NR | NR | |||
| c.1048_1049delAG b | p.Arg350GlyfsTer21 | NR | NR | |||
| c.2161C>T | p.Arg721Ter | 0.00002179 | 1/2093 | [38] | ||
| c.2269C>T | p.Gln757Ter | 0.0001494 | 1/2093 | [38] | ||
| c.2547_2548del b | p.(Phe850Profs*33) | NR | 1/2093 | [39] | ||
| c.3217del | p.(Thr1073Profs*8) | NR | NR |
| Gene | Reference | Nucleotide Change | Protein Change | Phenotype b | gnomAD | CSVS |
|---|---|---|---|---|---|---|
| RECQL5 | NM_004259.6 | c.130G>A a | p.Gly44Ser | BC, 49 years | NR | NR |
| c.657delC a | p.Cys220AlafsTer15 | BC, 34 years | 0.00005270 | 1/2037 | ||
| c.2308C>T | p.Arg770Ter | BC, 44 years | 0.00003567 | 2/2037 | ||
| c.2790C>T | p.(Lys931Serfs*14) | BC, 39 years | NR | NR | ||
| c.2874C>G a | p.Ser958Arg | BC, 48 years | 0.0001085 | 2/2037 | ||
| c.2874C>G | p.Ser958Arg | BC, 78 years | 0.0001085 | 2/2037 |
2.6. Case-Control Association Study
2.7. Combined Analysis
2.8. Splicing Studies
3. Results
3.1. RECQL1, BLM, WRN, and RECQL4 (Likely) Pathogenic Variants Are Not Associated with BC in Spanish BRCAX Cases
3.2. Analysis of RECQL5 (Likely) LoF Variants in 1993 BC-Only BRCAX Cases Shows a Tendency as a Moderate-Risk Gene Model
3.3. Selection of Potentially Damaging Missense and Synonymous Variants in the RECQ Helicases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fahad Ullah, M. Breast Cancer: Current Perspectives on the Disease Status. Adv. Exp. Med. Biol. 2019, 1152, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.P.; Menes, T.; Sonnenblick, A. Comparison of Patient Susceptibility Genes across Breast Cancer: Implications for Prognosis and Therapeutic Outcomes. Pharmgenom. Pers. Med. 2020, 13, 227–238. [Google Scholar] [CrossRef]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Dalivandan, S.T.; Plummer, J.; Gayther, S.A. Risks and Function of Breast Cancer Susceptibility Alleles. Cancers 2021, 13, 3953. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.R.; Bilgili, E.P.; Merner, N.D. A Review of Whole-Exome Sequencing Efforts Toward Hereditary Breast Cancer Susceptibility Gene Discovery. Hum. Mutat. 2016, 37, 835–846. [Google Scholar] [CrossRef]
- Sokolenko, A.P.; Suspitsin, E.N.; Kuligina, E.S.; Bizin, I.V.; Frishman, D.; Imyanitov, E.N. Identification of Novel Hereditary Cancer Genes by Whole Exome Sequencing. Cancer Lett. 2015, 369, 274–288. [Google Scholar] [CrossRef]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s Syndrome Gene Product Is Homologous to RecQ Helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of Rare Cancers in Werner Syndrome (Adult Progeria). Cancer Epidemiol. Biomark. Prev. 1996, 5, 239–246. [Google Scholar]
- Lindor, N.M.; Furuichi, Y.; Kitao, S.; Shimamoto, A.; Arndt, C.; Jalal, S. Rothmund-Thomson Syndrome Due to RECQ4 Helicase Mutations: Report and Clinical and Molecular Comparisons with Bloom Syndrome and Werner Syndrome. Am. J. Med. Genet. 2000, 90, 223–228. [Google Scholar] [CrossRef]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef]
- Cybulski, C.; Carrot-Zhang, J.; Klulniak, W.; Rivera, B.; Kashyap, A.; Wokolorczyk, D.; Giroux, S.; Nadaf, J.; Hamel, N.; Zhang, S.; et al. Germline RECQL Mutations Are Associated with Breast Cancer Susceptibility. Nat. Genet. 2015, 47, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, Y.; Xia, Y.; Xu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Mutations in RECQL Gene Are Associated with Predisposition to Breast Cancer. PLoS Genet. 2015, 11, e1005228. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Dumont, M.; Weber-Lassalle, N.; Joly-Beauparlant, C.; Ernst, C.; Droit, A.; Feng, B.J.; Dubois, S.; Collin-Deschesnes, A.C.; Soucy, P.; Vallée, M.; et al. Uncovering the Contribution of Moderate-Penetrance Susceptibility Genes to Breast Cancer by Whole-Exome Sequencing and Targeted Enrichment Sequencing of Candidate Genes in Women of European Ancestry. Cancers 2022, 14, 3363. [Google Scholar] [CrossRef] [PubMed]
- Bowden, A.R.; Tischkowitz, M. Clinical Implications of Germline Mutations in Breast Cancer Genes: RECQL. Breast Cancer Res. Treat. 2019, 174, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Abu-Libdeh, B.; Jhujh, S.S.; Dhar, S.; Sommers, J.A.; Datta, A.; Longo, G.M.C.; Grange, L.J.; Reynolds, J.J.; Cooke, S.L.; McNee, G.S.; et al. RECON Syndrome Is a Genome Instability Disorder Caused by Mutations in the DNA Helicase RECQL1. J. Clin. Investig. 2022, 132, e147301. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, A.P.; Iyevleva, A.G.; Preobrazhenskaya, E.V.; Mitiushkina, N.V.; Abysheva, S.N.; Suspitsin, E.N.; Kuligina, E.S.; Gorodnova, T.V.; Pfeifer, W.; Togo, A.V.; et al. High Prevalence and Breast Cancer Predisposing Role of the BLM c.1642 C>T (Q548X) Mutation in Russia. Int. J. Cancer 2012, 130, 2867–2873. [Google Scholar] [CrossRef]
- Prokofyeva, D.; Bogdanova, N.; Dubrowinskaja, N.; Bermisheva, M.; Takhirova, Z.; Antonenkova, N.; Turmanov, N.; Datsyuk, I.; Gantsev, S.; Christiansen, H.; et al. Nonsense Mutation p.Q548X in BLM, the Gene Mutated in Bloom’s Syndrome, Is Associated with Breast Cancer in Slavic Populations. Breast Cancer Res. Treat. 2013, 137, 533–539. [Google Scholar] [CrossRef]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome Instability Syndromes. Nat. Rev. Dis. Prim. 2019, 5, 64. [Google Scholar] [CrossRef]
- Anisimenko, M.S.; Kozyakov, A.E.; Paul, G.A.; Kovalenko, S.P. The Frequency of the BLM p.Q548X (c.1642C>T) Mutation in Breast Cancer Patients from Russia Is No Higher than in the General Population. Breast Cancer Res. Treat. 2014, 148, 689–690. [Google Scholar] [CrossRef]
- Sassi, A.; Popielarski, M.; Synowiec, E.; Morawiec, Z.; Wozniak, K. BLM and RAD51 Genes Polymorphism and Susceptibility to Breast Cancer. Pathol. Oncol. Res. 2013, 19, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Maksimenko, J.; Irmejs, A.; Trofimovičs, G.; Berziņa, D.; Skuja, E.; Purkalne, G.; Miklaševičs, E.; Gardovskis, J. High Frequency of Pathogenic Non-Founder Germline Mutations in BRCA1 and BRCA2 in Families with Breast and Ovarian Cancer in a Founder Population. Hered. Cancer Clin. Pract. 2018, 16, 12. [Google Scholar] [CrossRef]
- Kluźniak, W.; Wokołorczyk, D.; Rusak, B.; Huzarski, T.; Kashyap, A.; Stempa, K.; Rudnicka, H.; Jakubowska, A.; Szwiec, M.; Morawska, S.; et al. Inherited Variants in BLM and the Risk and Clinical Characteristics of Breast Cancer. Cancers 2019, 11, 1548. [Google Scholar] [CrossRef]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Oshima, J. Werner Syndrome. J. Biomed. Biotechnol. 2002, 2002, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ellingson, M.S.; Hart, S.N.; Kalari, K.R.; Suman, V.; Schahl, K.A.; Dockter, T.J.; Felten, S.J.; Sinnwell, J.P.; Thompson, K.J.; Tang, X.; et al. Exome Sequencing Reveals Frequent Deleterious Germline Variants in Cancer Susceptibility Genes in Women with Invasive Breast Cancer Undergoing Neoadjuvant Chemotherapy. Breast Cancer Res. Treat. 2015, 153, 435–443. [Google Scholar] [CrossRef]
- Oliver, J.; Quezada Urban, R.; Franco Cortés, C.A.; Díaz Velásquez, C.E.; Montealegre Paez, A.L.; Pacheco-Orozco, R.A.; Castro Rojas, C.; García-Robles, R.; López Rivera, J.J.; Gaitán Chaparro, S.; et al. Latin American Study of Hereditary Breast and Ovarian Cancer LACAM: A Genomic Epidemiology Approach. Front. Oncol. 2019, 9, 1429. [Google Scholar] [CrossRef]
- Lu, L.; Jin, W.; Wang, L.L. RECQ DNA Helicases and Osteosarcoma. Adv. Exp. Med. Biol. 2020, 1258, 37–54. [Google Scholar] [CrossRef]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-Gene Panel Analysis in a Case Series of 255 Women with Hereditary Breast and Ovarian Cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef]
- Bonache, S.; Esteban, I.; Moles-Fernández, A.; Tenés, A.; Duran-Lozano, L.; Montalban, G.; Bach, V.; Carrasco, E.; Gadea, N.; López-Fernández, A.; et al. Multigene Panel Testing beyond BRCA1/2 in Breast/Ovarian Cancer Spanish Families and Clinical Actionability of Findings. J. Cancer Res. Clin. Oncol. 2018, 144, 2495–2513. [Google Scholar] [CrossRef]
- Tavera-Tapia, A.; de la Hoya, M.; Calvete, O.; Martin-Gimeno, P.; Fernández, V.; Macías, J.A.; Alonso, B.; Pombo, L.; de Diego, C.; Alonso, R.; et al. RECQL5: Another DNA Helicase Potentially Involved in Hereditary Breast Cancer Susceptibility. Hum. Mutat. 2019, 40, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Benito-Sánchez, B.; Barroso, A.; Fernández, V.; Mercadillo, F.; Núñez-Torres, R.; Pita, G.; Pombo, L.; Morales-Chamorro, R.; Cano-Cano, J.M.; Urioste, M.; et al. Apparent Regional Differences in the Spectrum of BARD1 Pathogenic Variants in Spanish Population and Importance of Copy Number Variants. Sci. Rep. 2022, 12, 8547. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Peña-Chilet, M.; Roldán, G.; Perez-Florido, J.; Ortuño, F.M.; Carmona, R.; Aquino, V.; Lopez-Lopez, D.; Loucera, C.; Fernandez-Rueda, J.L.; Gallego, A.; et al. CSVS, a Crowdsourcing Database of the Spanish Population Genetic Variability. Nucleic Acids Res. 2021, 49, D1130–D1137. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Gayarre, J.; Martín-Gimeno, P.; Osorio, A.; Paumard, B.; Barroso, A.; Fernández, V.; De La Hoya, M.; Rojo, A.; Caldés, T.; Palacios, J.; et al. Characterisation of the Novel Deleterious RAD51C p.Arg312Trp Variant and Prioritisation Criteria for Functional Analysis of RAD51C Missense Changes. Br. J. Cancer 2017, 117, 1048–1062. [Google Scholar] [CrossRef]
- Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Rowley, S.M.; Choong, D.Y.H.; Tothill, R.W.; Thorne, H.; Barnes, D.R.; Li, J.; Ellul, J.; et al. Exome Sequencing Identifies Rare Deleterious Mutations in DNA Repair Genes FANCC and BLM as Potential Breast Cancer Susceptibility Alleles. PLoS Genet. 2012, 8, e1002894. [Google Scholar] [CrossRef]
- Cao, F.; Lu, L.; Abrams, S.A.; Hawthorne, K.M.; Tam, A.; Jin, W.; Dawson, B.; Shypailo, R.; Liu, H.; Lee, B.; et al. Generalized Metabolic Bone Disease and Fracture Risk in Rothmund-Thomson Syndrome. Hum. Mol. Genet. 2017, 26, 3046–3055. [Google Scholar] [CrossRef]
- Siitonen, A.H.; Sotkasiira, J.; Biervliet, M.; Benmansour, A.; Capri, Y.; Cormier-Daire, V.; Crandall, B.; Hannula-Jouppi, K.; Hennekam, R.; Herzog, D.; et al. The Mutation Spectrum in RECQL4 Diseases. Eur. J. Hum. Genet. 2009, 17, 151–158. [Google Scholar] [CrossRef]
- Newman, J.A.; Aitkenhead, H.; Savitsky, P.; Gileadi, O. Insights into the RecQ Helicase Mechanism Revealed by the Structure of the Helicase Domain of Human RECQL5. Nucleic Acids Res. 2017, 45, 4231–4243. [Google Scholar] [CrossRef]
- Shahi, R.B.; De Brakeleer, S.; Caljon, B.; Pauwels, I.; Bonduelle, M.; Joris, S.; Fontaine, C.; Vanhoeij, M.; Van Dooren, S.; Teugels, E.; et al. Identification of Candidate Cancer Predisposing Variants by Performing Whole-Exome Sequencing on Index Patients from BRCA1 and BRCA2-Negative Breast Cancer Families. BMC Cancer 2019, 19, 313. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, N.; Pfeifer, K.; Schürmann, P.; Antonenkova, N.; Siggelkow, W.; Christiansen, H.; Hillemanns, P.; Park-Simon, T.W.; Dörk, T. Analysis of a RECQL Splicing Mutation, c.1667_1667+3delAGTA, in Breast Cancer Patients and Controls from Central Europe. Fam. Cancer 2017, 16, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Rofes, P.; Del Valle, J.; Torres-Esquius, S.; Feliubadaló, L.; Stradella, A.; Moreno-Cabrera, J.M.; López-Doriga, A.; Munté, E.; De Cid, R.; Campos, O.; et al. Bard1 Pathogenic Variants Are Associated with Triple-Negative Breast Cancer in a Spanish Hereditary Breast and Ovarian Cancer Cohort. Genes 2021, 12, 150. [Google Scholar] [CrossRef] [PubMed]
- Diez, O.; Gutiérrez-Enríquez, S.; Balmaña, J. Heterogeneous Prevalence of Recurrent BRCA1 and BRCA2 Mutations in Spain According to the Geographical Area: Implications for Genetic Testing. Fam. Cancer 2010, 9, 187–191. [Google Scholar] [CrossRef]
- Díez, O.; Osorio, A.; Durán, M.; Martinez-Ferrandis, J.I.; de la Hoya, M.; Salazar, R.; Vega, A.; Campos, B.; Rodríguez-López, R.; Velasco, E.; et al. Analysis of BRCA1 and BRCA2 Genes in Spanish Breast/Ovarian Cancer Patients: A High Proportion of Mutations Unique to Spain and Evidence of Founder Effects. Hum. Mutat. 2003, 22, 301–312. [Google Scholar] [CrossRef]

| Gene | Cases-Heterozygotes/Non-Heterozygotes | Controls-Heterozygotes/Non-Heterozygotes a | Odds Ratio | p-Value | Confidence Interval |
|---|---|---|---|---|---|
| RECQL1 | 1/1992 | 132/51,061 b | 0.20 | 0.097 | 0.01–1.14 |
| BLM | 2/1991 | 125/51,199 | 0.42 | 0.338 | 0.05–1.55 |
| WRN | 5/1988 | 113/51,180 | 1.15 | 0.628 | 0.37–2.77 |
| RECQL4 | 9/1984 | 209/50,447 | 1.10 | 0.721 | 0.50–2.13 |
| RECQL5 | 6/1987 | 74/50,883 | 2.07 | 0.127 | 0.74–4.74 |
| RECQL5c | 10/2683 | 74/50,883 | 2.56 | 0.009 | 1.18–4.98 |
| Gene | Reference | Nucleotide Change | Protein Change | Phenotype | gnomAD | CSVS |
|---|---|---|---|---|---|---|
| RECQL5 | NM_004259.6 | c.130G>A | p.Gly44Ser | BC, 50 years | NR | NR |
| c.657delC | p.Cys220AlafsTer15 | BiBC, 34 years, 46 years | 0.00005270 | 1/2037 | ||
| c.2393dupC | p.Met799Aspfs*24 | BiBC, 37 years, 39 years | NR | NR | ||
| c.2874C>G | p.Ser958Arg | BC, 26 years | 0.0001085 | 2/2037 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchena-Perea, E.M.; Salazar-Hidalgo, M.E.; Gómez-Sanz, A.; Arranz-Ledo, M.; Barroso, A.; Fernández, V.; Tejera-Pérez, H.; Pita, G.; Núñez-Torres, R.; Pombo, L.; et al. A Large Case-Control Study Performed in Spanish Population Suggests That RECQL5 Is the Only RECQ Helicase Involved in Breast Cancer Susceptibility. Cancers 2022, 14, 4738. https://doi.org/10.3390/cancers14194738
Marchena-Perea EM, Salazar-Hidalgo ME, Gómez-Sanz A, Arranz-Ledo M, Barroso A, Fernández V, Tejera-Pérez H, Pita G, Núñez-Torres R, Pombo L, et al. A Large Case-Control Study Performed in Spanish Population Suggests That RECQL5 Is the Only RECQ Helicase Involved in Breast Cancer Susceptibility. Cancers. 2022; 14(19):4738. https://doi.org/10.3390/cancers14194738
Chicago/Turabian StyleMarchena-Perea, Erik Michel, Milton Eduardo Salazar-Hidalgo, Alicia Gómez-Sanz, Mónica Arranz-Ledo, Alicia Barroso, Victoria Fernández, Hugo Tejera-Pérez, Guillermo Pita, Rocío Núñez-Torres, Luz Pombo, and et al. 2022. "A Large Case-Control Study Performed in Spanish Population Suggests That RECQL5 Is the Only RECQ Helicase Involved in Breast Cancer Susceptibility" Cancers 14, no. 19: 4738. https://doi.org/10.3390/cancers14194738
APA StyleMarchena-Perea, E. M., Salazar-Hidalgo, M. E., Gómez-Sanz, A., Arranz-Ledo, M., Barroso, A., Fernández, V., Tejera-Pérez, H., Pita, G., Núñez-Torres, R., Pombo, L., Morales-Chamorro, R., Cano-Cano, J. M., Soriano, M. d. C., Garre, P., Durán, M., Currás-Freixes, M., de la Hoya, M., & Osorio, A. (2022). A Large Case-Control Study Performed in Spanish Population Suggests That RECQL5 Is the Only RECQ Helicase Involved in Breast Cancer Susceptibility. Cancers, 14(19), 4738. https://doi.org/10.3390/cancers14194738